

Abstract
The genomes of six Listeria bacteriophages were sequenced and analyzed. Phages A006, A500, B025, P35, and P40 are members of the Siphoviridae and contain double-stranded DNA genomes of between 35.6 kb and 42.7 kb. Phage B054 is a unique myovirus and features a 48.2-kb genome. Phage B025 features 3′ overlapping single-stranded genome ends, whereas the other viruses contain collections of terminally redundant, circularly permuted DNA molecules. Phages P35 and P40 have a broad host range and lack lysogeny functions, correlating with their virulent lifestyle. Phages A500, A006, and B025 integrate into bacterial tRNA genes, whereas B054 targets the 3′ end of translation elongation factor gene tsf. This is the first reported case of phage integration into such an evolutionarily conserved genetic element. Peptide fingerprinting of viral proteins revealed that both A118 and A500 utilize +1 and −1 programmed translational frameshifting for generating major capsid and tail shaft proteins with C termini of different lengths. In both cases, the unusual +1 frameshift at the 3′ ends of the tsh coding sequences is induced by overlapping proline codons and cis-acting shifty stops. Although Listeria phage genomes feature a conserved organization, they also show extensive mosaicism within the genome building blocks. Of particular interest is B025, which harbors a collection of modules and sequences with relatedness not only to other Listeria phages but also to viruses infecting other members of the Firmicutes. In conclusion, our results yield insights into the composition and diversity of Listeria phages and provide new information on their function, genome adaptation, and evolution.
The opportunistic pathogen Listeria monocytogenes is ubiquitous in nature and can become endemic in food processing environments, causing contamination of dairy products, meats, vegetables, and processed ready-to-eat food (14). L. monocytogenes is the causative agent of epidemic and sporadic listeriosis. The risk of infection is markedly increased among immunocompromised patients, newborns, pregnant women, and the elderly and is associated with a mortality rate of about 20 to 30% (37).
Although all strains of L. monocytogenes are considered potentially pathogenic, epidemiological evidence has shown that certain serovars are more frequently associated with both sporadic cases and larger food-borne outbreaks. However, genetic variation within the virulence genes of wild-type strains appears to be limited and could not be directly linked to differences in pathogenicity (30) or environmental distribution.
It is becoming increasingly clear that bacteriophages have an important role in bacterial biology, diversity, and evolution, as indicated by the advances in genome sequencing which revealed a high incidence of phage-related sequences in bacterial genomes. Many phages have been described for the genus Listeria, and lysogeny appears to be widespread (28). Availability of the genome sequences of different L. monocytogenes and L. innocua strains also revealed the presence of several different putative prophages in the genomes of these bacteria, constituting up to 7% of the coding capacities of the genomes (15, 32). Although the genomes of L. monocytogenes were found to be essentially syntenic, a significant portion of sequence variability is apparently based upon phage insertions and subsequent rearrangements. Investigations of prophage contributions to population dynamics in Salmonella suggest that prophages can improve the competitive fitness of the lysogenized host strains (5). This type of selective pressure also results in diversification and generation of new strains by lysogenic conversion. In the case of Listeria, however, the potential influence of prophages on their host strains, such as phenotypic variation or provision of selective benefits, has not been investigated. To gain more insight in bacteriophage-host interactions and the molecular biology and characteristics of Listeria phages, more information on the structure, information content, and variability of different Listeria phage genomes is required.
Although a number of Listeria phages have been isolated and described (25, 27, 42), only limited information was available at the sequence level for phages PSA, A118, A511, and P100 (9, 19, 26, 41). As the result of a comprehensive study to determine the diversity of this group of bacterial viruses, we here report the complete nucleotide sequences of a representative set of six different Listeria phages from the Siphoviridae (A006, A500, B025, P35, and P40) and Myoviridae (B054) families in the order Caudovirales. In addition to molecular and in silico analyses, we also determined the physical genome structures and attachment loci of the temperate phages, and we describe integration of the B054 prophage into a highly conserved elongation factor gene. Another interesting finding is the unusual decoding used by some of the phages, which use programmed frameshifting to generate C-terminally modified structural proteins required for assembly of the capsid and tail.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Listeria monocytogenes strains WSLC 1001 (serovar 1/2c) and WSLC 1042 (serovar 4b) and Listeria ivanovii WSLC 3009 (serovar 5) were used for phage propagation and as lysogenization hosts. Bacteria were cultivated at 30°C in tryptose medium supplemented with 1.25 mM CaCl2 (Merck, Darmstadt, Germany), or in brain heart infusion medium (Biolife, Milan, Italy). Escherichia coli strains DH5αMCR, XL1-Blue MRF′, and TOP10 (Invitrogen), used in recombinant DNA work, were grown in Luria-Bertani (LB) medium at 37°C. For selection of plasmid-bearing cells and blue-white screening, ampicillin and X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) were added to final concentrations of 100 μg/ml and 50 μg/ml, respectively.
Phage propagation and purification and DNA isolation.
Large-scale propagations to prepare high-titer phage stocks were performed using the agar plate method or liquid culture method as described earlier (1, 24, 29). Infections with P35 were performed on agar plates containing 5 mM CaCl2, and plates were incubated at room temperature due to the temperature sensitivity of this phage (18). The following host strains were used for phage propagation: L. monocytogenes 1001 for infections with A006 and P35; L. monocytogenes 1042 for A500; and L. ivanovii 3009 for B025, B054, and P40. Briefly, phage particles were precipitated from high-titer lysates using 10% polyethylene glycol 8000 in the presence of 1 M NaCl and incubation at 4°C for 16 h and were purified by centrifugation on stepped CsCl gradients (35). Genomic DNA was isolated after treatment with proteinase K (50 μg/ml; 1 h at 50°C) and extraction with phenol, phenol-chloroform, and chloroform using standard protocols (35).
Electron microscopy of phage particles.
Electron micrographs of purified P35 and P40 particles were taken as previously described (19). P35 was negatively stained with 2% phosphotungstic acid, and P40 was stained using 2% uranyl acetate.
Phage induction from lysogens.
