

Abstract
Adenylylsulfate reductase (adenosine 5′-phosphosulfate [APS] reductase [APSR]) plays a key role in catalyzing APS to sulfite in dissimilatory sulfate reduction. Here, we report the crystal structure of APSR from Desulfovibrio gigas at 3.1-Å resolution. Different from the α2β2-heterotetramer of the Archaeoglobus fulgidus, the overall structure of APSR from D. gigas comprises six αβ-heterodimers that form a hexameric structure. The flavin adenine dinucleotide is noncovalently attached to the α-subunit, and two [4Fe-4S] clusters are enveloped by cluster-binding motifs. The substrate-binding channel in D. gigas is wider than that in A. fulgidus because of shifts in the loop (amino acid 326 to 332) and the α-helix (amino acid 289 to 299) in the α-subunit. The positively charged residue Arg160 in the structure of D. gigas likely replaces the role of Arg83 in that of A. fulgidus for the recognition of substrates. The C-terminal segment of the β-subunit wraps around the α-subunit to form a functional unit, with the C-terminal loop inserted into the active-site channel of the α-subunit from another αβ-heterodimer. Electrostatic interactions between the substrate-binding residue Arg282 in the α-subunit and Asp159 in the C terminus of the β-subunit affect the binding of the substrate. Alignment of APSR sequences from D. gigas and A. fulgidus shows the largest differences toward the C termini of the β-subunits, and structural comparison reveals notable differences at the C termini, activity sites, and other regions. The disulfide comprising Cys156 to Cys162 stabilizes the C-terminal loop of the β-subunit and is crucial for oligomerization. Dynamic light scattering and ultracentrifugation measurements reveal multiple forms of APSR upon the addition of AMP, indicating that AMP binding dissociates the inactive hexamer into functional dimers, presumably by switching the C terminus of the β-subunit away from the active site. The crystal structure of APSR, together with its oligomerization properties, suggests that APSR from sulfate-reducing bacteria might self-regulate its activity through the C terminus of the β-subunit.
Sulfate-reducing bacteria (SRB) are a special group of prokaryotes that are found in sulfate-rich environments because of their ability to metabolize sulfate. SRB use sulfate as the final electron acceptor in various anaerobic environments, such as soil, oil fields, the sea, or the innards of animals or even human beings (10, 11, 19, 25, 33). Their ability to degrade sulfate offers protection against environmental pollution. SRB can remove sulfate and toxic heavy atoms from factory waste waters (12). The Desulfovibrio species is a much-studied representative of SRB, and Desulfovibrio gigas has been studied under many diverse conditions to elucidate metabolic pathways (23, 35).
Sulfate reduction is one of the oldest forms of cellular metabolism. The reduction can be either assimilatory or dissimilatory. Sulfate is the terminal electron acceptor in dissimilatory reduction and the raw material for the biosynthesis of cysteine in assimilatory reduction. The latter type of reduction occurs in archaebacteria, bacteria, fungi, and plants via various pathways (17). For example, in Escherichia coli, the reduction initially catalyzes sulfate to adenosine 5′-phosphosulfate (APS) by ATP sulfurylase. APS is then phosphorylated by APS kinase to 3′-phosphate APS, which is then further reduced to sulfite by 3′-phosphate APS reductase (APSR). Finally, sulfite is reduced by sulfite reductase to sulfide, which condenses with O-acetylserine by O-acetylserine lyase to form cysteine. For comparison, in dissimilatory sulfate reduction, sulfate is first catalyzed by ATP sulfurylase to APS, which is then directly reduced by APSR to sulfite. Sulfite is subsequently reduced by dissimilatory sulfite reductase to the following three possible products: trithionite (S3O62−), thiosulfate (S2O32−), or sulfide (S2−).
Adenylylsulfate reductase, also called APSR, plays an important role in catalyzing APS to AMP and sulfite in the dissimilatory sulfate reduction. APSR was first partially purified and characterized from Desulfovibrio desulfuricans (32). Multiple forms of APSR in Desulfovibrio vulgaris were observed in buffers under varied conditions (1) and were found in the cytoplasm of cells (18). APSR from D. gigas was first purified by Lampreia et al. (21) and showed a molecular mass of 400 kDa comprised of α- and β-subunits, corresponding to the molecular masses of 70 kDa and 23 kDa, respectively. One flavin adenine dinucleotide (FAD) and two [4Fe-4S] clusters per APSR have been observed and characterized by electron paramagnetic resonance and Mössbauer spectroscopy. The enzyme from D. gigas has been described as an α2β complex involving one FAD and two [4Fe-4S] clusters (20). In D. vulgaris, APSR is apparently an α2β2 complex with a molecular mass of 186 kDa; only one Fe-S cluster is found in the αβ-heterodimer (31). Thus, the subunit and quaternary structures of APSR and their constitution of cofactors in terms of FAD and iron-sulfur clusters are still under debate. Only the enzyme from Archaeoglobus fulgidus has benefited from having an X-ray crystal structure. In this APSR, the functional unit has been shown to be the 1:1 αβ-heterodimer, containing two iron-sulfur clusters and one FAD in the structure (7). However, crystal packing shows that the asymmetric unit is an α2β2-heterotetramer.
The catalytic mechanism of APSR can be divided into the transport of electrons and the cleavage of APS by FAD. Electron input to the FAD catalyzes the cleavage of APS, releasing AMP and sulfite. Although there have been a number of mechanisms proposed to explain the catalytic cleavage of APS to AMP and sulfite (7, 8, 13, 20, 34), many features of the postulated mechanism remain unsettled, including the proteinogenic hydrogen acceptor in the reaction, the conformational change in the enzyme induced by reduction/oxidation of the FAD cofactor, and the reasons for the observed multiple forms of APSR. The divergence between A. fulgidus and Desulfovibrio species also suggests an obvious distinction in the phylogeny of the α- and β-subunits of APSR.
To clarify the difference between APSR from A. fulgidus and that from Desulfovibrio species, we have undertaken a structural study of APSR from D. gigas for comparison with the A. fulgidus enzyme. We have isolated and purified APSR directly from massive, anaerobically grown D. gigas cells for structure determination and characterization. The comparison of the structures and sequences revealing the notable differences at the C termini, activity sites, and other regions for the function is discussed. The structure of oxidized APSR from D. gigas provides much direct evidence about the subunit interactions and the role of the quaternary structure in the regulation of the catalytic mechanism.
MATERIALS AND METHODS
Growth of the organism and preparation of crude extracts.
