

Abstract
Mycobacterium tuberculosis PhoP of the PhoP-PhoR two-component signaling system orchestrates a complex transcription program and is essential for the growth and virulence of the tubercle bacillus. PhoP comprises a phosphorylation domain at the amino-terminal half and a DNA-binding domain in the carboxy-terminal half of the protein. We show here that the protein recognizes a 23-bp sequence of the phoP upstream region comprising two adjacent direct repeat motifs believed to promote transcription regulation. DNA binding, which involves the recruitment of two monomeric PhoP molecules, was dependent on conserved adenines of the repeat sequences and the orientation of the repeat motifs relative to each other. Although response regulators such as PhoB and FixJ dimerize upon phosphorylation, we demonstrate here that PhoP dimerization can also be stimulated by DNA binding. Using the established asymmetric tandem binding model by members of the OmpR/PhoB protein family as a guide, we set out to examine intermolecular interactions between PhoP dimers by protein cross-linking. Our results are consistent with a model in which two PhoP protomers bind the duplex DNA with a symmetric head-to-head orientation to project their N termini toward one another, arguing against previously proposed head-to-tail tandem dimer formation for members of the OmpR/PhoB protein subfamily.
Mycobacterium tuberculosis demonstrates multiple phases of regulation in order to adapt to numerous harsh conditions within its host. Recent evidences suggest that M. tuberculosis adapts to its host environments to a large extent through signal transduction, leading to switching on complex transcriptional programs (8). Gene expression required for adaptive responses under specific environmental conditions is often controlled by two-component regulatory systems (TCSs) consisting of a membrane-associated sensor kinase and a cytoplasmic response regulator (RR) (1). Together, these protein pairs sense environmental stimuli and initiate complex transcriptional programs in the bacterium.
In a wide variety of organisms, PhoP plays a central regulatory role in controlling expression of diverse genes. A growing body of evidence in recent years has emphasized role(s) of phoP-phoR TCSs in M. tuberculosis growth and survival in animal and cellular models (12, 14, 26, 31, 34; for a review, see reference 28). A phoP knockout mutant of M. tuberculosis showed severe growth attenuation in humans, in mouse bone marrow-derived macrophages, and in mice (26), suggesting that phoP-phoR is essential for M. tuberculosis virulence and is required for the bacilli to multiply in a host. Furthermore, the mutant M. tuberculosis strain lacked sulfatides, diacyltrehaloses, and polyacyltrehaloses in the cell envelope, suggesting the involvement of the regulatory system in complex lipid biosynthesis (12, 34). Two recent studies further suggest that a point mutation in PhoP contributes to avirulence of M. tuberculosis H37Ra (21) and also accounts for the absence of polyketide-derived acyltrehaloses in M. tuberculosis H37Ra (5). More recently, PhoP has also been implicated in the ESAT-6 secretion and specific T-cell recognition during virulence regulation of M. tuberculosis (9). Although activation of phoP-phoR could not be detected under a variety of stress conditions examined, global gene expression profiling indicates that 44 genes are upregulated and another 70 genes are downregulated by PhoP in M. tuberculosis (34). However, a consensus DNA-binding motif within the PhoP-regulated promoters was not apparent.
PhoP, a member of the Escherichia coli OmpR/PhoB subfamily, consists of two functional domains: an N-terminal receiver domain and a C-terminal transactivation domain (also called an effector domain). The N-domain, like other members of the RR family, also shares a conserved doubly wound (α/β)5 topology with a phosphorylation site at the N terminus (D71 for PhoP [15]). The C domain of the protein has been structurally characterized (PDB ID: 2PMU [35]). The structural analysis revealed overall folds similar to those of four other OmpR family proteins, M. tuberculosis PrrA (24), E. coli PhoB (25), Thermotoga maritima DrrD (4), and Bacillus subtilis PhoP (2) with a winged-helix-turn-helix DNA-binding motif involved in DNA binding. Despite global functional diversity, members of the PhoP family share significant structural homology in their receiver domain, as well as in the basic mode of DNA binding. All of the family members utilize a winged helix-turn-helix DNA-binding motif, which has been experimentally shown to bind direct tandem repeat sites (3, 36) and inverted repeats of DNA (11). However, there are significant differences in the mechanism to regulate DNA-binding activity and modulate transcription. The only reported interaction of PhoP from M. tuberculosis H37Rv involves binding of the regulator to its own promoter (13). Previously, we demonstrated transcriptional autoregulation of phoP by sequence-specific interaction of PhoP from M. tuberculosis H37Ra to its own promoter (15). Strikingly, these two independent studies show largely similar DNA sequences being recognized by PhoP in the DNase I footprint. However, very little is known about the sequence motif recognized by PhoP and the orientation of the protein(s) on the target DNA to promote transcription regulation. As a step toward understanding how the regulator functions, we show here sequence-specific recognition of the 23-bp region of the phoP promoter by the protein. We further show that two molecules of monomeric PhoP are recruited on a phoP promoter-derived oligonucleotide-based substrate DNA comprising two direct repeat motifs. Although our results suggest that DNA binding stimulates the dimerization of PhoP, evidence is presented that PhoP, unlike other members of the subfamily of proteins, binds to DNA in a head-to-head orientation to project their N termini toward each other.
MATERIALS AND METHODS
Cloning and mutagenesis of PhoP: E. coli.
DH5α was used for all cloning procedures. To express PhoP in E. coli, a T7 lac-based expression system pET15b (Novagen) was used, and PhoP was modified by an amino-terminal His6 tag. To this end, plasmid pET-phoP was constructed as described previously (15). The coding region of phoP gene was amplified from the genomic DNA of M. tuberculosis H37Rv using primers that introduced an NdeI site (phoPstart) at the start codon and a BamHI site (phoPstop) 3′ of the stop codon (Table 1). The cloning strategy resulted in pET-phoP, a PhoP derivative (247 amino acid residues) with natural C termini and an N-terminal His tag. Plasmid pGEX-phoP was constructed as follows. PCR primers that included a BamHI site at the start (GST-phoPstart) and an XhoI site 3′ of the stop codon (GST-phoPstop) were used to amplify residues 1 to 247 of PhoP (Table 1). The PCR-derived phoP fragment was gel purified, digested with BamHI and XhoI, and ligated to the BamHI/XhoI backbone fragment of pGEX 4T-1 (GE Healthcare) to construct pGEX-phoP. The cloning strategy resulted in GST-PhoP, a PhoP derivative (247 amino acid residues) with natural C termini and an N-terminal glutathione S-transferase (GST) tag. The single cysteine mutations within PhoP were introduced in which the endogenous alanine or glycine is replaced with a cysteine. Site-directed mutagenesis of individual PhoP residues (described in Results) was carried out by a PCR-based two-stage overlap extension method (18) using complementary oligonucleotides (Table 1) with the mutated codon and Deep Vent DNA polymerase (New England Biolabs). Sequence analysis using an automated DNA sequencer (Applied Biosystems) with chain termination chemistry revealed that the constructions were error-free. The mutant proteins behaved as wild-type cysteine-less PhoP in all assays tested, and this ensured that any reactivity was due solely to the introduced cysteine. All enzymatic manipulations of DNA were performed according to standard procedures with reagents purchased from New England Biolabs. Polyacrylamide gel electrophoresis (PAGE)-purified oligonucleotides were synthesized by Sigma. Plasmid DNA isolation, recovery, and purification of DNA fragments or PCR products from agarose gels were carried out using Qiagen spin columns and procedures (Qiagen, Germany).
TABLE 1.
Primers and plasmids used in this study
| Primer or plasmid | Sequence (5′-3′) or descriptiona | Source or reference |
|---|---|---|
| Primersb | ||
| phoPstart | GTTTGCCATATGCGGAAAGGGGTTGAT | 15 |
| phoPstop | GTGGTGGATCCTCGAGGCTCCCGCAGTAC | 15 |
| GST-phoPstart | CCTGGATCCATGCGGAAAGGGGTT | This study |
| GST-phoPstop | GGTCTCGAGTCGAGGCTCCCGCAG | This study |
| FPphoPC21 | ACACCGGAGTGCCGTGTCCTCGTG | This study |
| RPphoPC21 | CACGAGGACACGGCACTCCGGTGT | This study |
| FPphoPC115 | CTGACCCTGGGTTGCGACGACTAT | This study |
| RPphoPC115 | ATAGTCGTCGCAACCCAGGGTCAG | This study |
| FPphoPC189 | TTCGTGATCAACTGCGGCACCGTG | This study |
| RPphoPC189 | CACGGTGCCGCAGTTGATCACGAA | This study |
| Plasmids | ||
| pET15b | E. coli cloning vector; Ampr | Novagen |
| pET-phoP | His6-tagged PhoP expression plasmid | This study |
| pGEX-4T-1 | E. coli cloning vector; Ampr | GE Healthcare |
| pGEX-phoP | GST-tagged PhoP expression plasmid | This study |
| pET-phoPC21 | Ala-21 codon mutated to Cys in pET-PhoP | This study |
| pET-phoPC115 | Gly-115 codon mutated to Cys in pET-PhoP | This study |
| pET-phoPC189 | Ala-189 codon mutated to Cys in pET-PhoP | This study |
Ampr, ampicillin resistance.