Overnight cultures of the lysogens were used to inoculate fresh cultures (1:10, vol/vol) and grown for 2 h at 30°C. Cultures were then transferred to sterile petri dishes and UV irradiated at a wavelength of 254 nm for 60 to 90 s, followed by 2 h of incubation at 30°C in the dark. Bacterial cells and debris were removed by centrifugation (6,000 × g, 10 min, 4°C), and the supernatant was sterile filtered (0.22-μm pore size). Liberated bacteriophages were enumerated by plating and plaque formation on susceptible host strains.
Cloning, sequencing, and bioinformatic analysis.
For the construction of genomic libraries, purified DNAs (approximately 10 μg) of B054, B025, and P40 were partially digested using Tsp509I (New England Biolabs), and the DNAs of A006, A500, and P35 were fragmented using a nebulizer (GATC Biotech, Constance, Germany). Fragments of the desired size (1 to 2.5 kb) were recovered from agarose gels, purified, and shotgun cloned into pBluescript II SK− (Stratagene) predigested with EcoRI or EcoRV as described earlier (26). Plasmids were transformed into E. coli DH5αMCR or E. coli XL-1-Blue MRF′, and plasmid-harboring cells were identified by blue-white screening. The phage DNA inserts of all libraries were sequenced, and the sequences were edited and aligned using the software Vector NTI Advance version 10.3 (Invitrogen) or DNASIS Max version 2.6 (Hitachi). Gaps between the contigs were closed by primer walking using the purified phage DNAs as templates (19). Open reading frames (ORFs) corresponding to a minimum size of 30 amino acids were predicted with Vector NTI or Artemis (http://www.sanger.ac.uk/Software/Artemis) using ATG, GTG, and TTG as possible start codons. The BLAST algorithms used for sequence homology searches are available through NCBI (http://www.ncbi.nlm.nih.gov), Vector NTI, or the HUSAR analysis package (http://genome.dkfz-heidelberg.de). Sequence manipulations and alignments were performed using CLC Main Workbench version 5 (CLC Bio, Aarhus, Denmark). Comparative DotPlots were generated using HUSAR, with a window size of 25 and a word size of 20.
Determination of the physical structure of phage DNA molecules.
Five hundred nanograms of phage DNA was used for each reaction, restriction enzymes were used according to the manufacturer's instructions (New England Biolabs or Fermentas), and digests were analyzed by agarose gel electrophoresis. To test whether single-stranded, cohesive ends (cos) were present in the phage DNA molecule, purified DNA was digested with suitable restriction enzymes and the reaction mixture heated to 60°C for 10 min. Immediately thereafter, fragments were gel separated, and nonheated digests served as the control. For B025, the sequences of the 3′ and 5′ overhanging single-stranded ends were determined by using runoff sequencing primers facing toward the genome ends, as previously described (41).
Determination of attP and attB prophage insertion sites.
Lysogenization of host strains was carried out essentially as described previously (26). Genomic DNAs of the lysogenic strains L. monocytogenes 1001::A006, L. monocytogenes 1042:A500, L. ivanovii 3009::B025, and L. ivanovii 3009::B054 were used for identification of the phage attachment sites via inverse PCR (33). Briefly, the attP location was expected in a noncoding region immediately downstream of the integrase. Restriction endonucleases with recognition sites within approximately 1 kb of the putative location were selected for treatment of the bacterial DNA, and fragments were ligated with T4 ligase. Divergent primers binding to the phage sequence were used for PCR amplification and sequencing of the phage-host junction.
Peptide mass fingerprinting.
Purified phage particles from the CsCl fractions were collected by centrifugation (10°C, 2 h, 76,000 × g), resuspended in 50 to 100 μl pure water, and mixed with an equal volume of sodium dodecyl sulfate (SDS) gel loading buffer prior to heating (95°C, 5 min). Proteins were separated on precast, horizontal, ultrathin SDS-polyacrylamide gels (ExcelGel gradient 8 to 18% polyacrylamide gels; GE Healthcare, Germany), followed by staining with Coomassie blue R-350 (Phastblue R; GE Healthcare). Major bands were excised from the gel and, after tryptic in-gel digestion, analyzed by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MS) to determine the peptide masses of the fragments as described elsewhere (41). Data were analyzed using Mascot (Matrix Science, London, United Kingdom) or Scaffold 2 (Proteome Software Inc., Portland, OR) software. Protein domains were predicted with InterProScan (http://www.ebi.ac.uk/InterProScan).
Nucleotide sequence accession numbers.
Sequences have been deposited in GenBank under the following accession numbers: A006, DQ003642; A500, DQ003637; B054, DQ003640; B025, DQ003639; P35, DQ003641; and P40, EU855793.
RESULTS
General features of Listeria phage genomes.
P35 and P40 belong to a previously unknown new class of small Listeria Siphoviridae, featuring an approximately 57-nm head diameter and 110-nm tail length (Fig. 1, Table 1). The complete genome sequences of six double-stranded DNA Listeria bacteriophages were determined with approximately fourfold redundancy and nucleotide sequences assembled and studied in silico. Of the Siphoviridae, P40 had the smallest genome, of 35.6 kb, followed by P35 (35.8 kb), A006 (38.1 kb), A500 (38.9 kb), and B025 (42.7 kb), whereas the genome of the temperate myovirus B054 was slightly larger (48.2 kb) (Table 1). Restriction maps predicted in silico were in agreement with patterns observed by restriction enzyme analysis, indicating correct assembly of the contigs (Fig. 2). The average G+C contents of 35 to 36 mol% are comparable to those reported for A118 and PSA (26, 41) but are slightly lower than those described for different L. monocytogenes strains (38 to 39 mol%) and L. innocua Clip11262 (37 mol%) (15, 32). Again, P35 and P40 are exceptions with average G+C contents of 40.8 mol% and 39.3 mol%, respectively.
FIG. 1.