Desulfovibrio gigas (ATCC 19364) was grown at 37°C for 22 h in a lactate-sulfate medium modified from the previously described procedure (23). The adjusted medium containing the following ingredients (in distilled water) were used: 60% sodium lactate, 12.5 ml/liter; Na2SO4, 4 g/liter; NH4Cl, 2 g/liter, MgSO4·7H2O, 2 g/liter; K2HPO4, 0.5 g/liter; Na2S·9H2O, 0.063 g/liter; CaCl2·2H2O, 0.1 g/liter; FeSO4·7H2O, 0.01 g/liter; and yeast extract, 1 g/liter. This medium was supplemented by the addition of a trace element solution (1 ml/liter), which contained the following: nitrilotriacetic acid, 1.5 g/liter; MgSO4·7H2O, 3 g/liter; MnSO4, 0.5 g/liter; NaCl, 1 g/liter; FeSO4·7H2O, 0.1 g/liter; CoCl2·6H2O, 0.12 g/liter; CoSO4·7H2O, 0.1 g/liter; NiCl2·2H2O, 0.1 g/liter; CaCl2·2H2O, 0.1 g/liter; ZnSO4·7H2O, 0.1 g/liter; CuSO4·5H2O, 0.01 g/liter; AlKSO4, 0.1 g/liter; H3BO3, 0.01 g/liter; Na2MoO4·2H2O, 0.1 g/liter; and Na2SeO3, 0.001 g/liter. The pH of the medium was adjusted to 7.6 before being autoclaved at 120°C for 20 min. Cells were suspended in Tris buffer (10 mM, pH 7.6) at a 1:1 (vol/vol) ratio at 4°C and frozen at −20°C for 12 h. Cells were centrifuged (8,000 rpm) to separate the periplasm after defrosting (18). The centrifuged cells were collected and resuspended with Tris buffer (10 mM) at a 1:1 (vol/vol) ratio. The cells were sonicated for 20 min after addition of small amounts of DNase I and DNase II. The crude extracts were ultracentrifuged at 40,000 rpm for 2 h to remove the membrane pellet. The solution was dialyzed with Tris buffer (10 mM, pH 7.6) at 4°C for 12 h for further purification.
Purification of APSR from D. gigas.
All purification procedures were performed in Tris buffer (pH 7.6) at 4°C. To minimize the duration of the purification, the mass of total protein was limited to less than 500 mg. The purity of the protein was examined by an activity assay and the UV-visible absorption ratio (A278/A392). The purification steps were performed as previously described, with some modification to minimize the time course of the process. The crude extract (210 mg) was loaded into a DE-52 column (2.5 cm by 8 cm) equilibrated in Tris buffer (20 mM, pH 7.6). The column was first washed with Tris buffer (20 mM, 100 ml) and then treated with 200 ml Tris with a gradient from 10 mM to 300 mM. After the fractions with a yellow color eluted between 150 mM and 200 mM were collected, the buffer was switched to Tris (10 mM) with a Centricon centrifugal filter device (Amicon). The total yellow fractions were then applied into the macro-DEAE column (2.5 cm by 4 cm) equilibrated previously with Tris buffer (10 mM). After washing (20 mM Tris, 80 ml) and elution (10 mM to 300 mM Tris, 150 ml) steps were performed, the fractions with APSR activity (eluted from 200 mM to 250 mM Tris) were loaded on to the hydroxyapatite (HTP) column (2.5 cm by 2.5 cm) equilibrated previously with 300 mM Tris. The HTP column was washed with step gradients of Tris concentrations (20 mM, 50 ml; 150 mM, 50 ml; 300 mM, 50 ml) to discard the unbound proteins. The active fractions were eluted after washing the HTP column with phosphate buffer (20 mM) until colorless fractions emerged. The active fractions were collected, and the buffer was changed to 10 mM Tris with Centricon (Amicon). The last purification step was performed on the fast protein liquid chromatography system with a macro-DEAE (1.5-cm by 3.5-cm) column. APSR was eluted between 200 mM and 250 mM Tris. The purified APSR protein was dialyzed, desalted, and concentrated before used in biochemical assays and crystallization.
Activity assay of APSR.
The activity of APSR was assayed in reverse by determining the formation of APS from AMP with a modified procedure described previously (32). The activity assay depends on the transfer of electrons from AMP to oxidized APSR to yield the reduced state. The reduced APSR can transfer electrons to the oxidizing agent ferricyanide to form ferrocyanide. The absorption at 420 nm was used to monitor the quantity of ferricyanide reduced. The reaction mixture containing AMP (3.3 mM) and 2-mercaptoethanol (0.5 mM) in Tris buffer (100 mM, pH 7.6) was first used as the control solution at 420 nm and was incubated with oxidized APSR and ferricyanide [K3Fe(CN)6, 1.3 mM] for ∼100 s at 25°C. After sodium sulfite (Na2SO3, 3 mM) in EDTA (5 mM) was added into the mixture, a decrease in absorption at 420 nm was recorded. The specific activity was expressed as μmol APS formed per min and per mg of APSR.
Amino acid sequencing.
The genome DNA of D. gigas was extracted with a Wizard genomic DNA purification kit. As the gene sequence of APSR from D. gigas was not available, we designed a series of primers based on the alignment of genes of other Desulfovibrio species (D. vulgaris strain Hildenborough and D. desulfuricans). The PCR product was sequenced by Mission Biotech Co. Forward and reverse sequencing confirmed the correctness of the DNA sequence. The plasmid was transformed into E. coli JM109 cells, and the cells were developed on an LB plate. One colony was selected and confirmed by colony PCR for the right plasmid. The selected colony was amplified and extracted with a high-speed plasmid minikit (Geneaid commercial production). The purified plasmid was sequenced by Mission Biotech Co.
DLS.
Dynamic light scattering (DLS) analysis was performed with Zetasizer Nano S (Malvern Instruments) at room temperature (25°C). About 1 mg/ml of APSR was analyzed with the DTS software for each set of DLS data in 50 mM Tris buffer (1 ml), pH 7.5. To examine the effect of AMP on the oligomerization of APSR, 0.35 mM AMP was added into APSR protein solution before measurement. All protein solutions were filtered through a membrane with 0.2 μm porosity to remove any dust prior to addition to the sampling cell (DTS0012 disposable sizing cuvette). For the experiment of investigating the effect of the disulfide bond Cys156-Cys162 on APSR oligomerization, 186 mM β-mercaptoethanol was added into the APSR protein solution before measurement. All the measurements were repeated at least three times.
Analytical ultracentrifugation.
The sedimentation velocity experiments were carried out on a Beckman Optima XL-A analytical ultracentrifuge equipped with a UV-visible optics detection system, using an An-60 Ti rotor and standard double-sector cells. The molecular weights of APSR multimers were measured at 20°C with a protein concentration of 5 μM in the absence or presence of AMP. Data analysis was performed using the SEDFIT85 program (22) to determine the sedimentation coefficient distribution.
Protein crystallization.