FP, forward primer; RP, reverse primer.
Expression and purification of PhoP.
Wild-type and single cysteine mutant PhoP proteins from M. tuberculosis H37Rv were expressed in E. coli BL21(DE3) as fusion proteins containing an N-terminal His6 tag (Novagen) and purified by immobilized metal-affinity chromatography (Ni-NTA; Qiagen) as described earlier (15, 29). GST-PhoP was produced by inducing 1-liter log-phase cultures in LB medium for overnight at 18°C with 0.4 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Cells were harvested by centrifugation, dissolved in 40 ml of lysis buffer (50 mM HEPES-Na+ [pH 7.20], 0.6 M NaCl, 10% glycerol), and lysed by sonication. The supernatant was cleared by centrifugation at 12,000 × g for 40 min at 4°C and then loaded onto a glutathione-Sepharose (GE Healthcare) affinity column (1-ml bed volume). The column was washed with 40 ml of lysis buffer and eluted with 10 ml of lysis buffer containing 20 mM glutathione (Sigma). The fusion protein eluted in the first 5-ml fraction. The molecular mass determined was in agreement with the predicted mass of the monomeric form of the protein. Purity of the proteins preparations was ≥95%, as judged by sodium dodecyl sulfate (SDS)-PAGE and subsequent staining with Coomassie blue. Protein concentration was determined by a BCA protein assay (Pierce) with bovine serum albumin as a standard. All protein concentrations are given in equivalents of protein monomers.
Gel filtration.
Size exclusion chromatography was performed at 25°C on a Protein Pak 300sw (0.75 by 30 cm; Waters) using a high-pressure liquid chromatography system (Shimadzu Corporation, Japan). Samples of PhoP were eluted at a flow rate 0.5 ml/min in 50 mM Na+-phosphate (pH 7.3), 10% glycerol, and 200 mM NaCl. The elution profiles were monitored by recording the absorbances at 280 and 220 nm. The column was extensively equilibrated with the same buffer prior to each run. Elution of RNase A (15.2 kDa), chymotrypsinogen A (21.2 kDa), ovalbumin (48.1 kDa), albumin (63.3 kDa), and aldolase (158 kDa) (all from GE Healthcare) were used to obtain the calibration curve.
Homology modeling of PhoP.
Amino acid sequence of PhoP within the protein data bank showed highest sequence identity of 45% with M. tuberculosis PrrA. To this end, a three-dimensional structural model of PhoP was predicted by using Modeler 8.0 with the M. tuberculosis PrrA structure (PDB accession no. 1YS6 [24]) as the template. Nineteen amino acids from the N terminus of PhoP were not modeled since no corresponding residues have been found in the alignment. The modeled structure was energy minimized using the AMBER force field, and the overall geometry of the structural model was assessed by using PROCHECK (20). The molecular image was generated by using PYMOL (6).
Cross-linking of mutant proteins.
Purified single cysteine mutants of PhoP (7.5 μM) were incubated in absence or presence of 1 μM bis-maleimidohexane (BMH; Pierce) for 60 min at 25°C in buffer containing 25 mM HEPES-Na+ (pH 7.3), 100 mM NaCl, and 5 mM EDTA. The reactions were quenched by the addition of 50 mM dithiothreitol, and the products were separated by SDS-PAGE and stained with Coomassie blue. The gels were scanned and quantified by densitometric analysis to determine the percent cross-linking. To study the effect of DNA on protein cross-linking, the DNA concentration was determined by trials. All data are presented as means ± the standard errors of the mean.
Oligodeoxyribonucleotide probes.
PAGE-purified oligonucleotides were purchased from Sigma. The oligonucleotide concentrations were determined from absorbance at 260 nm, using calculated extinction coefficients. The 60-bp DNA fragments used to assess sequence-specific DNA binding by PhoP or its variants are 5′-ATTTGGCGATTCCTGGCAGACTGTTAGCAGACTACTGGCAACGAGCTTTCAGGAATTACA-3′ and its complement 5′-TGTAATTCCTGAAAGCTCGTTGCCAGTAGTCTGCTAACAGTCTGCCAGGAATCGCCAAAT-3′ (the direct repeat sites are underlined). This sequence of the phoP upstream region is within the PhoP-protected DNase I footprint (13, 15) and is hereafter referred to as DR1,2. In DR2,1, the DR2 sequence is present upstream of DR1, as opposed to the natural sequence, where DR1 is located upstream of DR2. In contrast, DR1,2I contains wild-type DR1 site with the DR2 sequence turned around, yielding adjacent inverted repeats instead of adjacent direct repeats. DR1(A1C)DR2 and DR1(A7C)DR2 carried single substitutions of A1C and A7C, respectively, in the DR1 motif without any alteration in the DR2 motif. Likewise, DR1DR2(A1C) and DR1DR2(A7C) carried the substitutions A1C and A7C, respectively, in the DR2 motif without any alteration in the DR1 site. NSP represents a nonspecific probe of comparable base composition, 5′-CTGGCCAGCCGGTTGGCGCGGGCAATCGTGTCATCGATTCCCAGCATGTTGGCCATTGAG-3′, and its complement, 5′-CTCAATGGCCAACATGCTGGGAATCGATGACACGATTGCCCGCGCCAACCGGCTGGCCAG-3′, and is derived from a distal region of phoP promoter which lacks any PhoP binding site. The target binding site(s) in each set of oligonucleotides were flanked by matching nonspecific sequences chosen to avoid fortuitous binding. The relevant features of the various DNA substrates used are described in the Results and explained in the respective figure legends.
DNA-binding assays and Ferguson analysis.
5′-32P labeling of oligonucleotide probes was carried out using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (BRIT, India). Unincorporated nucleotides and labeled oligonucleotide were separated using a Sephadex-G50 quick-spin column (GE Healthcare). DNA probes for electrophoretic mobility shift assays (EMSAs) were generated by annealing the labeled strand to its unlabeled complement in 10 mM Tris (pH 8.0) containing 50 mM NaCl after slow cooling to room temperature from being heated to 95°C for 5 min. Annealing efficiency was verified by native PAGE. Binding reactions (10 μl) contained DNA (20 nM) and PhoP in 20 mM HEPES-Na+ (pH 7.5), 50 mM NaCl, 200 μg of bovine serum albumin/ml, 10% glycerol, 1 mM dithiothreitol, and 200 ng of sheared herring sperm DNA. Incubation was performed for 10 min at 20°C. GST-PhoP containing the GST tag was only used in the competitive EMSA experiment whose results are shown in Fig. 4C. The reactions were analyzed by electrophoresis through nondenaturing 6% (unless otherwise mentioned) polyacrylamide gels (acrylamide-bisacrylamide [40:1.1] in 1× Tris-EDTA buffer). The gels were run at 70 V for 6 to 8 h at 4°C, dried, and visualized by autoradiography. After electrophoresis and gel drying, radioactive bands were quantified by using a phosphorimager. The data were converted to the percent labeled DNA bound and plotted versus the protein concentration. The concentration of PhoP required for 50% binding to DNA is presented as the C50 value.
FIG. 4.

Stoichiometry of PhoP-DNA interactions. (A) Gel filtration chromatography was performed with ∼20 μg of purified PhoP. The inset shows a calibration curve of protein standards generated to determine approximate molecular mass of PhoP. Elution time of PhoP is indicated by an upward arrow. (B) PhoP-DNA complexes were resolved on a series of native polyacrylamide gels alongside nondenatured protein molecular mass standards, as described in Materials and methods. The retardation coefficient (Kr) values for protein standards were defined by Ferguson analysis and plotted as a function of molecular mass. Interpolation of −Kr (solid lines) indicated a molecular mass of ∼99.2 kDa for the PhoP-DNA complex. (C) DR1,2 comprising two 9-bp direct repeat motifs in tandem binds two molecules of PhoP. For the competitive EMSA experiment, ∼20 nM end-labeled DR1,2 DNA was incubated in binding reactions with recombinant forms of PhoP. Binding reactions contained no protein (lane 1), 4 μM PhoP alone (lane 2), 4 μM GST-PhoP alone (lane 13), and mixtures of indicated concentrations of PhoP and GST-PhoP (lanes 3 to 12). Protein-DNA complexes were analyzed as described for Fig. 1. HH, two molecules of PhoP; GG, two molecules of GST-PhoP; HG, one molecule each of PhoP and GST-PhoP bound to end-labeled DR1,2.