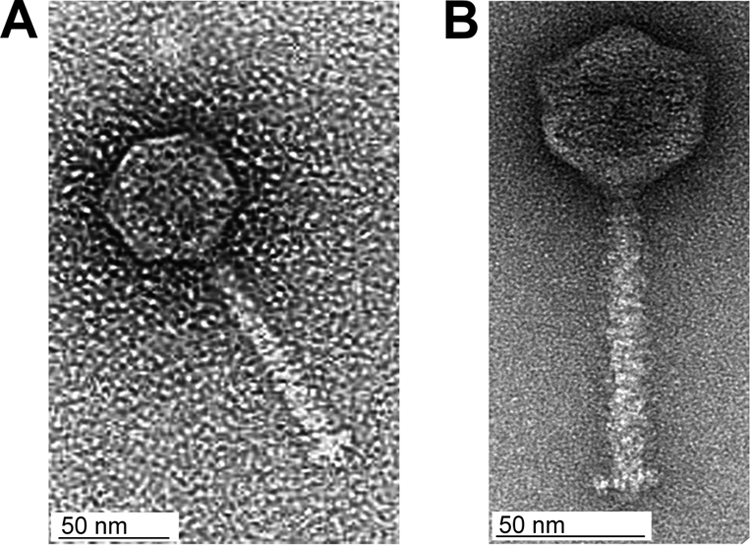
Electron micrographs of P35 (A) and P40 (B).
TABLE 1.
General characteristics and genome features of Listeria bacteriophages
| Bacteriophage | Virus family | Capsid diam (nm) | Tail length (nm) | G+C content (mol %) | Genome size (bp) | Genome structurea | No. of ORFs predicted/no. with function assigned | attB integration site | Lifestyle | Host serovar group(s) | Reference(s) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| B054 | Myoviridae | 64 | 244 | 36.2 | 48,172 | t.r., c.p. | 80/17 | EF-Ts | Temperate | 5, 6 | 23, 42; this study |
| B025 | Siphoviridae | 63 | 252 | 35.1 | 42,653 | cos | 65/17 | tRNAArg | Temperate | 5, 6 | 24, 42; this study |
| PSA | Siphoviridae | 61 | 180 | 34.8 | 37,618 | cos | 57b/18 | tRNAArg | Temperate | 4 | 25, 41 |
| A500 | Siphoviridae | 62 | 274 | 36.7 | 38,867 | t.r., c.p. | 64/23 | tRNALys | Temperate | 4 | 24, 42,; this study |
| A118 | Siphoviridae | 61 | 298 | 36.1 | 40,834 | t.r., c.p. | 72b/27 | comK | Temperate | 1/2 | 23, 26, 42 |
| A006 | Siphoviridae | 62 | 280 | 35.5 | 38,124 | t.r., c.p. | 62/19 | tRNAArg | Temperate | 1/2 | 23, 42; this study |
| P35 | Siphoviridae | 58 | 110 | 40.8 | 35,822 | t.r., c.p. | 56/13 | Virulent (strictly lytic) | 1/2 | 18; this study | |
| P40 | Siphoviridae | 56 | 108 | 39.3 | 35,638 | t.r., c.p. | 62/12 | Virulent (strictly lytic) | 1/2, 4, 5, 6 | This study |
t.r., c.p., terminally redundant, circularly permuted; cos, complementary single-stranded overlapping (cohesive) ends.
Predicted ORFs include those resulting from programmed translational frameshifts.
FIG. 2.

Restriction profiles of phage DNA molecules correlate with sequence assemblies and correspond to the proposed genome structures. Lanes: M, λ HindIII marker; A, ClaI; B, BglII; C, PstI; D, ClaI; E, ClaI; F, BspTI; G, EcoRV; H, PvuII; I, BamHI; K, SphI; L, BspHI; N, PacI.
All but one of the phages feature terminally redundant genomes, which was deduced from the restriction profiles of the phage DNA (Fig. 2) and also agrees with a phylogenetic analysis of the large terminase amino acid sequences (see below). Phage B025 revealed overlapping, single-stranded cohesive genome ends (cos sites), which was shown by typical breakoffs in runoff sequencing and alteration of the restriction pattern in restriction enzyme analysis after heat treatment of the sample (data not shown). The precise sequence of the single-stranded 3′ overhang (cos site) was confirmed by sequencing of closed, ligated ends (5′-CGGTGTGGGG-3′). Table 1 presents a synopsis of the genome features and other characteristics of all Listeria bacteriophages studied so far (except the large myoviruses P100 and A511) (9, 19).
Modular phage genomes.
As expected, the numbers of genes predicted by bioinformatics were approximately proportional to the phage genome sizes, with P35 containing 56 putative ORFs, the smallest number, and B054 having 80 ORFs (Table 1). Predicted ORFs having a minimum of 30 codons and preceded by a recognizable ribosome binding site with the consensus sequence 5′-AGGAGGTG-3′ (15) were considered potential protein-coding sequences. The majority of them initiate translation with an ATG start codon, whereas only a few ORFs per genome feature alternative start codon GTG or TTG. In P35, only two ORFs start with TTG, whereas in P40, TTG is used nine times and GTG occurs four times.
In general, bioinformatics revealed an organization of the genomes into functional modules (Fig. 3) typical for temperate bacteriophages (26, 41). In a standardized genome map, the left-arm, rightward-transcribed cluster represents “late genes” coding for structural proteins, DNA packaging, and the lysis systems. It is followed by a second cluster transcribed mostly in the opposite direction, which encodes the lysogeny functions (integrase, repressor, and similar) and the prophage attachment and integration locus attP. The third module is again transcribed rightwards; it comprises the “early genes” required for the early state of phage reproduction and encodes products for replication, recombination, and modification of the viral DNA (Fig. 3).
FIG. 3.

Relatedness of sequenced Listeria bacteriophages on the level of gene products. Genomes are drawn to approximate scales, and predicted ORFs are displayed as arrows, which also indicate the direction of transcription. The ORFs are numbered consecutively; the detailed results from BLAST homology searches are listed in Tables S4 to S9 in the supplemental material. The different functional modules are shown in different colors, and the genes encoding proteins with significant levels of identity (>25%) are connected by shaded areas. SV, serovar; aa, amino acid.
P35 and P40 lack lysogeny control functions.