Crystallization of the protein was accomplished by the hanging-drop vapor diffusion method at 18°C using drops (2 μl) of the purified protein (∼4.6 mg/ml) in Tris buffer (0.1 M, pH 7.4) mixed with equal volumes of the reservoir solution containing PEG 6000 (20% [wt/vol]) and ammonium sulfate (60 mM) in Tris buffer (0.1 M, pH 7.0) and equilibrated against the reservoir solution (0.5 ml) in a 24-well ADX plate (Hampton Research Corp.). The yellowish crystals of APSR appeared after 5 days and continued to grow to a final size of 0.12 by 0.12 by 0.2 mm3 after 3 weeks.
X-ray data collection and processing.
A single crystal of APSR was transferred from the crystallization drop to the cryoprotectant solution containing mother liquid with 25% glycerol in 5 μl, mounted on a nylon loop (0.1 to 0.3 mm; Hampton Research Corp.), and frozen in liquid nitrogen. The preliminary crystal quality was examined at beamline BL12B2 of SPring-8 in Japan. Data collection was completed at SPXF beamline BL13B1 equipped with a charge-coupled-device detector (Q315; ADSC) at National Synchrotron Radiation Research Center (NSRRC) in Taiwan. The diffraction data were measured, for a total rotation of 150° with 0.5° of oscillation for an exposure duration of 30 s per frame using X-ray wavelength at 1.0 Å and a distance of 460 mm from the crystal to the detector at 110 K in a nitrogen stream provided by a cryosystem (X-stream; Rigaku/MSC, Inc.). Overlap diffraction occurred in the data processing because of the long c axis of the unit cell. The data set was indexed, integrated, scaled, and merged using the program HKL2000 (31). Details of data statistics appear in Table 1.
TABLE 1.
Statistics of crystallographic data and structure refinement
| Statistics | Value(s)a |
|---|---|
| Crystal diffraction statistics | |
| Wavelength (Å) | 1.00 |
| Resolution range (Å) | 30.0-3.10 (3.21-3.10) |
| Space group | P3121 |
| Unit cell parameters (Å) | |
| a | 199.63 |
| c | 317.42 |
| Completeness (%) | 91.9 (93.9) |
| I/σ(I) | 13.63 (3.64) |
| Average redundancy | 4.6 |
| No. of unique reflections | 105,084 |
| Rsym (%) | 10.2 (44.6) |
| Mosaicity | 0.39 |
| No. of molecules per ASUb | 6 |
| Solvent content (%) | 48.06 |
| Structure refinement statistics | |
| Resolution range (Å) | 30-3.1 |
| Rwork/Rfree (%) | 19.3/24.6 |
| No. of water molecules | 69 |
| RMSD from ideal geometry | |
| Bond length (Å) | 0.06 |
| Bond angle (°) | 1.05 |
| Chirality (°) | 0.08 |
| Average B-factors (Å2) | 45.37 |
| Main chain | 45.48 |
| Side chain and water | 45.25 |
Values in parentheses are for the highest-resolution shell.
ASU, asymmetric unit.
Structure determination and refinement.
The Matthews coefficient was calculated as 3.27 Å3 Da−1, corresponding to a solvent content of 62.43% and six APSR molecules per asymmetric unit. The phase was solved by the molecular replacement method with the program MOLREP (36) in the CCP4 suite (4) using the monomer structure of APSR from A. fulgidus (Protein Data Bank [PDB] accession number, 1JNR) as a search model, with sequence similarities of 64% for the α-subunit and 82% for the β-subunit (7). The molecular replacement solution was found and confirmed that the space group is P3121 and that there are six molecules in an asymmetric unit. After rigid-body refinement using the CNS version 1.2 program (2) in a resolution range of 30 to 3.5 Å, the R and Rfree factors were 50.6% and 51.8%, respectively. Throughout the refinement, a random selection (5%) of the data was set aside as a “free data set,” and the model was refined against the remaining data as a working data set. The structure refinement was performed using the programs CNS version 1.2 and REFMAC5 (30) in the CCP4 suite. Composite omit maps of electron density with coefficients 2Fo-Fc were calculated with CNS version 1.2 and visualized using O software (15), and the model was rebuilt and adjusted iteratively as required. Several cyclic model refinements were performed, and the refinement converged to a final R factor of 19.3% and Rfree of 24.6% for all data to a resolution of 3.1 Å. The correctness of the stereochemistry of the model was verified, and the calculations of root mean square deviations (RMSD) from ideality for bonds, angles, and dihedral and improper angles performed in CNS version 1.2 showed the stereochemistry to be satisfactory. In a Ramachandran plot, all main-chain dihedral angles were in the most favored and additionally allowed regions, except for glycines. Model statistics for APSR are given in Table 1.
Protein structure accession number.
The atomic coordinates and structure factors have been deposited in the PDB with the accession number 3GYX.
RESULTS AND DISCUSSION
Purification and characterization of APSR.
APSR from D. gigas was purified in four steps. The purity of the elutions following each purification step was examined with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and an activity assay. The molecular mass of oxidized APSR was measured at 20°C in Tris-HCl buffer (pH 7.6, 25°C) by ultracentrifugation, which showed a major peak at about 500 kDa, together with other impurities. The ratio of the UV absorption (A278/A392) of the protein obtained from the final step was about 4.89, which differed slightly from the previous report of that of 4.97 (21). SDS-PAGE indicated the presence of two subunits with molecular masses of 70 kDa and 20 kDa, respectively. The elution from the last purification step using a macro-DEAE column showed a specific activity of 4.85 units (1 unit = 1 μmol APS formed per min and per mg of APSR). Interestingly, applying an additional ion-exchange column (Mono Q) after the DEAE column separated the purified enzyme further into one major fraction and one minor fraction (Fig. 1A). Activity assays of fractions 25, 27, and 28 showed relatively low activity (less than 1 unit), but fraction 26 exhibited a significant higher specific activity of 3.6 units. Surprisingly, the mixture of fractions 25 and 28 at a ratio of 1:1 shows about six times the activity of the corresponding individual fractions. Since SDS-PAGE showed that APSR from separate collected fractions (approximately fraction 25 to 28) all contained bands of mass at 70 kDa and 20 kDa (Fig. 1B), the observations clearly represented various multiple forms of the enzyme with distinct compositions in terms of the numbers of α- and β-subunits. The oligomerization of APSR from D. vulgaris had previously been observed during purification of the protein under low-salt conditions (37). With the ion-exchange column chromatography used in our work, the gradient concentration of Tris-HCl buffer might affect the composition of APSR from D. gigas.
FIG. 1.