To determine the stoichiometry of PhoP binding to its substrate, a gel electrophoresis-based method as described by Ferguson (7) was used to determine the size of the retarded complex. Purified PhoP was combined with the labeled DNA in binding reactions as described above, and the substrate and complexes were resolved on a series of native polyacrylamide gels (6, 7, 8, and 9% polyacrylamide in 1× Tris-EDTA) alongside 5 μg of nondenatured protein molecular mass standards (Sigma). The standards included α-lactalbumin (14.2 kDa), carbonic anhydrase (29 kDa), chicken egg ovalbumin (45 kDa), bovine serum albumin (66 kDa), and urease (272 kDa). A retardation coefficient (Kr) was derived from migration of the species, as described elsewhere (27), and a standard curve was generated by the plotting retardation coefficient of each species against the corresponding molecular mass.
RESULTS
PhoP binding to the phoP upstream region is dependent on adjacent direct repeat motifs and orientation between the repeat motifs.
To determine whether PhoP from M. tuberculosis H37Rv recognizes the repeat motifs identified within the phoP promoter region (13), the binding of purified PhoP to its promoter was investigated using synthetic oligonucleotides comprising two adjacent direct repeat motifs. Note that nucleotide sequences outside the direct repeats included 5 bp of intervening spacer sequence along with 19- and 18-bp extensions of natural sequence at the 5′ and 3′ ends, respectively. In EMSA experiments, PhoP bound efficiently to oligonucleotide-based DR1,2 probe consisting of DR1 and DR2 repeat motifs (Fig. 1A, lanes 2 to 7) with an apparent dissociation constant of 3.5 ± 0.15 μM. However, oligonucleotide-based DNA probes comprising single repeat motifs (DR1 or DR2 alone) with identical flanking sequences were found to be inefficient in forming PhoP-DNA complexes stable to gel electrophoresis (data not shown). These observations suggest that two direct repeats in tandem significantly contribute to formation of a complex stable to gel electrophoresis.
FIG. 1.

PhoP binding to phoP promoter region is dependent on two direct repeat motifs and their orientation. (A) EMSA for binding of indicated concentrations of PhoP to 5′-end-labeled 20 nM oligonucleotide-based DNA probe DR1,2 (lanes 1 to 7). Reaction conditions were as described in Materials and Methods. DR1,2 contains repeat motifs DR1 (open arrowhead) and DR2 (filled arrowhead) with a natural intervening spacer sequence of five bases. Protein-DNA complexes were detected by autoradiography. Open arrowheads indicate the origins of the native polyacrylamide gel, and filled arrowheads indicate band shifts produced in the presence of PhoP. The gels are representative of at least three independent experiments. (B) The specificity of DNA binding by PhoP (5.2 μM) with end-labeled DR1,2 was carried out in the presence of increasing concentrations of specific or nonspecific competitor DNA. Protein-DNA complex was analyzed as described for panel A. Lane 1, labeled DNA without any protein; lane 2, no competitor DNA added; lanes 3 to 6, binding mix containing 12.5-, 25-, 50-, and 100-fold molar excesses, respectively, of specific competitor; lanes 7 to 10, binding mix containing 12.5-, 25-, 50-, and 100-fold molar excesses, respectively, of nonspecific competitor. (C) Sequences and organizations of oligonucleotide-based DNA probes consisting of direct-repeat motifs DR1 (open arrow) and DR2 (filled arrow). (D) PhoP binding to phoP upstream region is dependent on the orientation of direct repeat motifs. EMSA of oligonucleotide-based DNA probes (∼20 nM) DR2,1 (lanes 1 to 7) and DR1,2I (lanes 9 to 14), as described in the Results was carried out in the absence or the presence of indicated concentrations of PhoP. Protein-DNA complexes were analyzed as described for panel A. Note that nucleotides outside of the direct repeat sequences comprise 19- and 18-bp extensions of natural sequence at the 5′ and 3′ ends, respectively, and are identical in all of the DNA probes used.
To test the specificity of PhoP binding to the DR1,2 duplex, DNA binding was carried out in the presence of different concentrations of unlabeled DR1,2 (as specific competitor) or unlabeled nonspecific competitor (hereafter referred to as NSP [whose sequence is described in Materials and Methods]), which lacks a consensus PhoP binding site but has a comparable base composition. Although unlabeled DR1,2 at a 50-fold molar excess efficiently competed for PhoP binding (only 13% ± 1.5% binding; Fig. 1B, lane 5), an identical fold excess of nonspecific competitor DNA resulted in a minor variation of DNA-binding efficiency (50.5% ± 2% binding; Fig. 1B, lane 9) compared to the no-competitor control (64% ± 2.5% binding; Fig. 1B, lane 2), reflecting that PhoP binding to DR1,2 is sequence specific.
In order to investigate the importance of the orientation of repeat motifs in sequence-specific DNA binding by PhoP, control oligonucleotides carrying wild-type direct repeat motifs that were altered in the order and orientation of repeat motifs (DR2,1 and DR1,2I, Fig. 1C) were synthesized and used in binding assays with PhoP. Interestingly, if the oligonucleotide is altered by placing DR2 upstream of DR1, binding of PhoP is hardly affected (DR2,1; Fig. 1D, lanes 2 to 7). In contrast, if the oligonucleotide is changed by turning around one of the direct repeat sites so that they are inverted repeats instead of direct repeats, the binding of PhoP is severely repressed (DR1,2I [Fig. 1D, lanes 9 to 14]; <10% binding was observed at the highest PhoP concentration, based on the limits of detection in this assay and based on other gels [data not shown]). Thus, binding of PhoP to the phoP promoter region is dependent on the direct repeat motifs and their orientation relative to each other.
Conserved nucleotides within the repeat sequences are important for DNA binding by PhoP.
We noticed that four bases A1, A7, C2, and C9 (shaded in gray in Fig. 2A) are conserved in the repeat motifs identified within the phoP promoter (13, 15). To examine contribution of each of the residues in PhoP-DNA interaction, oligonucleotides were synthesized where conserved nucleotides were modified in both direct repeat motifs, one at a time. The changes were generated by interchanging A's with C's and G's with T's and vice versa. Interestingly, A1C substitution at the DR1 motif (Fig. 2B, lanes 2 to 7) showed a significant decrease in PhoP binding compared to the wild-type site. Although PhoP at a protein concentration of 5 μM showed an efficient DNA binding of 93% ± 3% with DR1,2, DR1(A1C)DR2 carrying A1C substitution in the DR1 repeat at an identical PhoP concentration showed 42% ± 5% DNA binding (compare lane 6 in Fig. 1A to lane 6 in Fig. 2B). A quantitative analysis of the binding data reveals (2.2 ± 0.5)-fold lowering of the C50 for PhoP binding to the mutant oligonucleotide compared to the wild-type DNA. These results suggest that A1 of the DR1 motif is likely to be important for PhoP-DNA interactions. In sharp contrast, a C2A change (lanes 2 to 7, see also Fig. S1A in the supplemental material) at comparable concentrations of PhoP displayed overall DNA binding comparable to that of DR1,2. Also, substitution of A7 and C9 of the DR1 site (lanes 9 to 14 in Fig. 2B and lanes 9 to 14 in Fig. S1A in the supplemental material, respectively) did not show any significant effect on binding of PhoP compared to the wild-type DR1,2 (Fig. 1A, lanes 2 to 7).
FIG. 2.

Roles of conserved adenines of DR1 and DR2 repeat motifs in PhoP-DNA interactions. (A) Sequence of the 23-bp core binding region comprising two direct repeat motifs (indicated in uppercase letters) with a 5-bp intervening spacer region (indicated in lowercase letters). The nucleotides in both repeat motifs are numbered from 5′ to 3′ (at the bottom), and the nucleotides conserved in all three direct repeat motifs are indicated by gray shadings. (B and C) EMSA of ∼20 nM oligonucleotide-based DNA probes with the indicated concentrations of PhoP using duplexes carrying substitutions at the A1 (lanes 1 to 7) and A7 (lanes 8 to 14) of the DR1 (B) or DR2 motifs (C). Direct-repeat specific substitutions are shown above each panel. The reaction condition is as described in Materials and Methods. Protein-DNA complexes were analyzed as described for Fig. 1A. The gels are representative of three independent experiments.