Analyses of the comparatively small genomes of P35 and P40 revealed not only their relationship but also that they both lack any lysogeny-related functions (Fig. 3; see Tables S8 and S9 in the supplemental material) and therefore represent true virulent (i.e., obligately lytic) phages. This agrees well with our finding that P35 was unable to lysogenize its host strain and with their rather broad host ranges: P35 can infect most Listeria serovar 1/2 strains, whereas P40 infects the majority of all tested serovar 4, 5, and 6 strains.
Another unusual observation unique among the Listeria phages was that the genes encoding the putative holin and the endolysin (experimentally confirmed [M. Schmelcher et al., unpublished data]) in the P35 and P40 genomes appear to be separated by several other small ORFs with yet-unknown functions.
Comparative genome and sequence analysis shows extensive mosaicism.
Peptide sequences deduced from putative ORFs were screened for similarities with proteins from the databases using the BlastP algorithm (2). The basic characteristics of the predicted gene products, significant database matches, and preliminary functional assignments are summarized in Tables S4 to S9 in the supplemental material. In general, a large proportion of the predicted gene products show no obvious biological function, but the majority of them at least revealed homologies to proteins from the databases.
The genomes of A006 and A500 revealed an overall relatedness to Listeria phage A118, the first published Listeria phage sequence (26). However, the regions of homology comprise different parts of the genomes (Fig. 3). The early gene modules are almost identical in A006 and A118, whereas there are only two structural (late) gene products with significant similarity. The other gene products primarily resemble proteins of prophage-like sequences identified in the genomes of L. monocytogenes (strains H7858, F6854, and EGDe) and L. innocua Clip11262 (15, 32). In contrast, A500 and A118 show high similarity among the structural genes (Fig. 3).
Interestingly, B054 late genes showed significant homology to proteins encoded by Enterococcus faecalis V583 (34), with amino acid identities of up to 57% (see Table S7 in the supplemental material).
Most of the predicted gene products of B054, especially the structural components, show high similarity/identity scores to phage-like sequences which have been identified in the genome of L. innocua Clip11262 (15). In preliminary experiments, using PCR primers specific for the five prophage-related sequences, upon UV induction of putative prophage (results not shown), we were able to demonstrate viability of only one phage, corresponding to Clip11262 locus tags lin1700 to lin1765. The other four prophage-like sequences in the genome of this strain did not respond to the UV induction and may therefore be cryptic.
In contrast to the genomic synteny of B054 and the Clip11262 prophage, the genome of B025 appears to be highly mosaic. Predicted gene products revealed high protein similarities to Listeria phage PSA (and to a lesser extent to A118) and to L. innocua Clip11262 (locus tags lin2585 to lin2610) and other L. monocytogenes genomes. Many of the structural components are also related to proteins of Staphylococcus phages 77 and 3A (20).
The situation is strikingly different for the novel virulent phages P35 and P40. Approximately half of their sequences revealed no significant match to known database entries, and the other gene products revealed weak similarities mostly to E. faecalis phage V583 (34) but also to Clostridium thermocellum, Oenococcus oeni, and Leuconostoc mesenteroides phages. Only two gene products of P35, one of which is the phage endolysin, showed weak similarities to proteins of L. innocua Clip11262.
Pairwise alignments of the phage proteomes (Fig. 3) indicated the best matches between P35 and P40 (26 common gene products with 35 to 61% sequence identity) and between A006 and A500 (15 gene products with 35 to 98% identity). The proteins of the latter two phages represent homologues of A118 and are encoded predominantly by the module of “early” genes. Similarly, homologies between B025 and A006 are restricted to 10 proteins (30 to 100% identity), which are also found in A118, PSA, and Clip11262 prophages.
Such “indirect linkage” or “secondary homology” is common among the Listeria phages investigated. Overarching similarities between A006, A500, B025, and B054 can be observed among gene products with homology to L. innocua and L. monocytogenes prophages and to Listeria phages A118 and PSA. The main capsid protein gp6 of P35 and P40, the terminase gp2 of P40, and the endolysin gp28 of P35 weakly resemble those of A006 and A500.
In conclusion, A006, A118, and A500 form one more closely related group, while PSA and B025 are contained in a separate cluster. These two groups are linked to each other by a few homologous sequences among the late genes. Phages P35 and P40 are clearly different from the above and form a group of their own. This is also evident from a rooted phylogenetic tree based on whole-genome alignments (Fig. 4A), which clearly shows P35 and P40 clustering in a separate branch of the tree, whereas A500, A006, and A118 as well as B025 and PSA seem to belong to the same subgroup. In addition, nucleotide sequence-based DotPlot analyses (see Fig. S1 in the supplemental material) also underline the close relationships between A500 and A118 (see Fig. S1B in the supplemental material) over the full sequence and between B025 and PSA (see Fig. S1D in the supplemental material) and A006 and A118 (see Fig. S1A in the supplemental material) among late genes and indicate a more distant relationship between A006 and A500 (see Fig. S1C in the supplemental material). The same results are obtained by a phylogenetic tree analysis based on a Muscle alignment (13) of the large terminase amino acid sequences of the available Listeria phages (Fig. 4B). Again, the relationships between P35 and P40, A500 and A118, and PSA and B025 are evident. Interestingly, the large terminase sequences reliably indicate not only the packaging strategy of the terminase holoenzyme (10) but also the physical structure of the phage DNA molecule, and the results confirm the DNA packaging mode assumed for the Listeria phages investigated here. The large SPO1-like phage A511 forms a distant branch of the tree, in line with its packaging strategy, which is completely different from that of the other Listeria phages (19).
FIG. 4.

(A) Phylogenetic tree constructed on the basis of the complete phage genome sequences using ClustalW alignment of phage genomes and the unweighted pair group method with arithmetic mean (36) with bootstrap analysis (1,000 replicates). (B) Phylogenetic tree constructed using the neighbor-joining method and bootstrap analysis (1,000 replicates) of a Muscle (13) alignment of large terminase amino acid sequences. cos, cohesive genome ends; cp, tr, circularly permuted, terminally redundant genome ends.
Identification of attP/attB sites.