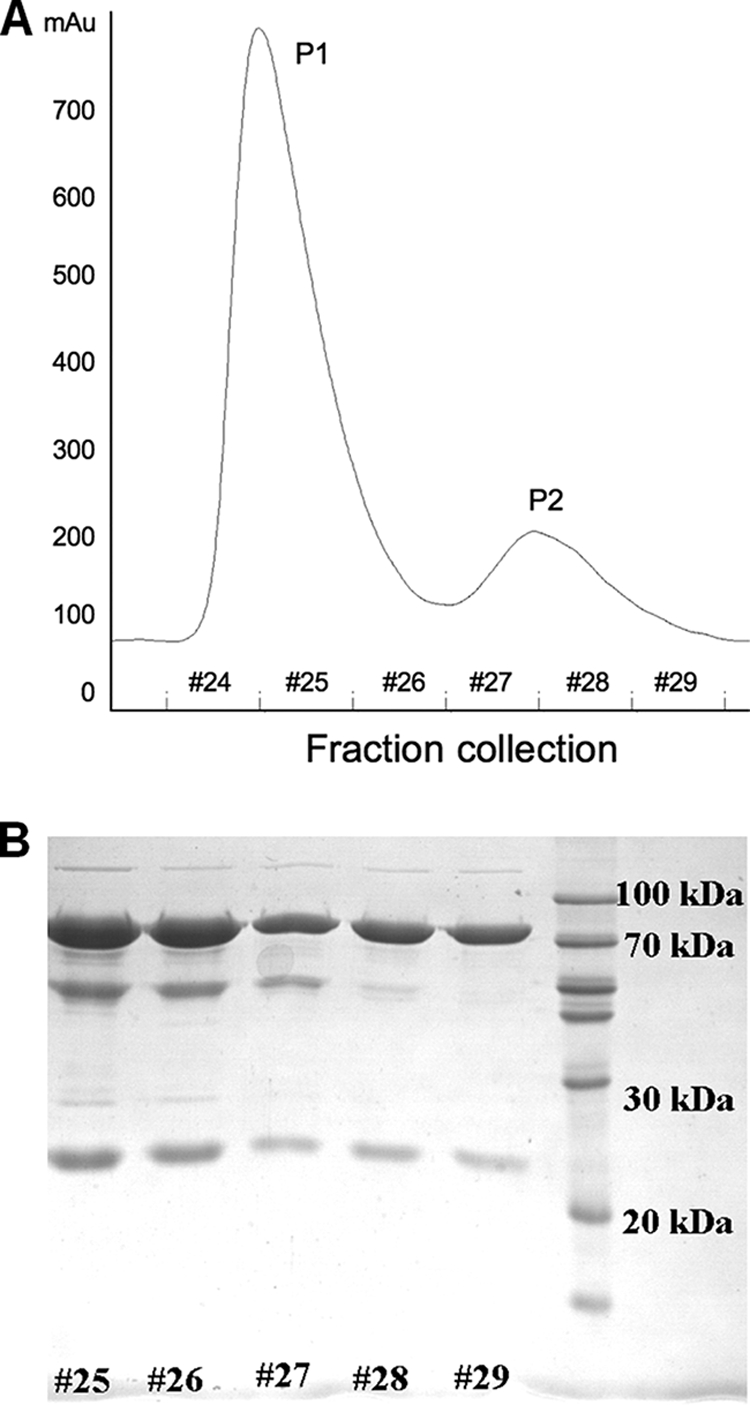
Separated fractions of APSR. (A) Two fractions from a Mono Q ion-exchange column. P1 represents the major fraction, and P2 represents the minor fraction. Fractions 24 to 29 denote the collection fractions. (B) Analysis (12% SDS-PAGE) of various collections with fractions 24 to 29 from P1 and P2. The molecular masses of the α- and β-subunits are about 70 and 20 kDa, respectively.
The UV-visible spectrum between 340 nm and 500 nm of oxidized APSR from D. gigas showed the general protein absorption at 278 nm and the absorption maximum at 392 nm, with a shoulder at 445 nm. The absorption features between 340 nm and 500 nm are due to the [4Fe-4S] clusters and FAD. These features in the UV-visible spectrum are similar to those previously reported for APSR from various Desulfovibrio species and A. fulgidus (1, 8).
Sequence of APSR from D. gigas.
The DNA sequence of APSR from D. gigas was determined. As reported for other Desulfovibrio species (27), the locus of the aprB (β-subunit) gene appears before that of the aprA (α-subunit) gene. A DNA segment containing 79 nucleotides is inserted between the aprB and aprA genes. The molecular masses of the α- and β-subunits are calculated to be 74.7 kDa and 18.5 kDa, respectively, after translating DNA to a protein sequence, which is presented in Fig. 2.
FIG. 2.

Sequence alignment of APSRs. Sequence alignment of α-subunits of APSRs (A) and that of β-subunits of APSRs (B). The abbreviations DESGI, DESVH, THBPB, and ARCFU represent the different species D. gigas, D. vulgaris strain Hildenborough, T. plumbophilus, and A. fulgidus, respectively. The secondary structures are defined according to the APSR structure from D. gigas.
The DNA-translated protein sequence of the α-subunit in D. gigas is homologous to the sequences of the corresponding proteins in D. vulgaris strain Hildenborough (93% similarity), in the sulfur-oxidizing bacteria “Thiobacillus plumbophilus” (fragmental sequence, 73% similarity), and in Archaeoglobus fulgidus (64% similarity). The sequence of the β-subunit in D. gigas is homologous to those of D. vulgaris Hildenborough (94% similarity), Thiobacillus plumbophilus (fragmental sequence, 78% similarity), and Archaeoglobus fulgidus (82% similarity). The sequence alignments of the α- and β-subunits from various species are shown in Fig. 2A and B, respectively. Eight cysteine residues in the β-subunit form two Fe-S cluster-binding motifs, Cys-X1-X2-Cys-X3-X4-Cys-…-Cys-Pro and Cys-X1-X2-Cys-X3-X4-…-X8-X9-Cys-…-Cys-Pro (X, any amino acid; ellipses, missing amino acids). Two other cysteine residues (Cys156 and Cys162) in D. gigas and D. vulgaris Hildenborough differ from those in other bacteria, according to the alignment. The enzyme from Thiobacillus plumbophilus has been postulated to operate in the reverse direction in which the sulfite is oxidized to APS (28). Although APSR performs in the reverse direction to sulfate-oxidizing bacteria, the alignment between T. plumbophilus and D. gigas notably shows a high homology.
The sequence alignment of the α-subunits in D. gigas and A. fulgidus reveals two large gaps, both in the FAD-binding domains. The sequence of D. gigas lacks the segment of amino acids (aa) 83 to 89 in A. fulgidus, whereas the sequence of A. fulgidus lacks the segment of aa 133 to 153 in D. gigas. However, the major difference between D. gigas and A. fulgidus lies in the C termini of the β-subunits, where the C terminus is notably shorter in A. fulgidus.
Overall structure of APSR.