To examine the importance of conserved nucleotides of DR2 site, a similar strategy was used to construct substrate oligonucleotides carrying changes in the conserved nucleotides (A1, A7, C2, and C9) of DR2, leaving DR1 site unaltered. The end-labeled substrates were analyzed for their ability to generate complexes with purified PhoP. Interestingly, an oligonucleotide carrying an A1C substitution in the DR2 motif efficiently bound to PhoP with a binding affinity comparable to that of wild-type DNA (compare lanes 2 to 7 in Fig. 2C to lanes 2 to 7 in Fig. 1A). These results reflect the repeat motif-specific importance of A1 for PhoP-DNA interaction(s). Similar binding experiments of PhoP using oligonucleotides carrying substitutions of C2 with A2 in DR2 (see Fig. S1B, lanes 2 to 7, in the supplemental material) and C9 with A9 (see Fig. S1B, lanes 9 to 14, in the supplemental material) showed binding of PhoP comparable to that of wild-type DR1,2. However, a striking inhibition of DNA binding was observed when A7 of DR2 site was substituted by C7 (Fig. 2C, lanes 9 to 14). A quantitative analysis revealed that under identical binding condition, at least 10-fold reduction in DNA binding by PhoP was observed with the mutant DNA (based on the limits of detection in this assay and based on other gels [data not shown]). Note that under identical binding conditions, the substitution of A7 of DR1 by C7 (Fig. 2B, lanes 9 to 14) did not influence the PhoP binding efficiency to the mutant DNA. Thus, the conserved A7 residue of DR2 repeat motif appears to be essential for PhoP-DNA interactions. From these results, we surmise that the conserved adenines (A1 of DR1 and A7 of DR2) are important for forming PhoP-DNA complexes that are stable to gel electrophoresis. Since A1 of DR1 and A7 of DR2 are located in the 5′ and the 3′ ends, respectively, of the substrate DNA, our results suggest the importance of the 5′ end of DR1 and the 3′ end of DR2 in PhoP-DNA interactions.
We next examined whether the effects of substitutions on DNA-protein interaction are direct repeat specific or location dependent. To this end, we synthesized oligonucleotides with DR2 repeat motifs upstream of DR1 carrying substitutions in A1 and A7 of both repeat motifs. Identical DNA-binding experiments with PhoP showed that A1 of DR2 affected PhoP binding (compare lanes 2 to 7 in Fig. 3B to lanes 2 to 7 in Fig. 1D) with an ∼1.5-fold lower C50 of binding for the mutant substrate compared to DR2,1. However, A1 of DR1 (Fig. 3C, lanes 2 to 7), as well as A7 of DR2 (Fig. 3B, lanes 9 to 14), did not show any detectable effect on PhoP binding (as measured by the C50 of protein binding) to the mutant oligonucleotides. This is in sharp contrast to what we observed for identical substitutions in the oligonucleotide-containing natural sequence with DR1 site upstream of DR2 (Fig. 2). Interestingly, substitution at A7 of the DR1 motif (Fig. 3C, lanes 9 to 14) completely abrogated PhoP binding with <5% detectable complex formation even at the highest concentration of PhoP examined. This is strikingly similar to what we observed with a substitution at A7 in DR2 (Fig. 2C, lanes 9 to 14). Together, our observations suggest that PhoP binding to oligonucleotides containing two adjacent direct repeat sites involve the 5′ end of the upstream and the 3′ end of the downstream repeat motif.
FIG. 3.

Roles of conserved adenines in repeat motifs (DR2 and DR1) of DR21 in PhoP-DNA interactions. (A) Sequence of the 23-bp core binding region comprising two direct repeat motifs (indicated in uppercase letters) with the DR2 site upstream of DR1 site, including the 5-bp intervening spacer region (indicated in lowercase letters) of the natural sequence. The nucleotides in both repeat motifs are numbered from 5′ to 3′ (at the bottom), and the conserved adenines are indicated by gray shadings. (B and C) EMSA of oligonucleotide-based DNA probes (∼20 nM) with various concentrations of PhoP using duplexes carrying the indicated substitutions at the A1 (lanes 1 to 7) and A7 (lanes 8 to 14) positions of the DR2 (B) or DR1 motifs (C). Reaction condition was as described in Materials and Methods. Protein-DNA complexes were analyzed as described in the legend to Fig. 1A.
Two adjacent repeat sequences recruit two molecules of monomeric PhoP.
To determine quaternary organization of PhoP in solution when prepared in buffer that is similar to the one used in in vitro DNA-binding assays, we next performed gel filtration chromatography on purified PhoP. Single-peak elution was observed for multiple replicates of PhoP from the Protein Pak 300sw column (Fig. 4A). Compared to the protein standards, the estimated molecular mass of PhoP was calculated to be 29 ± 1 kDa (Fig. 4A, inset). This is in good agreement with the predicted molecular mass of 29.5 kDa for the monomeric form of PhoP. To estimate the number of PhoP molecules binding to the DNA fragments, we undertook gel electrophoresis-based Ferguson analysis as described by Robbins et al. (27). This gel retardation-based approach involves the electrophoretic separation of protein-DNA complexes alongside protein size standards on native polyacrylamide gels of various concentrations. To this end, a series of protein standards under native condition was run parallel to PhoP-DNA complex. PhoP in complex with end-labeled DR1,2 yielded an estimated mass of 99.2 kDa (Fig. 4B). After subtracting the mass of the 60-bp oligonucleotide (39 kDa), the complex resulted in an estimated mass of 60.2 kDa for the protein component. This translates into ∼2.04 molecules of PhoP per molecule of DR1,2 substrate with a PhoP monomer mass of 29.5 kDa.
We next undertook Ferguson analysis of PhoP binding to DNA substrate of different size. We observed that the binding of two PhoP protomers to substituted DR1 site (2CTG4 of DR1 changed to 2AAA4) adjacent to the wild-type DR2 sequence (sDR1DR2) was inhibited (29). In contrast, substitution of the DR2 (2CTG4 of DR2 changed to 2AAA4) sequence adjacent to the wild-type DR1 site (DR1sDR2) did not prevent the recruitment of two PhoP molecules (29). PhoP-DNA complexes, parallel to a series of protein standards run at various acrylamide concentrations under native conditions, yielded estimated masses of 50.8 and 76 kDa with sDR1DR2 and DR1sDR2, respectively (data not shown). Subtracting the mass of the 36-bp oligonucleotide (23.4 kDa), the complexes resulted in estimated masses of 27.4 and 52.6 kDa, respectively, for the protein component. This translates into ∼0.93 and ∼1.80 molecules, respectively, of PhoP per molecule of substrate DNA with PhoP monomer mass of 29.5 kDa. Thus, it is likely that a single molecule of PhoP binds to sDR1DR2, whereas two molecules bind DR1sDR2. These results suggest that Ferguson analysis, at least under the conditions examined, yields a reliable stoichiometry of PhoP-DNA interactions.
We have previously shown that PhoP forms a retarded complex with end-labeled sDR1DR2 (with substituted DR1) comprising a single PhoP molecule. However, the complex of PhoP with DR1sDR2 (with substituted DR2 site) consists of two PhoP protomers (29). These observations suggest that (i) the upstream repeat motif (DR1 in the case of DR1,2 DNA) binding appears to be the prerequisite for recruitment of two PhoP protomers and (ii) “half-site” binding of PhoP is only possible at the intact DR2 (downstream repeat motif) motif with substitutions in the DR1 site. These results further suggest that a PhoP monomer binding to DR1 site is a requirement for the second monomer, which would then bind to DR2 with an incredibly high affinity. However, in the present study we show that A7 of the downstream repeat motif (DR2 in this case) is likely to be the key contacting base important for the recruitment of a second PhoP monomer at the DR2 site. The fact that DR1DR2(A7C) or DR2DR1(A7C) fails to show any significant binding suggests that only a singly bound PhoP-DNA complex of upstream repeat motif (DR1 or DR2, respectively) appears to be unstable to gel electrophoresis and thus is not observed in EMSA experiments under the conditions examined. It is noteworthy that A7 of the DR2 repeat motif remains unchanged in DR1sDR2 (with a substitute DR2 site).