The phage attachment and integration sites (attP) are located mostly in the short intergenic noncoding regions upstream of the integrase int genes and were identified using PCR-based approaches (41). Phages A006, B025, and B054 revealed integration sites with core sequences of similar length (16 and 17 nucleotides, respectively), whereas the region of homology between attP of A500 and attB of its L. monocytogenes host extends over 45 bp (Fig. 5). Three of the four phages integrate into the 3′ ends of single-copy tRNA genes. Interestingly, the attP sites of B025 and PSA (21) are identical, the predicted integrases of both phages show 95% amino acid identity, and both prophages occupy an identical tRNAArg locus in the genomes of their different L. innocua and L. monocytogenes (21) hosts. In contrast, B054 was found to integrate into the 3′ end of tsf (lin1766; nucleotide positions 1763134 to 1762250 of the L. innocua Clip11262 genome) and maintains its functionality by reconstitution of the terminal six codons including the stop codon by phage sequence. This gene encodes the translation elongation factor EF-Ts and represents a novel and unique phage integration locus not previously reported.
FIG. 5.

Integration sites of four temperate Listeria phages. The nucleotide sequences of the phage attachment sites (attP), the corresponding bacterial attachment sites (attB) in lysogenized host strains, and the corresponding sequences retrieved from sequenced Listeria genomes are aligned. The core sequences of the attachment sites are indicated with light gray boxes. The genome positions (nucleotide coordinates) of the displayed fragment are indicated on the right. The attachment sites of phages A500, A006, and B025 are all located at the 3′ ends of tRNA genes, shown in panels A, B, and C (underlined). The predicted cloverleaf structures of the tRNAs encoded by attB site genes are shown in panels E (A500), F (A006), and G (B025). The anticodons are circled, and attP/attB core sequences are indicated by dotted lines. Phage B054 (D) integrates into the 3′ end of translation elongation factor gene tsf (dashed line).
Programmed translational frameshifting.
Protein profiles of the structural components were mapped by SDS-polyacrylamide gel electrophoresis (Fig. 6), and the identity of individual protein species was analyzed by peptide mass fingerprinting. The most prominent bands generally represent major head (Cps) and major tail (Tsh) protein. In several cases, however, these proteins could be identified in more than a single band, which could suggest posttranslational processing of the polypeptides.
FIG. 6.

Listeria phage structural proteomes and peptide fingerprint identification of protein bands by mass spectrometry. Identified phage proteins are indicated. SDS-polyacrylamide gel electrophoresis molecular mass marker sizes are indicated in kDa. Abbreviations: cps, major capsid protein; tsh, tail sheath protein; tmp, tape measure protein; gp, gene product. Detailed information on these and other gene products is listed in Tables S4 to S9 in the supplemental material.
In A500, both the major capsid protein (Cps) and the major tail protein (Tsh) are represented by two proteins of different size (Fig. 6). A similar situation was found in phage A118 (26, 42). Bioinformatic analysis indicated that in both phages both major protein types are made using programmed translational (ribosomal) frameshifts (−1 in Cps and +1 in Tsh) at the 3′ ends of the implicated genes, which results in the synthesis of a normal-length and a larger polypeptide species (41). The in silico-calculated molecular masses of Cps-L and Tsh-L nicely corresponded to the observed band sizes (Fig. 6) (26, 42). This situation could then be confirmed by peptide mass fingerprinting. Tandem MS (matrix-assisted laser desorption ionization MS/MS and electrospray ionization MS/MS) allowed identification of the exact amino acid sequences of several of the tryptic fragments from Tsh-L and Cps-L of both phages (see Tables S2 and S3 in the supplemental material) and yielded peptides and sequences corresponding to products predicted from −1 or +1 frameshifts (see Tables S2 and S3 in the supplemental material). This enabled us to determine the precise location and mode of the frameshift (Fig. 7). In both cases, the cps frameshift occurs at a locus close to the 3′ end of the transcripts, at the mRNA sequence GCG′GGA′AAC. When it reaches the GGA (glycine) codon, the ribosome apparently slips backwards one nucleotide in the 5′ direction and continues from the overlapping glycine codon GGG in the −1 frame until it reaches the stop codons at positions 6238 in A118 and 6215 in A500. Thus, in both phages, Cps-L contains 53 extra C-terminal amino acids from the alternate reading frame.
FIG. 7.

Programmed translational frameshifts near the 3′ ends of the genes result in synthesis of two different-length products for Cps and Tsh major structural proteins in the two Listeria phages A118 and A500. (A) The −1 frameshift in cps of A118, leading to an extended version of the Cps. (B) The +1 frameshift in the sequence coding for the Tsh of phage A118. (C and D) Similar to A118, the Cps-L product of phage A500 is made as the result of a −1 frameshift, whereas the Tsh-L of phage A500 is produced through a +1 decoding event.
With respect to the tsh mRNA, a slippery sequence, AAA′CCC′UGA, is located at the 3′ end of the gene. In contrast to the −1 frameshift in cps, the ribosome here slips one nucleotide position forward toward the 3′ end and continues translation in the +1 frame ending at positions 8440 (A118) and 8420 (A500). As a result of the alternative decoding, the longer Tsh-L species both have an additional 87 C-terminal amino acids.
DISCUSSION
Genome sequencing and computational analysis of six Listeria bacteriophages revealed basic and important information about the DNA structure, genome organization and layout, phage relatedness, and phage attachment and host insertion sites. Integration of the viral DNA into the host chromosome by site-specific recombination is essential for propagation of the prophage. A006, A500, and B025 integrate into their hosts at the 3′ ends of tRNA genes, which are single-copy genes for specific codons in each case, indicating that they have to be reconstituted to keep them functional. Integration close to the 3′ ends of tRNA genes also has been shown for P4-related phages, whereas others, such as the lambdoid P22, integrate into the anticodon loop (8). The bacterial attachment site of B054 is located at the 3′ end of tsf, a translation elongation factor gene (15). Surprisingly, the attachment sites of B025 (which infects Listeria innocua only) and PSA (which infects Listeria monocytogenes serovar 4b only) (21) were found to be identical. This correlates well with the overall relatedness of the phage genomes (Fig. 3) and the physical structures of their DNA molecules, featuring overlapping, single-stranded, and cohesive ends. The observation that Listeria phages with a nonoverlapping host range show such a close relationship suggest coevolution and adaptation of ancestral phages and bacteria along the divergent development of the two species and along the existing genetic lineages (12, 31, 38).