Structure refinement of APSR yielded an R factor of 19.3% and Rfree of 24.6% (Table 1). There are, in total, six αβ-heterodimers in the asymmetric unit, and each αβ-heterodimer contains one α-subunit and one β-subunit. In the αβ-heterodimers, the C-terminal segment of the β-subunit wraps around the α-subunit, and the globular domain of the β-subunit is embedded in a shallow hollow of the α-subunit. The αβ-heterodimer performs a rotation about a pseudo-twofold axis with another αβ-heterodimer to form a tightly contacted α2β2-heterotetramer (Fig. 3A). The β-subunit C termini of the α2β2-heterotetramer exhibit anchor-like hooks on the α-subunits of another two α2β2-heterotetramers (Fig. 3B). Three α2β2-heterotetramers connect to each other through the C termini of the β-subunits to form a hexamer structure containing six αβ-heterodimers (Fig. 3C). The overall structure of APSR can be grouped into the structure family of fumarate reductase (9, 14).
FIG. 3.

Structure of the APSR hexamer from D. gigas. (A) Structure of the α2β2-heterotetramer from D. gigas. The αβ-heterodimer performs a rotation around a twofold axis (dashed line) with another αβ-heterodimer to form a tightly contacted α2β2-heterotetramer. (B) Interactions between the αβ-heterodimers and the C terminus of another β-subunit. The long C-terminal loop of the β-subunit (shown in green ribbon) plugs into the active channel of another αβ-heterodimer (shown in electrostatic surface; the positive potential is in blue, and the negative potential is in red). (C) View of the hexamer structure. Three α2β2-heterotetramers contact each other through the C-termini of the β-subunits to form a hexamer containing six αβ-heterodimers. (D) B-factor-labeled structure of the αβ-heterodimer. The structure exhibits a high B-factor in red and the larger caliber of the cartoon style. The blue box shows the high B-factor at the C terminus (aa 144 to 167) of the β-subunit. The electron-accepting site on the β-subunit that is suggested to interact with an unknown electron donor shows a higher B-factor in the red box. The capping domain in the α-subunit also shows a higher B-factor in the yellow box.
The crystal packing of APSR molecules from D. gigas differs from that of A. fulgidus (7). The asymmetric unit of APSR from D. gigas consists of six αβ-heterodimers (Fig. 3C), but the unit consists of an α2β2-heterotetramer in the case of A. fulgidus. APSR has notably variable molecular masses in different buffers and in the presence of substrates (1). The molecular mass of APSR from D. vulgaris (strain Hildenborough) is about 440 kDa in the phosphate buffer and 220 kDa in the Tris-maleate buffer. The different crystal packings between the D. gigas and the A. fulgidus proteins might be caused by the C-terminal tail of the β-subunit and the Tris-HCl buffer effect. The functional unit for APSR from A. fulgidus is the αβ-heterodimer (6, 7).
The structure of the αβ-heterodimer labeled with temperature B-factors shows an overview of the flexibility of the structure (Fig. 3D). The tail domain of the β-subunit that tightly wraps around the α-subunit exhibits no secondary structure with small B-factors, but the terminal loop of the tail domain in the β-subunit that connects the adjacent αβ-heterodimer shows large B-factors. A region of broken electron density is present between residues Ile148 and Ala151 on the C terminus of the β-subunit, which implies that the flexibility at the terminal region of the tail domain is related to the connection of the adjacent αβ-heterodimer. The residues surrounding the [4Fe-4S] cluster II on the surface of the β-subunit also exhibit large B-factors; this region presumably serves as a contact with an unknown electron donor (7).
Subunit structures.
The structure of the α-subunit is grouped into three parts, as follows: FAD-binding (3 to 280 and 410 to 503), capping (281 to 409) and helical (504 to 664) domains (Fig. 4A). The FAD-binding domain shows a noncovalent bond association with FAD, covered partially by the capping domain for exposure to APS. There is a channel with a diameter of ∼13 Å that is formed by the FAD-binding domain and the capping domain presumably to transport a substrate. The helical and FAD-binding domains form a shallow hollow in which the β-subunit is embedded. The α-subunit of APSR can be grouped into the fumarate reductase family, and the sequence identities when aligned with the sequences of aspartate oxidase and fumarate reductase from E. coli are 23% and 26%, respectively. A superimposition of the structure of APSR from D. gigas with those of aspartate oxidase (PDB accession number 1CHU) (26) and fumarate reductase from E. coli (PDB accession number 1KFY) (14) reveals the RMSD of the Cα atoms to be 10.7 Å and 6.0 Å, respectively. A superimposition of the structures shows notable differences among these three structures; the largest deviations are observed in the FAD-binding site near FAD (Fig. 4B). Structural comparison shows that the α-subunit structure of APSR from D. gigas might be homologous with the structures of the ancestor of flavoenzymes because of the early appearance of the anaerobic sulfur metabolism in their evolution.
FIG. 4.

Subunit structures of APSR from D. gigas. (A) α-Subunit. The structure of the α-subunit is grouped into the following three parts: the FAD-binding domain (red), capping domain (yellow), and helical domain (green). (B) Superimposition of FAD-binding domains. The structure of the FAD-binding domain of the α-subunit (red) is superimposed with aspartate oxidase (green) and fumarate reductase (blue) from E. coli. The superimposition shows that less similarity is exhibited in the partial FAD-binding domain that is near FAD. (C) β-Subunit. The β-subunit structure is grouped into the following three segments: [4Fe-4S] cluster-binding domain (red), β-sheet domain (yellow), and C-terminal tail domain (green). (D) Structural alignment of the cluster-binding domain in the β-subunit. The cluster-binding domain in the β-subunit of APSR from D. gigas (green) is superimposed with ferredoxin II from D. gigas (yellow) and ferredoxin from Chromatium vinosum (blue).
The β-subunit is divisible into the following three segments: [4Fe-4S] cluster-binding (aa 2 to 68), β-sheet (aa 69 to 104), and C-terminal tail (aa 105 to 167) domains (Fig. 4C). The β-subunit belongs to the ferredoxin family. The structure of the cluster-binding domain can be superimposed on ferredoxin II from D. gigas (PDB accession number 1FXD) (16) and ferredoxin from Chromatium vinosum (PDB accession number 1BLU) (29) with the RMSDs of 3.2 Å and 3.4 Å of the main-chain atoms, respectively (Fig. 4D). The folding of the cluster-binding domain is similar to the that of the ferredoxin structure and enwraps two [4Fe-4S] clusters. The cluster-binding domain is embedded in a shallow hollow of the α-subunit, with the other two domains interacting with the α-subunit to stabilize the heterodimer (Fig. 3A). The total contact area between the α- and β-subunits is calculated to be ∼4,400 Å2. In particular, the interface between the tail domain (aa 105 to 167) and the α-subunit is about 2,139 Å2, which is larger than the contact area between the other two domains (the cluster-binding and the β-sheet domains) and the α-subunit. The tail domain uses 47 residues (aa 103 to 149) to wrap around the α-subunit and 16 residues (aa 152 to 167) to form the loop that plugs into the adjacent αβ-heterodimers. The only disulfide bond (Cys156 and Cys162) of the αβ-heterodimers is formed at the loop of the tail domain. The sequence of APSR from D. vulgaris (Hildenborough) also contains two Cys residues at the same positions, which implies that the disulfide bond can also be formed in the D. vulgaris.