To further confirm the number of PhoP molecules bound to the repeat sequences of the phoP promoter, we next performed competitive EMSA with end-labeled DR1,2. To this end, GST-tagged PhoP (GST-PhoP) and His-tagged PhoP (PhoP) proteins were used for the experiments because the difference in molecular masses between these recombinant proteins carrying affinity tags of different sizes could easily be distinguished by EMSA. The recombinant GST-PhoP was purified to homogeneity in a single-step procedure by glutathione-Sepharose affinity chromatography that resulted in a single homogeneous species with a relative molecular mass of 56 kDa on an SDS-polyacrylamide gel, in agreement with the predicted mass (data not shown). In competitive EMSA experiments with GST-PhoP and PhoP using end-labeled DR1,2, addition of the PhoP alone to the binding mix yielded a single protein-DNA complex (indicated as HH in Fig. 4C, lane 2). Expectedly, a single protein-DNA complex with a molecular mass greater than that observed with PhoP was also observed in a binding reaction containing GST-PhoP alone (indicated as GG in Fig. 4C, lane 13). However, the addition of increasing concentrations of GST-PhoP to binding reactions containing decreasing amounts of PhoP (lanes 3 to 12) resulted in the generation of a third protein-DNA complex that migrated at a position between those observed with PhoP and GST-PhoP (indicated as HG in Fig. 4C). Since GST-PhoP eluted as a monomeric protein in solution (data not shown), this pattern of mobility is consistent with one PhoP molecule being recruited by each direct repeat motif. However, our gel retardation assays under the conditions examined could not resolve HG and GG complexes in the same lane of the gel. Perhaps the presence of GST tag in GST-PhoP stimulates GG complex formation over HG, since Fig. 4C (lanes 9 to 10) shows the formation of GG complex in the presence of 0.5 and 0.4 μM PhoP, even when there is substantial unbound DNA available for HG complex formation. Together, in conjunction with results from the Ferguson analysis, we concludeed that two tandem repeat motifs of DR1,2 recruit two PhoP protomers.
DNA binding stimulates the dimerization of PhoP.
Recombinant PhoP is shown to be monomeric in solution, and yet two PhoP molecules bind DNA (Fig. 4). To examine the effect of DNA binding on PhoP oligomerization, we introduced three unique cysteine substitutions at disparate positions within the PhoP protein. Introduction of a cysteine residue in the primary sequence of the protein provides an opportunity to study intermolecular protein-protein cross-linking using thiol-specific cross-linkers. Conveniently, there is no cysteine residue in the primary sequence of PhoP. We chose to introduce unique cysteine residues by replacing endogenous alanine, glycine, and alanine at positions 21, 115, and 189 of PhoP, respectively. The positions of the cysteine substitutions were based on an M. tuberculosis PrrA (PDB accession no. 1YS6 [24])-dependent homology modeled structure of PhoP (see Fig. S2 in the supplemental material). Note than while residue 21 is at the extreme N terminus preceding the β1 strand, residue 115 is within the α4-β5-α5 face, which consists of the proposed site of dimerization in the OmpR/PhoB subfamily of proteins (24). However, Cys-189 is at the beginning of the loop connecting α6 and α7 of the C-terminal DNA-binding domain (PDB ID: 2PMU [35]). The three mutant proteins, each containing a unique cysteine, were expressed and purified to homogeneity as described in Materials and Methods.
To investigate PhoP oligomerization, we used the homobifunctional cross-linker BMH, which contains two sulfhydryl-reactive maleimides separated by a six-carbon spacer arm (13.1 Å). For protein cross-linking experiments, the proteins were incubated in the absence or presence of BMH. In the absence of BMH, all three mutant proteins migrated in SDS-PAGE at ∼29 kDa, which is consistent with PhoP's molecular mass (Fig. 5). Also, no higher-molecular-mass species was observed for any of the single cysteine mutants of PhoP in the absence of the cross-linker (Fig. 5A, lanes 1 and 4, and Fig. 5B, lane 1). However, in the presence of BMH, all of the three single cysteine mutants displayed cross-linked dimer formation. Although PhoPC21 formed ca. 29% ± 0.8% of dimer in the presence of BMH (Fig. 5A, lane 2), PhoPC189 and PhoPC115, under identical conditions, generated 35% ± 0.7% (Fig. 5A, lane 5) and 33% ± 1% (Fig. 5B, lane 2) of cross-linked dimer, respectively.
FIG. 5.
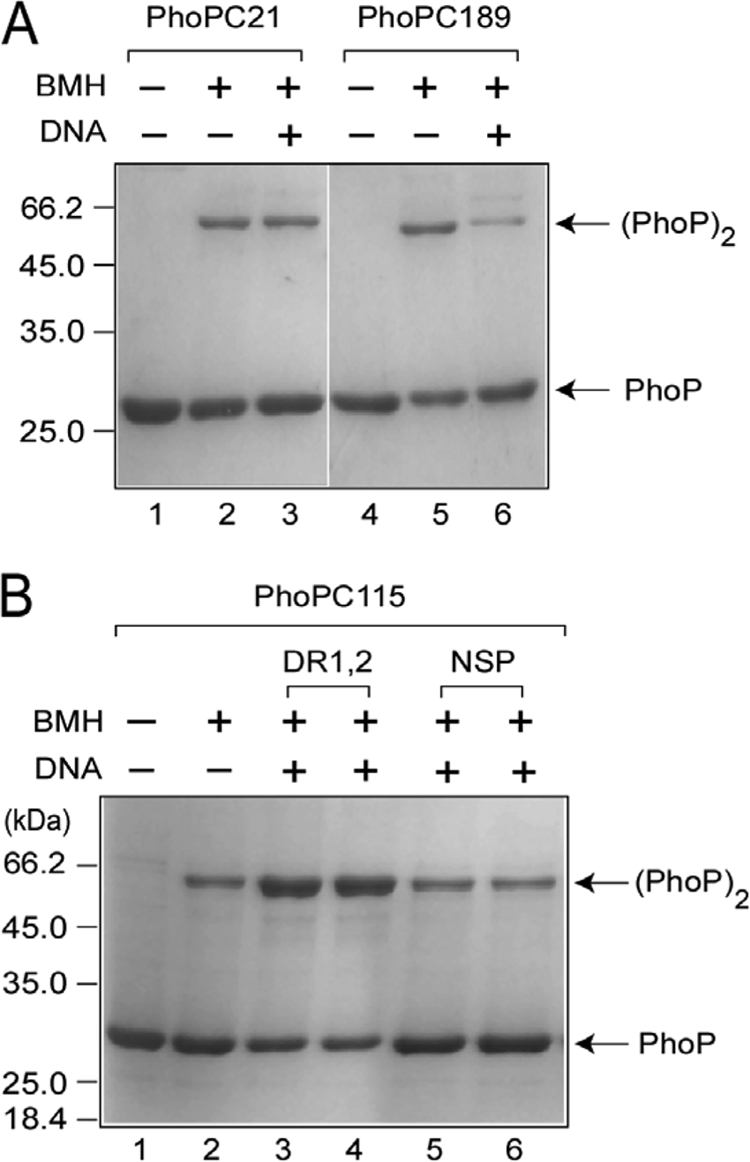
Cross-linking of single cysteine mutants of PhoP. (A and B) The indicated proteins (7.5 μM) were cross-linked as described in Materials and Methods to assess their ability to form BMH cross-linked dimers in the absence or presence of cognate DNA. Above the figure, the absence or presence of BMH and the oligonucleotide-based DNA probes is indicated by “−” or “+,” respectively. For the experimental results shown in panel A, 5 μM DR1,2 was included in the cross-linking reaction. For the experimental results shown in panel B, two oligonucleotide-based DNA probes, DR1,2 and NSP (a nonspecific DNA lacking any PhoP binding site), were used at 5 and 10 μM in lanes 3 and 4 and in lanes 5 and 6, respectively. The sizes of molecular mass markers are indicated in kilodaltons to the left of the figure. The PhoP monomer and PhoP dimer are indicated to the right as PhoP and (PhoP)2, respectively.
We next examined the cross-linking of unique cysteine mutants of PhoP in the presence of target DNA to study the effect of DNA on PhoP dimerization. PhoPC21 showed no significant change in BMH-mediated protein cross-linking when compared in the absence (29% ± 0.8% of cross-linked dimer) or presence (27% ± 1% of cross-linked dimer) of DR1,2 (compare lanes 2 and 3 of Fig. 5A). In the case of PhoPC189, the presence of DNA in the cross-linking reaction showed an inhibition of cross-linked dimer formation from 35% ± 0.7% to 22% ± 0.9% (compare lanes 5 and lane 6 of Fig. 5A). This observation suggests that the presence of DNA (or DNA binding by the C-terminal domain) presumably renders Cys-189 of PhoP unavailable for cross-linking. This is consistent with the proposed structural model of the PhoP C-domain-DNA complex, with residue 189 located within the loop connecting α6-α7 (35). However, a striking stimulation of dimer formation was observed for PhoPC115 from 33% ± 1% to 63% ± 0.5% in the presence of DR1,2 (compare lanes 3 and 2 of Fig. 5B). Two different concentrations of DR1,2 (5 and 10 μM) DNA showed comparable stimulation of protein cross-linking of PhoPC115 (compare lanes 3 and 4 to lane 2 of Fig. 5B). To examine the specificity of the DNA-mediated stimulation of dimerization, BMH cross-linking experiments were carried out in the presence of nonspecific DNA NSP (see Materials and Methods) as a control. Interestingly, NSP at comparable concentrations and under identical conditions failed to show a DNA-dependent stimulation of cross-linked dimer formation by PhoPC115 (compare lanes 5 and 6 to lane 2 of Fig. 5B). Although we observed an ∼2-fold stimulation of dimerization of PhoP by cognate DNA, the fact that a nonspecific DNA lacking any PhoP binding sequence did not show any stimulation underscores the importance of the DNA-dependent dimerization of PhoP.