Site-specific recombination between attB and attP is mediated by the phage integrases, which can be grouped into two major families, the tyrosine recombinases and the serine recombinases (16). InterProScan analyses indicated that the Listeria phage integrases are mostly members of the first family, which utilize a catalytic tyrosine residue to mediate strand cleavage and tend to recognize longer attP sequences. These integrases have similarity to XerC and XerD, tyrosine recombinases that can recognize simple identical sequences of <50 bp. In contrast, A118 features a serine integrase, which can recognize very short regions of identity (26).
We have shown programmed ribosomal frameshifting to occur in A500 and A118 decoding. Both phages utilize +1 as well as −1 programmed translational frameshifting for generation of Cps and Tsh proteins with C termini of different lengths (see Tables S2 and S3 in the supplemental material). Similar to the situation in phage PSA (41), A118 and A500 feature a capsid structure with an assumed T = 7 triangulation number (11). Such capsids contain pentameric and hexameric protein ring-shaped capsomeres (39). The proportions of Cps and Cps-L could be explained by the ratio of these subunits required for building the capsid structures (41), and a similar situation can be assumed for A500 and A118. Frameshifts within Tsh are assumed to play a role in correct assembly of the tail, as was demonstrated for B. subtilis phage SPP1 (3). In bacteriophage λ, a frameshift controls the production of gpG and gpGT, which are also involved in tail assembly (22). Accordingly, variable decoding achieved through translational frameshifting seems to be important for the biological function, since they are widespread among phages and are highly conserved (4, 40).
The overall genome organization into defined functional modules appears to be conserved among the temperate Listeria phages and nicely matches the findings for PSA and A118 (26, 41) (Fig. 3). Similar genomic arrangements have also been established for other phages infecting the Firmicutes (7). A representative example for the frequently observed mosaicism within single gene products is the endolysin (Ply) of B054: the two separate functional domains of the enzyme show similarity to PlyPSA (41) (C terminal) and Clostridium tetani (6) (N terminal), respectively.
P35 and P40 strikingly differ in genome size and organization compared to the other siphoviruses investigated here, due to the complete absence of a lysogeny module, including the attachment site and all functions required for establishment and maintenance of lysogeny. Therefore, both P35 and P40 have a virulent (i.e., obligately lytic) lifestyle. This finding also explains why our previous attempts to use P35 for lysogenization of host cells have always failed (unpublished data). In addition, it has been shown that transduction using P35 is an infrequent event (18). The lack of the lysogeny control functions appears to be advantageous in avoiding homoimmunity suppression, providing a wider host range including the frequent lysogens. This correlates well with the rather broad host range of both P35 and P40 compared to other Listeria phages of this type. On a molecular basis, P35 and P40 also reveal no noticeable homology to the other Listeria phages and therefore form a group of their own (Fig. 3 and 4). One possible explanation would be that these phages originated from a specific lineage in the distant evolutionary past of the Listeriaceae and, due to their strictly virulent lifestyle, only few opportunities exist for recombination with resident prophages or other coinfecting phages.
Phage B054 was classified into the Myoviridae family, featuring a contractile tail. Of particular interest is the rather unusual contraction mechanism of the tail, where along with the conformational change of the sheath, it retracts away from the capsid toward the base plate, thereby exposing part of the inner tail tube just below the neck (42). Reflecting this more complex morphology, the B054 genome contains an extended cluster of structural genes (Fig. 3; see Table S7 in the supplemental material). Database searches indicated a convincing synteny between the genomes of B054 and a prophage resident in L. innocua Clip11262 (nucleotide positions 1715216 to 1762089) (15). We found evidence that this prophage is the only one inducible by UV radiation, whereas the other four prophages in this L. innocua genome probably are nonfunctional. However, the genomes of B054 and L. innocua prophage 7/12 are not identical; 34 putative gene products of B054 show sequence homology to another prophage of L. innocua Clip11262, contained in segment 6/12 (nucleotide positions 1248769 to 1294845).
B025 features single-stranded 3′ overhangs of 10 nucleotides length, which corresponds nicely with cluster alignment of large terminase subunit sequences (Fig. 4B) and also correlates nicely with the mode of DNA packaging in phages (10). About half of the gene products of B025 revealed similarity to products of the other cos end phage PSA; most of them are located in the lysogeny control region and the early genes, encoding the elements required for genome replication (Fig. 3).
Overall, the B025 genome is strikingly mosaic, featuring patches of homology to A118, PSA, B054, L. innocua genomic prophages, and phages infecting Staphylococcus aureus and Streptococcus pyogenes. In this respect, B025 appears to be exemplary for the modular evolution of phage genomes, which presumes DNA exchange between tailed phages by nonhomologous and homologous recombination that frequently occurs among phylogenetically related local groups (17). Staphylococci and listeriae are grouped in the taxon Bacillales. However, we have to consider that, despite all recognizable homologies, Listeria phages are strictly genus specific and the cross-genus sequence homologies are rather low, suggesting that any genetic exchange probably occurred in the distant past.
Considering the information currently available, it seems to be time to revise the previous classification system used for Listeria phages, which was based solely on virus morphology (42). Although a significant correlation between ultrastructure and overall DNA homology exists, it does not always correlate with molecular data. Although this discrepancy has been recognized earlier (27), the lack of complete sequences prevented a better recognition of genetic relationships. However, it will still remain a difficult task to reclassify the (many) known Listeria phages, because not only sequence relatedness and genome structure but also soft criteria such as morphology, lifestyle, and host range may be taken into consideration for the establishment of a revised (and more universal) phage taxonomy.
In conclusion, comparative genomic analysis of Listeria phages revealed the heavily mosaic nature of their genomes. Such gradual relatedness is reflected by more closely related groups such as (i) A006, A500, and A118 and (ii) B025 and PSA. B054 seems more distant and, despite its morphology, does not form a link to the unique A511 group of the SPO1-like phages (9, 19). P35 and P40 are also strikingly different and form a group of their own. The incorporation of more and new sequence data into the classification scheme established here will be required to close the gaps in phylogeny and provide an even better and more complete insight into the variability and evolution of Listeria phages.