Cofactor structures.
The cofactors FAD and [4Fe-4S] clusters are important for the transfer of the electrons. The isoalloxazine ring of FAD in oxidized and reduced APSR from A. fulgidus was observed to be substantially bent by about 25° and 10°, respectively, with consequences on the reduction potential of the flavin (7, 34). Such bending of FAD was, however, not clearly revealed in the structure of oxidized APSR from D. gigas, presumably because of the resolution (3.1 Å) of our structure. The FAD-binding domain of the α-subunit in oxidized APSR from D. gigas exhibits six hydrogen bonds with FAD (Table 2).
TABLE 2.
Interactions between the FAD-binding domain and FADa
| Atom | Contact residueb | Contact atom | Distance (Å) |
|---|---|---|---|
| N3 | Ala76 | O | 2.82 |
| O1P | Gly35 | N | 3.02 |
| O2 | Asn78 | N | 3.01 |
| O2 | Ser472 | OG | 2.71 |
| O2 | Ser472 | N | 2.81 |
| O3P | Met34 | N | 3.46 |
FAD is the cofactor for all data given.
Contact residues are in the FAD-binding domain of the α-subunit.
There are two [4Fe-4S] clusters in the β-subunit from D. gigas. Cluster I is embedded in the β-subunit, and the binding residues for cluster I are Cys25, Cys47, Cys50, and Cys53 of the β-subunit. Cluster II, coordinated by residues Cys10, Cys13, Cys21, and Cys57, is near the surface of the β-subunit and is thought to accept electrons from an unknown electron donor to transfer electrons further to cluster I. The reduction potentials of clusters I and II are reported to be 0 mV and less than −400 mV, respectively (21). Electrons are transferred from the negative potential to the positive potential, and this driving force determines the efficiency of the electron transfer. The large difference in reduction potentials between clusters I and II is due to the number of local dipoles in close proximity to the clusters. The most-negative charges are localized on the acid-labile sulfur and cysteinyl sulfur atoms, where a cluster receives electrons from the donor. The hydrogen bonds between the amide and sulfur atoms stabilize the reduced state, and the backbone amide dipole can shift the reduction potential to a more-positive state (3, 5, 7, 24). There are 13 hydrogen bonds (NH—S) between the amide and cluster I, with distances of less than 3.6 Å, whereas cluster II shares only 9 hydrogen bonds with the amide (Table 3), which may determine the difference in reduction potentials between clusters I and II. In addition to the interactions of hydrogen bonding NH—S, the carboxylate oxygen of Asp11 of the β-subunit near the sulfur of cluster II offers an extra negative charge to stabilize the oxidized state of cluster II.
TABLE 3.
Interactions among cluster-binding motifs and residuesa
| Residue/cofactor | Atom | Contact residue | Contact atom | Distance (Å) |
|---|---|---|---|---|
| Cluster II binding domain | ||||
| Cluster II | S4 | Cys21 | N | 3.30 |
| Cluster II | S3 | Asp11 | OD2 | 3.55 |
| Cys10 | SG | Asp11 | N | 3.48 |
| Cys10 | SG | Gly12 | N | 3.23 |
| Cys13 | SG | Ala20 | N | 3.52 |
| Cys21 | SG | Met22 | N | 3.24 |
| Cys57 | SG | Gln59 | N | 3.52 |
| Cys57 | SG | Gly60 | N | 3.50 |
| Cys57 | SG | Pro58 | N | 3.50 |
| Cys57 | SG | Ala61 | N | 3.25 |
| Cluster I binding domain | ||||
| Cluster I | S1 | Cys53 | N | 3.48 |
| Cluster I | S2 | Trp48 | N | 3.54 |
| Cluster I | S3 | Asn41 | ND2 | 3.45 |
| Cluster I | S4 | Cys50 | N | 3.17 |
| Cluster I | S4 | Tyr51 | N | 3.18 |
| Cys25 | SG | Pro26 | N | 3.59 |
| Cys25 | SG | Asn27 | N | 3.50 |
| Cys25 | SG | Leu29 | N | 3.37 |
| Cys25 | SG | Asp28 | N | 3.57 |
| Cys47 | SG | Glu49 | N | 3.42 |
| Cys50 | SG | Ser52 | N | 3.59 |
| Cys50 | SG | Tyr51 | N | 3.58 |
| Cys53 | SG | Ile54 | N | 3.13 |
All residues are in the β-subunit.
Structural comparison of APSR from A. fulgidus and D. gigas.
The superimposed oxidized APSR structures from D. gigas and A. fulgidus show a RMSD of 0.87 Å for Cα atoms and reveal the structural differences between the enzymes from the two species. Two segments (aa 83 to 84 and aa 131 to 154 in D. gigas APSR) in the sequence alignment (Fig. 2A) of the α-subunits reveal large gaps and exhibit notable structural variations. Segments (aa 83 to 89, aa 326 to 332, and aa 289 to 299) in the α-subunit of D. gigas APSR provide an altered entrance of the substrate-binding channel relative to the structure from A. fulgidus, as discussed below. Another structural difference is evident in the FAD-binding site, where the segment (aa 127 to 156) forms a loop, following a short α-helix, to allow residue Lys143 in the α-subunit to form a hydrogen bond with residue Thr106 in the β-subunit to offer increased interactions between α- and β-subunits (see Fig. S1A in the supplemental material).
The largest structural difference in the β-subunit is observed in the C terminus, where the longer segment (aa 122 to 167) in the D. gigas structure affects the substrate-binding site in the α-subunit, as discussed earlier. The two segments (aa 121 to 124 and aa 132 to 135) in the β-subunit from D. gigas form a smaller area of contact with the α-subunit than with the β-subunit from A. fulgidus. In the latter structure, there are two loops (aa 121 to 125 and aa 134 to 143 from A. fulgidus) in the tail domain, which might imply some flexibility of the C terminus in the β-subunit from D. gigas (see Fig. S1B in the supplemental material).
Substrate-binding channel and the binding site.