The fact that BMH cross-linking of PhoPC21 was not influenced in the presence of DNA led us to speculate that the Cys-21 residues of two PhoP molecules presumably face away from each other (both in the absence and in the presence of DNA), and thus only a minor fraction of the protein is available for cross-linking in the presence of BMH. To investigate whether the DNA-binding properties of PhoPC115 or PhoPC189 contribute to the observed stimulation or inhibition of cross-linking in the presence of DR1,2 DNA, respectively, all three single cysteine mutants were tested for their DNA- binding ability. To this end, EMSA experiments were carried out with purified single cysteine mutants using end-labeled DR1,2 and compared to the wild-type PhoP. The DNA-binding activities of the mutant proteins were indistinguishable from that of the wild-type protein (see Fig. S3 in the supplemental material), suggesting that none of the cysteine residues is critically required for DNA binding. From these results, we conclude that sequence-specific DNA binding promotes the dimerization of PhoP.
PhoP binds head-to-head to DNA.
To investigate whether cross-linking of the protein could influence DNA binding, we compared the DNA-binding efficiencies of non-cross-linked and BMH cross-linked single cysteine mutants of PhoP at relatively lower protein concentrations (Fig. 6A). Although cross-linking of PhoPC21 could not significantly influence the DNA binding of the protein, BMH cross-linked PhoPC115 showed significant stimulation of DNA binding compared to the non-cross-linked protein (compare the open columns and filled columns in Fig. 6A). Quantification of the DNA-binding data reveals that cross-linked PhoPC115 (filled columns) at 0.5 and 1.0 μM showed, respectively, 7.4% ± 0.8% and 15.2% ± 1.2% DNA binding. Strikingly, no detectable DNA binding was observed for the non-cross-linked PhoPC115 at comparable concentrations of the protein. A significant (∼3.5-fold) stimulation of DR1,2 binding by cross-linking of PhoPC115 (1.5 μM) is consistent with the previous result showing the stimulation of cross-linking in the presence of DR1,2 DNA (Fig. 5B). This observation clearly indicates that cross-linked PhoP has a higher DNA-binding affinity than the non-cross-linked form. In sharp contrast, while non-cross-linked PhoPC189 (open columns) showed 10.4 ± 1 and 37% ± 2% DNA binding at 1.5 and 2 μM, respectively, the cross-linked protein did not show any detectable DNA binding at the comparable protein concentrations examined. These data suggest that PhoP cross-linked at Cys189 is unable to bind DNA. This finding is in agreement with our previous data showing the presence of DR1,2 DNA inhibiting PhoP cross-linking by PhoPC189 (Fig. 5A). Together, these results highlight that cross-linking of PhoP only in a specific orientation (head-to-head) significantly stimulates DNA binding.
FIG. 6.

PhoP binds head-to-head to DNA. (A) Effect of BMH cross-linking of single cysteine mutants of PhoP on DNA-binding compares percent DR1,2 DNA bound of non-cross-linked (empty columns) and BMH cross-linked (filled columns) proteins at the indicated protein concentrations. (B) Relative efficiency of cross-linking (∼7.5 μM) of PhoPC115 in the absence or presence of the indicated DNA substrates. Above the figure, the absence or presence of BMH and various oligonucleotide based DNA probes (see Materials and Methods for details) is indicated by “−” or “+,” respectively. The sizes of the molecular mass markers, along with the non-cross-linked and cross-linked PhoPs, are indicated as for Fig. 5. (C) BMH cross-linking in the absence or presence of the indicated oligonucleotides was analyzed by SDS-PAGE and quantified by densitometric analysis, and the percent protein cross-linking is shown. (D) Relative ability of ∼7.5 μM PhoPC115 to form BMH cross-linked dimers in absence or presence of the indicated DNA substrates (sDR1DR2 and DR1sDR2). The absence or presence of BMH and DNA substrates are as described for Fig. 5. (C) The sizes of the molecular mass markers and the non-cross-linked and cross-linked PhoPs are indicated for Fig. 5.
To further study the effect of target DNA on PhoP cross-linking, several oligonucleotides with altered order and orientation of repeat motifs and substitutions in repeat sequences were used to examine their effect on PhoPC115 cross-linking. Expectedly, DR2,1 containing DR2 site upstream of DR1 (which binds to PhoP as efficiently as does DR1,2) displayed stimulation of cross-linking from 33 ± 1 to 62% ± 5% (compare lanes 2 and lane 3 in Fig. 6B). However, DR1DR2I with DR1 and DR2 sites as inverted repeats failed to show a stimulation of cross-linking (compare lane 4 [36% ± 3% dimer] and lane 2 [33% ± 1% dimer] in Fig. 6B). This observation is consistent with very weak DNA binding of PhoP to DR1DR2I with DR2 site turned around to generate two inverted repeats (Fig. 1D, lanes 9 to 14). To further examine the effect of mutant target DNA on cross-linking of PhoP, oligonucleotide substrates carrying substitutions at the conserved adenine residues of both direct repeat motifs were tested for their ability to promote PhoPC115 cross-linking in the presence of BMH. Oligonucleotide-based substrate DNA with A1C substitution at the DR1 site showed almost wild-type DR1,2-like stimulation of cross-linking of PhoPC115 (Fig. 6B, lane 5; 57% ± 4% cross-linking [Fig. 6C]). These data are in agreement with a modest (∼2-fold) inhibition of DNA binding of PhoP due to A1C substitution at the DR1 site (Fig. 2B). Expectedly, the presence of oligonucleotides carrying substitution A7C at the DR1 site or substitution A1C at the DR2 site (lanes 6 and 7, respectively, in Fig. 6B) did not display a significant difference in cross-linking compared to wild-type DR1,2 DNA (59% ± 5% and 60% ± 1% of cross-linking, respectively). In sharp contrast, oligonucleotide-based DNA carrying substitutions A7C at the DR2 site (Fig. 6B, lane 8) failed to show a DNA-mediated stimulation of PhoP cross-linking (32% ± 2% of cross-linking [Fig. 6C]; compare lanes 2 and 8 in Fig. 6B).
The observation that substitution A1C at the DR1 site modestly inhibits DNA binding of PhoP compared to wild-type DR1,2 (∼2-fold difference in C50, compare Fig. 2B and Fig. 1A) and stimulates PhoP cross-linked dimer formation almost as efficiently as DR1,2 (compare Fig. 6B, lane 5, and Fig. 5B, lane 3) led us to speculate that a few adjacent nucleotides at the 5′ end of DR1 may be involved in PhoP-DNA interaction(s). In agreement with this proposal, strikingly, the first three nucleotides following A1 of the DR1 repeat motif but not that of the DR2 site were shown to be critical for multiple DNA binding by PhoP (29). To examine the role of mutant DNA substrates in DNA-mediated cross-linking of PhoP, protein cross-linking experiments were carried out with PhoPC115 in the presence of sDR1DR2 and DR1sDR2 (sequence as described in reference 29). Interestingly, the sDR1DR2 oligonucleotide with substitution at the DR1 site failed to influence PhoP cross-linking (35% ± 3% cross-linking in the presence of sDR1DR2 versus 33% ± 1% cross-linking in the absence of DNA [compare lanes 3 and 2 in Fig. 6D]). However, DR1sDR2 showed a clear stimulation of cross-linking to 65.8% ± 2% (compare lanes 4 and 2 in Fig. 6D). From these results we surmise that DNA-mediated cross-linking of PhoP involves the 5′ end of the upstream and the 3′ end of downstream repeat motifs. Together, there are three lines of evidence: (i) the PhoP-DNA complex formation involves C-terminal DNA-binding domain of the protein (35) and terminal adenine residues of the upstream (A1 at the 5′ end of DR1) and the downstream repeat motifs (A7 at the 3′ end of DR2) of the DNA, (ii) sequence-specific DNA binding stimulates cross-linking of two PhoP proteins via its N domains, and (iii) PhoP cross-linked at the N-terminal Cys-115 binds to DNA with higher affinity than its uncross-linked form, strongly suggesting that PhoP likely binds head-to-head to DNA.