Supplementary Material
Acknowledgments
We are grateful to Siegfried Scherer (Technical University Munich, Germany) and the BMBF-funded PathoGenoMik Competence Network (Würzburg, Germany) for financial support to J.D.
We thank Patrick Schiwek and Ursula Schuler for their excellent technical assistance, and we are grateful to the late Ross B. Inman (Madison, WI) for preparing the electron micrograph of P35 and to Rudi Lurz (Berlin, Germany) for providing electron micrographs and morphological data for P40.
Footnotes
Published ahead of print on 25 September 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Adams, M. H. 1959. Bacteriophages. Interscience Publishers Inc., New York, NY.
- 2.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 3.Auzat, I., A. Droge, F. Weise, R. Lurz, and P. Tavares. 2008. Origin and function of the two major tail proteins of bacteriophage SPP1. Mol. Microbiol. 70:557-569. [DOI] [PubMed] [Google Scholar]
- 4.Baranov, P. V., O. Fayet, R. W. Hendrix, and J. F. Atkins. 2006. Recoding in bacteriophages and bacterial IS elements. Trends Genet. 22:174-181. [DOI] [PubMed] [Google Scholar]
- 5.Bossi, L., J. A. Fuentes, G. Mora, and N. Figueroa-Bossi. 2003. Prophage contribution to bacterial population dynamics. J. Bacteriol. 185:6467-6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruggemann, H., S. Baumer, W. F. Fricke, A. Wiezer, H. Liesegang, I. Decker, C. Herzberg, R. Martinez-Arias, R. Merkl, A. Henne, and G. Gottschalk. 2003. The genome sequence of Clostridium tetani, the causative agent of tetanus disease. Proc. Natl. Acad. Sci. USA 100:1316-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brussow, H., and F. Desiere. 2001. Comparative phage genomics and the evolution of Siphoviridae: insights from dairy phages. Mol. Microbiol. 39:213-222. [DOI] [PubMed] [Google Scholar]
- 8.Campbell, A. 2003. The future of bacteriophage biology. Nat. Rev. Genet. 4:471-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlton, R. M., W. H. Noordman, B. Biswas, E. D. de Meester, and M. J. Loessner. 2005. Bacteriophage P100 for control of Listeria monocytogenes in foods: genome sequence, bioinformatic analyses, oral toxicity study, and application. Regul. Toxicol. Pharmacol. 43:301-312. [DOI] [PubMed] [Google Scholar]
- 10.Casjens, S. R., E. B. Gilcrease, D. A. Winn-Stapley, P. Schicklmaier, H. Schmieger, M. L. Pedulla, M. E. Ford, J. M. Houtz, G. F. Hatfull, and R. W. Hendrix. 2005. The generalized transducing Salmonella bacteriophage ES18: complete genome sequence and DNA packaging strategy. J. Bacteriol. 187:1091-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caspar, D. L., and A. Klug. 1962. Physical principles in the construction of regular viruses. Cold Spring Harbor Symp. Quant. Biol. 27:1-24. [DOI] [PubMed] [Google Scholar]
- 12.Doumith, M., C. Cazalet, N. Simoes, L. Frangeul, C. Jacquet, F. Kunst, P. Martin, P. Cossart, P. Glaser, and C. Buchrieser. 2004. New aspects regarding evolution and virulence of Listeria monocytogenes revealed by comparative genomics and DNA arrays. Infect. Immun. 72:1072-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farber, J. M., and P. I. Peterkin. 1991. Listeria monocytogenes, a food-borne pathogen. Microbiol. Rev. 55:476-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glaser, P., L. Frangeul, C. Buchrieser, C. Rusniok, A. Amend, F. Baquero, P. Berche, H. Bloecker, P. Brandt, T. Chakraborty, A. Charbit, F. Chetouani, E. Couve, A. de Daruvar, P. Dehoux, E. Domann, G. Dominguez-Bernal, E. Duchaud, L. Durant, O. Dussurget, K. D. Entian, H. Fsihi, F. Garcia-del Portillo, P. Garrido, L. Gautier, W. Goebel, N. Gomez-Lopez, T. Hain, J. Hauf, D. Jackson, L. M. Jones, U. Kaerst, J. Kreft, M. Kuhn, F. Kunst, G. Kurapkat, E. Madueno, A. Maitournam, J. M. Vicente, E. Ng, H. Nedjari, G. Nordsiek, S. Novella, B. de Pablos, J. C. Perez-Diaz, R. Purcell, B. Remmel, M. Rose, T. Schlueter, N. Simoes, A. Tierrez, J. A. Vazquez-Boland, H. Voss, J. Wehland, and P. Cossart. 2001. Comparative genomics of Listeria species. Science 294:849-852. [DOI] [PubMed] [Google Scholar]
- 16.Groth, A. C., and M. P. Calos. 2004. Phage integrases: biology and applications. J. Mol. Biol. 335:667-678. [DOI] [PubMed] [Google Scholar]
- 17.Hendrix, R. W. 2002. Bacteriophages: evolution of the majority. Theor. Popul. Biol. 61:471-480. [DOI] [PubMed] [Google Scholar]
- 18.Hodgson, D. A. 2000. Generalized transduction of serotype 1/2 and serotype 4b strains of Listeria monocytogenes. Mol. Microbiol. 35:312-323. [DOI] [PubMed] [Google Scholar]
- 19.Klumpp, J., J. Dorscht, R. Lurz, R. Bielmann, M. Wieland, M. Zimmer, R. Calendar, and M. J. Loessner. 2008. The terminally redundant, nonpermuted genome of Listeria bacteriophage A511: a model for the SPO1-like myoviruses of gram-positive bacteria. J. Bacteriol. 190:5753-5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwan, T., J. Liu, M. DuBow, P. Gros, and J. Pelletier. 2005. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 102:5174-5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lauer, P., M. Y. Chow, M. J. Loessner, D. A. Portnoy, and R. Calendar. 2002. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol. 184:4177-4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin, M. E., R. W. Hendrix, and S. R. Casjens. 1993. A programmed translational frameshift is required for the synthesis of a bacteriophage lambda tail assembly protein. J. Mol. Biol. 234:124-139. [DOI] [PubMed] [Google Scholar]