The electrons are presumably transferred from an unknown electron donor through the two [4Fe-4S] clusters I and II in the β-subunit to the FAD in the α-subunit over a linear distance of about 22 Å. The substrate channel is formed by the FAD-binding and the capping domains. The channel opening is about 19.5 Å in linear distance from the N5 atom of FAD (Fig. 5). Trp48 of the β-subunit is sandwiched between FAD and cluster I, in van der Waals contact with both cofactors, and presumably serves as a conduit for electron transfer from cluster I to FAD. The tryptophan side chain is locked into position by π interactions with Arg249 and hydrogen bonding with the carbonyl oxygen of Ala250 on the α-subunit. According to a superimposition of APSR structures from D. gigas and A. fulgidus, there is only a slight shift in the position of Trp48 between the two enzymes, underscoring the importance of this aromatic residue in the electron transfer of reducing equivalents from the iron-sulfur cluster I to FAD.
FIG. 5.
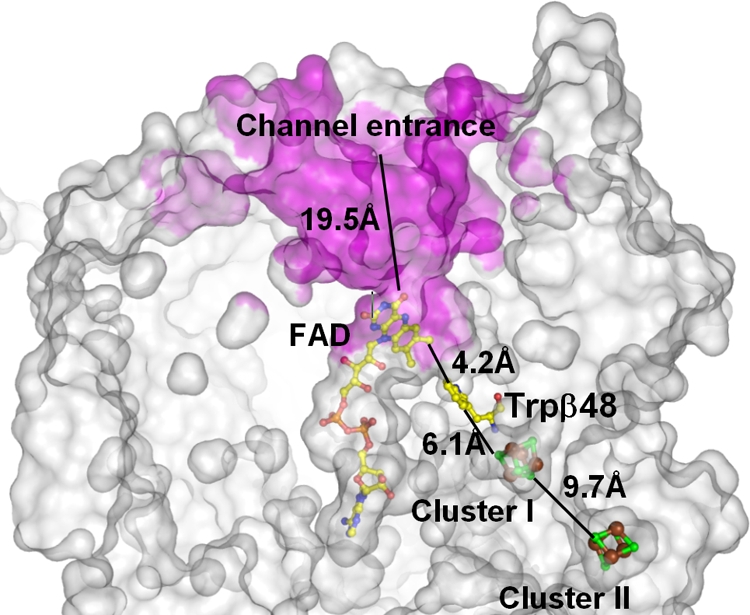
Active-site channel of APSR. The clipped molecular surface shows the active-site channel (magenta) and the positions of cofactors, with the related distances. The substrates arrive in the channel with a linear distance of about 19.5 Å from the entrance of the channel to atom N5 of FAD. The electrons are transferred from an unknown electron donor through two [4Fe-4S] clusters and the conserved Trpβ48 to FAD, with a total linear distance of about 22 Å.
There is a substantial difference in the structure of the substrate channel between APSR from D. gigas and that from A. fulgidus. Compared to the A. fulgidus enzyme, the D. gigas enzyme is lacking the segment 82LTGRSER on the α-subunit. This sequence contains two Arg residues and one Glu residue. The positively charged side chain of Arg83 in A. fulgidus was thought to play a role in recognizing the substrates (34). A structural comparison of APSR between D. gigas and A. fulgidus reveals that the segment (aa 80 to 89) forms a shorter loop in APSR structure from D. gigas (see Fig. S1C in the supplemental material). None of the side chains of the residues that form this short loop are positively charged in D. gigas, and all are, in fact, facing in the direction opposite to the channel entrance. An inspection of the superimposed APSR structures of A. fulgidus and D. gigas shows that residue Arg160 of the α-subunit from D. gigas is another positively charged residue facing the channel entrance, located between the positively charged Arg83 and the negatively charged Glu143 of the α-subunit from A. fulgidus (see Fig. S1C in the supplemental material). Thus, it is likely that the positively charged residue Arg160 of D. gigas replaces the role of Arg83 in the structure of A. fulgidus for the recognition of substrates. Similar positively charged residues arginine and lysine are notably present in APSR from D. vulgaris strain Hildenborough and T. plumbophilus, respectively, according to sequence alignments (Fig. 2A).
The Arg160 identified above, together with the conserved residues Lys298, Lys300, and Arg336 in the α-subunit of D. gigas, provides a positively charged surface at the channel entrance, with a diameter of about 13 Å, for the recognition of substrates. Interestingly, in the crystal structure of the D. gigas APSR protein, the channel entrance is occupied by the C terminus of the β-subunit from another αβ-heterodimer (Fig. 6A). There are 15 hydrogen bonds formed by interactions between the α-subunit and the loop of the C terminus of the β-subunit from the second αβ-heterodimer. On the loop of the C terminus of the β-subunit, the negatively charged residues Gluβ152 and Gluβ163 interact with the positively charged residues Lysα300 and Argα160, respectively, at the entrance of the substrate-binding channel of the α-subunit in the first αβ-heterodimer (Table 4). Similarly, residue Aspβ159 interacts with Argα282, and the terminal residue Argβ167 of the β-subunit inserts into the negatively charged space formed by Glu377, Glu380, and Glu381 in the α-subunit of the first αβ-heterodimer (Fig. 6A).
FIG. 6.

(A) Interactions between the α- and β-subunits at the active site. Arg160, Lys298, Lys300, and Arg366 form a positively charged surface at the entrance of the substrate-binding channel in the α-subunit. Surface areas with positive and negative electrostatic potentials are shown in blue and red, respectively. The negatively charged Glu152 and Glu163 on the C-terminal loop of the β-subunit from another αβ-heterodimer interact with the positively charged surface of the channel entrance, and Arg167 and Asp159 are located at the oppositely charged environment. (B) The electron density of the 2Fo-Fc composite omit map shows that the side chain of Argα336 (black; σ = 1.5) in the α-subunit faces the entrance of the substrate-binding channel (“arginine out”), which is occupied by the loop of the C terminus (magenta; σ = 1.5) of the β-subunit. The disulfide bond between Cysβ156 and Cysβ162 is clearly revealed. Argα282 (black; σ = 1.5) at the substrate-binding site interacts with Aspβ159 on the C terminus. (C) The C-terminal loop of the β-subunit blocks the substrate-binding site. APSR structures soaked with substrates APS and AMP from A. fulgidus are colored in yellow and purple, and the structures of the α- and β-subunits from D. gigas are shown in green and cyan, respectively. The C-terminal loop of the β-subunit from another αβ-heterodimer from D. gigas overlaps with the soaked substrates APS and AMP in A. fulgidus. There is a hydrogen bond interaction between the substrate-binding residue Arg282 of the α-subunit and Asp159 of the C-terminal loop of the β-subunit. The negatively charged Glu152 and Glu163 on the C terminus of the β-subunit interact with Lys300 and Arg160, respectively, at the channel entrance of the α-subunit.
TABLE 4.