DISCUSSION
There is accumulating evidence that PhoP plays a global regulatory role in M. tuberculosis, including its critical involvement in the regulation of unknown virulence determinants (12, 14, 34; for a review, see reference 28). Despite extensive genetic analyses, a detailed description of dimerization and DNA binding has not been determined for the key regulator. phoP has been previously shown to be autoregulated through direct binding of the PhoP protein to the phoP upstream region (13, 15). In the present study, we have probed regulator-promoter interactions using PhoP from M. tuberculosis H37Rv and the phoP promoter-derived oligonucleotide-based substrate DNA comprising two adjacent direct repeat motifs. We further show the importance of the conserved adenines of the repeat motifs in PhoP-DNA complex formation. While our biochemical experiments suggest that DNA binding promotes dimerization of PhoP, in conjunction with protein cross-linking experiments, we propose that two PhoP protomers bind the duplex DNA in a head-to-head orientation with the N-terminal receiver domains pointing to each other.
We had reported earlier that phosphorylation of PhoP from M. tuberculosis H37Ra was not required for DNA binding (15). However, phosphorylation displayed enhanced DNA binding by protein-protein interactions in DNase I footprinting experiments (29). A recent study showed phosphorylation-dependent binding of PhoP from M. tuberculosis H37Rv to the phoP promoter region (5). Although differences in in vitro phosphorylation and DNA-binding conditions, as well as substrate DNA used in the binding experiments, appear to account for the variability, the results reported here involved studies with PhoP from M. tuberculosis H37Rv in the unphosphorylated form. Our results are in agreement with another recent study showing phosphorylation-independent binding of PhoP to its own promoter (13). Phosphorylation-independent low-affinity binding of PhoP to its own promoter also finds support from the structural studies of the DNA-binding domain, which demonstrates specific DNA binding by the C domain of PhoP (lacking the N-terminal phosphorylation domain) to form a complex stable to gel electrophoresis (35).
Although PhoP remains predominantly monomeric in solution (Fig. 4A), our data show recruitment of two PhoP protomers on DR1,2, a result suggested by Ferguson analysis (Fig. 4B) and confirmed by competitive EMSA (Fig. 4C). In agreement with these observations, OmpR has been shown to be monomeric in solution and to bind multiple repeat elements upstream of target genes in a tandem fashion (23). Using affinity cleavage experiments, two molecules of OmpR have been proposed to bind asymmetrically to DNA, with each monomer having its recognition helix in the major groove (16). However, protein cross-linking experiments suggest head-to-head symmetric binding of two OmpR molecules on the target DNA (22). Other than the contradictory results on the orientation of the OmpR monomers within an OmpR-DNA complex, the only available structural data regarding the DNA protein complexes indicate that E. coli PhoB-DNA (3) and B. subtilis Spo0A-DNA (36) display tandem binding of the protein on two adjacent repeat motifs, where a single protomer recognizes each repeat sequence. Two or more tandemly arranged PhoP binding sites have also been reported in the pstS promoter of Streptomyces coelicolor (30). Recruitment of two PhoP protomers on two adjacently arranged direct repeat sites provides an interesting example of how similar sequence modules with minor variations of nucleotide sequence enable functional diversification by the same family of RR.
In order to establish the specific nature of the PhoP recruitment, oligonucleotides with various orientations between two repeat motifs were synthesized (Fig. 1C), and the binding was compared to DR1,2 containing the natural sequence (Fig. 1A). Notably, the very little change in the overall DNA-binding efficiency (5% ± 0.5% change of C50) of PhoP with DR2,1 (where the DR2 site is upstream of DR1; Fig. 1D, lanes 2 to 7) suggests simultaneous binding of PhoP to both the upstream and the downstream repeat motifs. What is particularly interesting and we think mechanistically important is that when the orientation of DR2 sequence of DR1,2 is changed by turning around (in DR1,2I) so that the DR1 and DR2 sites are inverted instead of direct repeats, binding of PhoP is virtually abolished (Fig. 1D, lanes 9 to 14). The failure to see significant amounts of complex stable to gel electrophoresis with DR1,2I probably reflects the steric interference from the singly bound PhoP.
PhoP is a member of the subfamily of RR proteins, which in most cases appear to recognize repeat DNA sequences. Recently, M. tuberculosis MprA, a regulator of stress, was shown to recognize two tandemly arranged, loosely conserved, 8-bp direct repeat motifs, a sequence present upstream of the mprA and pepD genes (17). However, M. smegmatis RegX3 has been shown to recognize and bind inverted repeat sites present upstream of the phosphate-regulated genes phoA and pstS (11). In contrast to M. tuberculosis MprA and M. smegmatis RegX3, we show here that PhoP recognizes a 23-bp nucleotide sequence comprising two 9-bp direct repeat motifs. Thus, it appears that the difference in the orientation of repeat motifs, the spacing between the repeats, and the nucleotide sequence of the repeat motifs in a given promoter regulate transcription factors of the same family to control diverse biological responses.
Biochemical and structural studies with many members of the PhoP subfamily of proteins indicate that these RRs bind DNA as a dimer. Consistently, the DNA-binding domain of E. coli PhoB, which shares a significant structural homology and DNA-binding mechanism with those of the C-terminal domain of PhoP (35), in complex with pho site target DNA reveals a tandem dimer (3). Also, the crystal structure of the N-terminal domain of B. subtilis PhoP shows a tandem association (2). Although recent studies suggest that the PhoP C-terminal domain predominantly exists as a monomer in solution, the crystal packing in the domain structure displayed two types of symmetric dimers (35). It has been proposed that these dimeric interfaces can potentially participate in bringing different regions of DNA together for the regulation of gene expression. However, we provide evidences here showing (i) a differential requirement of the 5′ end of the upstream and the 3′ end of the downstream repeat motif, respectively, to form PhoP-DNA complex stable to gel electrophoresis (Fig. 2 and 3); (ii) stimulation of PhoPC115 cross-linking (but not of other cysteine mutants) only by specific DNA (Fig. 5); and (iii) increased DNA-binding affinity by pre-cross-linked PhoPC115 (and not other cysteine mutants) compared to its uncross-linked form (Fig. 6). Strikingly, Cys-115 of PhoP is located within the conserved α4-β5 loop (see Fig. S2 in the supplemental material), a region which is part of the α4-β5-α5 face of the receiver domain and is known to be the proposed site of dimerization in OmpR/PhoB subfamily of RR proteins (24). It is noteworthy that the highly conserved α4-β5-α5 region of N domains has been shown to act as an interface to stabilize symmetric dimer formation on DNA for E. coli ArcA, TorR, and KdpE proteins (32, 33). Together, these results reflect a head-to-head orientation of two PhoP molecules on target DNA. This is consistent with a structure-based model of the C-terminal DNA-binding domain of PhoP, where critical DNA-binding interfaces such as the N-terminal end of α6 and one side of the α8 helix would remain blocked in a tandem association of the proteins and, as a consequence, the recognition helix would be unavailable for inserting into the major groove of DNA (35). The fact that PhoP appears to bind symmetrically (head-to-head) on tandem direct repeats suggests that one of the two direct repeats is being recognized backward (i.e., one recognizes 5′ to 3′ and the other recognizes 3′ to 5′). However, reversing DR2 in DR1DR2I so that the two repeats are symmetrical abrogates binding significantly (Fig. 1D). Note that if DR2 is recognized 3′ to 5′ (3′-ACGGTCATCaga-5′; the lowercase letters represent the spacer sequence between two direct repeats), there is considerable sequence conservation, including the nucleotides that have the most significant effect on binding (A1 of the DR1 and A7 of the DR2 repeat motifs, respectively).
In conclusion, our results identify sequence determinants recognized by PhoP and provide new insights into the substrate specificity of the key regulator. We show that PhoP-mediated autoregulation involves sequence-specific recognition of two adjacent 9-bp direct repeat motifs by two PhoP protomers. We also identify several DNA-binding features that contribute to the observed pattern of DNA binding. Whereas the structural data could only predict tandem binding as an unlikely possibility, the results reported here demonstrate that a tandem dimer of PhoP simply cannot fit with our biochemical results. However, the head-to-head model, a striking exception from the previous model of E. coli PhoB (3) and B. subtilis Spo0A (36), accounts for the EMSA and cross-linking results. Together, the study provides critical insight into the mechanism of DNA binding by the key regulator and suggests a model of the PhoP-DNA binary complex where proteins are predicted to bind the duplex DNA with a symmetric head-to-head orientation to project their N termini toward one another.