- 23.Loessner, M. J. 1991. Improved procedure for bacteriophage typing of Listeria strains and evaluation of new phages. Appl. Environ. Microbiol. 57:882-884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loessner, M. J., and M. Busse. 1990. Bacteriophage typing of Listeria species. Appl. Environ. Microbiol. 56:1912-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loessner, M. J., L. A. Estela, R. Zink, and S. Scherer. 1994. Taxonomical classification of 20 newly isolated Listeria bacteriophages by electron microscopy and protein analysis. Intervirology 37:31-35. [DOI] [PubMed] [Google Scholar]
- 26.Loessner, M. J., R. B. Inman, P. Lauer, and R. Calendar. 2000. Complete nucleotide sequence, molecular analysis and genome structure of bacteriophage A118 of Listeria monocytogenes: implications for phage evolution. Mol. Microbiol. 35:324-340. [DOI] [PubMed] [Google Scholar]
- 27.Loessner, M. J., I. B. Krause, T. Henle, and S. Scherer. 1994. Structural proteins and DNA characteristics of 14 Listeria typing bacteriophages. J. Gen. Virol. 75:701-710. [DOI] [PubMed] [Google Scholar]
- 28.Loessner, M. J., and C. E. Rees. 2005. Listeria phages: basics and applications, p. 362-379. In M. K. Waldor, D. I. Friedman, and L. A. Sankar (ed.), Phages: their role in bacterial pathogenesis and biotechnology. ASM Press, Washington, DC.
- 29.Loessner, M. J., C. E. Rees, G. S. Stewart, and S. Scherer. 1996. Construction of luciferase reporter bacteriophage A511::luxAB for rapid and sensitive detection of viable Listeria cells. Appl. Environ. Microbiol. 62:1133-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLauchlin, J., R. T. Mitchell, W. J. Smerdon, and K. Jewell. 2004. Listeria monocytogenes and listeriosis: a review of hazard characterisation for use in microbiological risk assessment of foods. Int. J. Food Microbiol. 92:15-33. [DOI] [PubMed] [Google Scholar]
- 31.Nadon, C. A., D. L. Woodward, C. Young, F. G. Rodgers, and M. Wiedmann. 2001. Correlations between molecular subtyping and serotyping of Listeria monocytogenes. J. Clin. Microbiol. 39:2704-2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson, K. E., D. E. Fouts, E. F. Mongodin, J. Ravel, R. T. DeBoy, J. F. Kolonay, D. A. Rasko, S. V. Angiuoli, S. R. Gill, I. T. Paulsen, J. Peterson, O. White, W. C. Nelson, W. Nierman, M. J. Beanan, L. M. Brinkac, S. C. Daugherty, R. J. Dodson, A. S. Durkin, R. Madupu, D. H. Haft, J. Selengut, S. Van Aken, H. Khouri, N. Fedorova, H. Forberger, B. Tran, S. Kathariou, L. D. Wonderling, G. A. Uhlich, D. O. Bayles, J. B. Luchansky, and C. M. Fraser. 2004. Whole genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen Listeria monocytogenes reveal new insights into the core genome components of this species. Nucleic Acids Res. 32:2386-2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ochman, H., A. S. Gerber, and D. L. Hartl. 1988. Genetic applications of an inverse polymerase chain reaction. Genetics 120:621-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulsen, I. T., L. Banerjei, G. S. Myers, K. E. Nelson, R. Seshadri, T. D. Read, D. E. Fouts, J. A. Eisen, S. R. Gill, J. F. Heidelberg, H. Tettelin, R. J. Dodson, L. Umayam, L. Brinkac, M. Beanan, S. Daugherty, R. T. DeBoy, S. Durkin, J. Kolonay, R. Madupu, W. Nelson, J. Vamathevan, B. Tran, J. Upton, T. Hansen, J. Shetty, H. Khouri, T. Utterback, D. Radune, K. A. Ketchum, B. A. Dougherty, and C. M. Fraser. 2003. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071-2074. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook, J., and D. W. Russell. 2001. Molecular cloning, 3rd ed., vol. 1 to 3. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 36.Sokal, R. R., and C. D. Mitchener. 1958. A statistical method for evaluating systematic relationships. Univ. Kansas Sci. Bull. 38:1409-1438. [Google Scholar]
- 37.Vazquez-Boland, J. A., M. Kuhn, P. Berche, T. Chakraborty, G. Dominguez-Bernal, W. Goebel, B. Gonzalez-Zorn, J. Wehland, and J. Kreft. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14:584-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ward, T. J., L. Gorski, M. K. Borucki, R. E. Mandrell, J. Hutchins, and K. Pupedis. 2004. Intraspecific phylogeny and lineage group identification based on the prfA virulence gene cluster of Listeria monocytogenes. J. Bacteriol. 186:4994-5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wikoff, W. R., L. Liljas, R. L. Duda, H. Tsuruta, R. W. Hendrix, and J. E. Johnson. 2000. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289:2129-2133. [DOI] [PubMed] [Google Scholar]
- 40.Xu, J., R. W. Hendrix, and R. L. Duda. 2004. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol. Cell 16:11-21. [DOI] [PubMed] [Google Scholar]
- 41.Zimmer, M., E. Sattelberger, R. B. Inman, R. Calendar, and M. J. Loessner. 2003. Genome and proteome of Listeria monocytogenes phage PSA: an unusual case for programmed + 1 translational frameshifting in structural protein synthesis. Mol. Microbiol. 50:303-317. [DOI] [PubMed] [Google Scholar]
- 42.Zink, R., and M. J. Loessner. 1992. Classification of virulent and temperate bacteriophages of Listeria spp. on the basis of morphology and protein analysis. Appl. Environ. Microbiol. 58:296-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.