Supplementary Material
Acknowledgments
This study was supported in part by the Council of Scientific and Industrial Research (CSIR) and by research grants (to D.S.) from the Department of Science and Technology (SR/SO/BB-16/2007) and the Department of Biotechnology (BT/PR8545/Med/14/1262/2006) of the Government of India. A.P. and A.S. are predoctoral students supported by research fellowships from the CSIR.
We thank Manish Dutt and Balwinder Singh of the Bioinformatics Division for homology modeling studies of PhoP; Karthikeyan Subramanian for helpful discussions; Sunil Mukherjee of the International Center for Genetic Engineering and Biotechnology, New Delhi, India, for critical comments on the manuscript; and Renu Sharma for technical assistance.
Footnotes
Published ahead of print on 9 October 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Av-Gay, Y., and V. Deretic. 2005. Two-component systems, protein kinases, and signal transduction in Mycobacterium tuberculosis, p. 359-367. In S. T. Cole (ed.), Tuberculosis and the tubercle bacillus. ASM Press, Washington, DC.
- 2.Birck, C., Y. Chen, F. M. Hulett, and J. P. Samama. 2003. The crystal structure of the phosphorylation domain in PhoP reveals a functional tandem association mediated by an asymmetric interface. J. Bacteriol. 185:254-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanco, A. G., M. Sola, F. X. Gomis-Ruth, and M. Coll. 2002. Tandem DNA recognition by PhoB, a two-component signal transduction transcriptional activator. Structure 10:701-713. [DOI] [PubMed] [Google Scholar]
- 4.Buckler, D. R., Y. Zhou, and A. M. Stock. 2002. Evidence of intradomain and interdomain flexibility in an OmpR/PhoB homolog from Thermotoga maritima. Structure 10:153-164. [DOI] [PubMed] [Google Scholar]
- 5.Chesne-Seck, M. L., N. Barilone, F. Boudou, J. A. Gonzalo, et al. 2008. A point mutation in the two-component regulator PhoP-PhoR accounts for the absence of polyketide-derived acyltrehaloses but not that of phthiocerol dimycocerosates in Mycobacterium tuberculosis H37Ra. J. Bacteriol. 190:1329-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeLano, W. L. 2002. The PyMOL user's manual. DeLano Scientific, Palo Alto, CA.
- 7.Ferguson, K. A. 1964. Starch-gel electrophoresis-application to the classification of pituitary proteins and polypeptides. Metabolism 13:985-1002. [DOI] [PubMed] [Google Scholar]
- 8.Fontan, P. A., S. Walters, and I. Smith. 2004. Cellular signaling pathways and transcriptional regulation in Mycobacterium tuberculosis: stress control and virulence. Curr. Sci. 86:122-134. [Google Scholar]
- 9.Frigui, W., D. Bottai, L. Majlessi, M. Monot, et al. 2008. Control of M tuberculosis ESAT-6 secretion and specific T-cell recognition by PhoP. PLoS Pathog. 4:e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reference deleted.
- 11.Glover, R. T., J. Kriakov, S. J. Garforth, A. D. Baughn, et al. 2007. The two-component regulatory system senX3-regX3 regulates phosphate-dependent gene expression in Mycobacterium smegmatis. J. Bacteriol. 189:5495-5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalo Asensio, J., C. Maia, N. L. Ferrer, N. Barilone, et al. 2006. The virulence-associated two-component PhoP-PhoR system controls the biosynthesis of polyketide-derived lipids in Mycobacterium tuberculosis. J. Biol. Chem. 281:1313-1316. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalo Asensio, J., C. Y. Soto, A. Arbues, J. Sancho, et al. 2008. Mycobacterium tuberculosis phoPR operon is positively autoregulated in the virulent strain H37Rv. J. Bacteriol. 190:7068-7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalo Asensio, J., S. Mostowy, J. Harders-Westerveen, K. Huygen, et al. 2008. PhoP: a missing piece in the intricate puzzle of Mycobacterium tuberculosis virulence. PloS ONE 3:e3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta, S., A. Sinha, and D. Sarkar. 2006. Transcriptional autoregulation of Mycobacterium tuberculosis PhoP involves recognition of novel direct repeat sequences in the regulatory region of the promoter. FEBS Lett. 580:5328-5338. [DOI] [PubMed] [Google Scholar]
- 16.Harrison-McMonagle, P., N. Denissova, E. Martinez-Hackert, R. H. Ebright, et al. 1999. Orientation of OmpR monomers within an OmpR:DNA complex determined by DNA affinity cleaving. J. Mol. Biol. 285:555-566. [DOI] [PubMed] [Google Scholar]
- 17.He, H., and T. C. Zahrt. 2005. Identification and characterization of a regulatory sequence recognized by Mycobacterium tuberculosis persistence regulator MprA. J. Bacteriol. 187:202-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, et al. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51-59. [DOI] [PubMed] [Google Scholar]
- 19.Reference deleted.
- 20.Laskowski, R. A., D. S. Moss, and J. M. Thornton. 1993. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 231:1049-1067. [DOI] [PubMed] [Google Scholar]
- 21.Lee, J. S., R. Krause, J. Schreiber, H. J. Mollenkopf, et al. 2008. Mutation in the transcriptional regulator PhoP contributes to avirulence of Mycobacterium tuberculosis H37Ra strain. Cell Host Microbe 3:97-103. [DOI] [PubMed] [Google Scholar]
- 22.Maris, A. E., D. Walthers, K. Mattison, N. Byers, et al. 2005. The response regulator OmpR oligomerizes via β-sheets to form head-to-head dimers. J. Mol. Biol. 350:843-856. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Hackert, E., and A. M. Stock. 1997. Structural relationships in the OmpR family of winged helix transcription factors. J. Mol. Biol. 269:301-312. [DOI] [PubMed] [Google Scholar]
- 24.Nowak, E., S. Panjikar, P. Konarev, D. I. Svergun, et al. 2006. The structural basis of signal transduction for the response regulator PrrA from Mycobacterium tuberculosis. J. Biol. Chem. 281:9659-9666. [DOI] [PubMed] [Google Scholar]
- 25.Okamura, H., S. Hanaoka, A. Nagadoi, K. Makino, et al. 2000. Structural comparison of the PhoB and OmpR DNA-binding/transactivation domains and the arrangement of PhoB molecules on the phosphate box. J. Mol. Biol. 295:1225-1236. [DOI] [PubMed] [Google Scholar]
- 26.Perez, E., S. Samper, Y. Bordas, C. Guilhot, et al. 2001. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol. Microbiol. 41:179-187. [DOI] [PubMed] [Google Scholar]
- 27.Robbins, J. B., M. Stapleton, M. J. Stanger, D. Smith, et al. 2007. Homing endonuclease I-TevIII: dimerization as a means to a double-strand break. Nucleic Acids Res. 35:1589-1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryndak, M., S. Wang, and I. Smith. 2008. PhoP, a key player in Mycobacterium tuberculosis virulence. Trends Microbiol. 16:528-534. [DOI] [PubMed] [Google Scholar]
- 29.Sinha, A., S. Gupta, S. Bhutani, A. Pathak, et al. 2008. PhoP-PhoP interaction at adjacent PhoP binding sites is influenced by protein phosphorylation. J. Bacteriol. 190:1317-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sola-Landa, A., A. Rodriguez-Garcia, E. Franco-Dominguez, and J. F. Martin. 2005. Binding of PhoP to promoters of phosphate regulated genes in Streptomyces coelicolor: identification of PHO boxes. Mol. Microbiol. 56:1373-1385. [DOI] [PubMed] [Google Scholar]
- 31.Soto, C. Y., M. C. Menendez, E. Perez, S. Samper, et al. 2004. IS6110 mediates increased transcription of the phoP virulence gene in a multidrug-resistant clinical isolate responsible for tuberculosis outbreaks. J. Clin. Microbiol. 42:212-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toro-Roman, A., T. R. Mack, and A. M. Stock. 2005. Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: a symmetric dimer mediated by the α4-β5-α5 face. J. Mol. Biol. 349:11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toro-Roman, A., T. Wu, and A. M. Stock. 2005. A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci. 14:3077-3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walters, S. B., E. Dubnau, I. Kolesnikova, F. Laval, et al. 2006. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol. Microbiol. 60:312-330. [DOI] [PubMed] [Google Scholar]
- 35.Wang, S., J. Engohang-Ndong, and I. Smith. 2007. Structure of the DNA-binding domain of the response regulator PhoP from Mycobacterium tuberculosis. Biochemistry 46:14751-14761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao, H., T. Msadek, J. Zapf, T. Madhusudan, et al. 2002. DNA complexed structure of the key transcription factor initiating development in sporulating bacteria. Structure 10:1041-1050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.