Interactions between the β-subunit and the active channela
| Source residue | Atom | Contact residue | Contact atom | Distance (Å) |
|---|---|---|---|---|
| Glu152 | CB | Lys300 | NZ | 3.52 |
| Glu152 | CG | Lys300 | NZ | 3.94 |
| Glu152 | CD | Lys298 | CG | 3.89 |
| Lys300 | NZ | 4.00 | ||
| Glu152 | OE1 | Lys298 | CE | 3.48 |
| Lys298 | CG | 3.68 | ||
| Glu152 | OE2 | Lys298 | CG | 3.47 |
| Lys300 | CE | 3.79 | ||
| Lys300 | NZ | 3.18 | ||
| Ser154 | C | Arg336 | NH1 | 3.86 |
| Ser154 | O | Arg336 | CZ | 3.80 |
| Arg336 | NH1 | 2.88 | ||
| Arg336 | NH2 | 3.82 | ||
| Gln155 | CA | Arg336 | NH1 | 3.68 |
| Gln155 | OE1 | Lys300 | CB | 3.49 |
| Lys300 | CA | 3.69 | ||
| Lys300 | C | 3.73 | ||
| Gln155 | C | Arg336 | NH1 | 3.68 |
| Cys156 | N | Arg336 | CZ | 3.96 |
| Arg336 | NH1 | 3.33 | ||
| Cys156 | CB | Arg336 | CZ | 3.41 |
| Arg336 | NH1 | 3.67 | ||
| Arg336 | NH2 | 3.64 | ||
| Arg336 | NE | 3.66 | ||
| Cys156 | O | Arg336 | CD | 3.91 |
| Asp159 | CG | Arg282 | NH2 | 3.38 |
| Asp159 | OD1 | Arg282 | CZ | 3.76 |
| Arg282 | NH2 | 2.57 | ||
| His418 | NE2 | 3.49 | ||
| Asp159 | OD2 | Arg282 | NH2 | 3.43 |
| Glu163 | CG | Arg160 | NH1 | 3.17 |
| Glu163 | CD | Arg160 | NH1 | 3.26 |
| Glu163 | OE2 | Arg160 | CZ | 3.98 |
| Arg160 | NH1 | 2.70 | ||
| Gly164 | N | Arg160 | NH1 | 3.77 |
| Arg160 | N | 3.50 | ||
| Gly164 | CA | Arg160 | CZ | 3.68 |
| Arg160 | NH1 | 3.92 | ||
| Arg160 | NH2 | 3.54 | ||
| Arg160 | CB | 3.89 | ||
| Arg160 | N | 3.44 | ||
| Arg160 | CA | 3.73 | ||
| Gly164 | C | Arg160 | N | 3.57 |
| Gly165 | N | Arg160 | N | 3.64 |
The interactions with a distance within 4 Å between the contact residues and the source residues from the α-subunit and β-subunit, respectively, are given.
In addition to differences in the amino acid residues lining the entrance of the substrate channel, the channel entrance in the APSR structure from D. gigas is larger in diameter relative to that in the A. fulgidus enzyme. Superimposition of the APSR structures from D. gigas and A. fulgidus indicates that the larger opening in the substrate channel is formed by shifting the loop (aa 326 to 332) and the helix (aa 289 to 299) by about 3 Å in the α-subunit, with concomitant movements of the adjacent helices (aa 352 to 336 and aa 289 to 299) (see Fig. S1D in the supplemental material). This conformational alteration is needed to accommodate the C terminus of the β-subunit from another αβ-heterodimer to form the α2β2-heterotetramer. The larger channel entrance would also facilitate more rapid uptake of substrates to increase the activity of APSR in D. gigas relative to A. fulgidus.
Based on the substrate-soaked structures of A. fulgidus (34), residues Asn78, His418, and Arg282 of the α-subunit from D. gigas provide the direct contacts with the substrates at the reface of FAD. These three residues show displacements of less than 1 Å in the superimposition of the oxidized APSR structures from D. gigas and A. fulgidus, with the substrate-soaked structures from A. fulgidus. Residue Arg317 was proposed to have a role in fixing the adenine ring of APS with a π-π interaction, and exhibited features of “arginine out” and “arginine in” in oxidized APSR from A. fulgidus (34). This feature of “arginine out” for residue Arg336 is also observed in the oxidized substrate-free APSR structure from D. gigas, where the side chain of Arg336 faces the entrance of the substrate-binding channel in the α-subunit (Fig. 6B). The side chain conformation of Arg336 might potentially switch into the channel (“arginine in”) once APS binds. In the APSR structure from D. gigas, residue Arg282 at the substrate-binding site shares hydrogen bonds with Asp159 on the C terminus of the β-subunit provided by a second αβ-heterodimer (Fig. 6B and C). The loop of the β-subunit C terminus from D. gigas overlaps with the substrates when the APSR structures from D. gigas and A. fulgidus are superimposed (Fig. 6C). These observations suggest that the active site on the α-subunit is obstructed by the loop of the β-subunit C terminus of the other αβ-heterodimer within the α2β2-heterotetramer.
AMP has been reported to affect the apparent molecular weight of APSR from D. vulgaris (1), and substrate-soaked APSR structure has revealed binding residues that stabilize AMP in the channel (34). As the activity channel is blocked by the loop of the β-subunit C terminus in the D. gigas structure, the B-factors of both main chains and side chains for the C terminus of the β-subunit are large in the structure, suggesting that the conformation of the β-subunit is relatively flexible, even when bound. Accordingly, AMP might affect the binding of the C terminus of the β-subunit of one αβ-heterodimer to the activity channel of the other within the α2β2-heterotetramer. To test this hypothesis, we have performed DLS and ultracentrifugation experiments to examine the effect of AMP on the oligomerization of APSR in solution.
Interactions between αβ-heterodimers.
The two αβ-heterodimers in the α2β2-heterotetramer are related by a twofold rotation (Fig. 3A). The contact area between the two heterodimers is about 3,600 Å2, and the α2β2-heterotetramer is stabilized by 37 hydrogen bonds. Although this area is smaller than the contact area between the α- and β-subunits within the αβ-heterodimer, the B-factors for the overall α2β2-heterotetramer structure suggest less flexibility, and presumably, greater stability, in the contact area between two heterodimers (Fig. 7A). In addition, the α2β2-heterotetramers associate further to give an overall aggregate of six αβ-heterodimers. The major interactions between two α2β2-heterotetramers involve mainly the C-terminal loops of the β-subunit, although there are minor interactions arising from the contacts between separate α-subunits as well. These interactions between contiguous α2β2-heterotetramers in the overall APSR hexameric structure of D. gigas obstruct the substrate-binding site and results in the “facing out” of the turn (aa 14 to 19 of the β-subunit) that is supposed to be associated with the electron transfer function of the β-subunit.
FIG. 7.
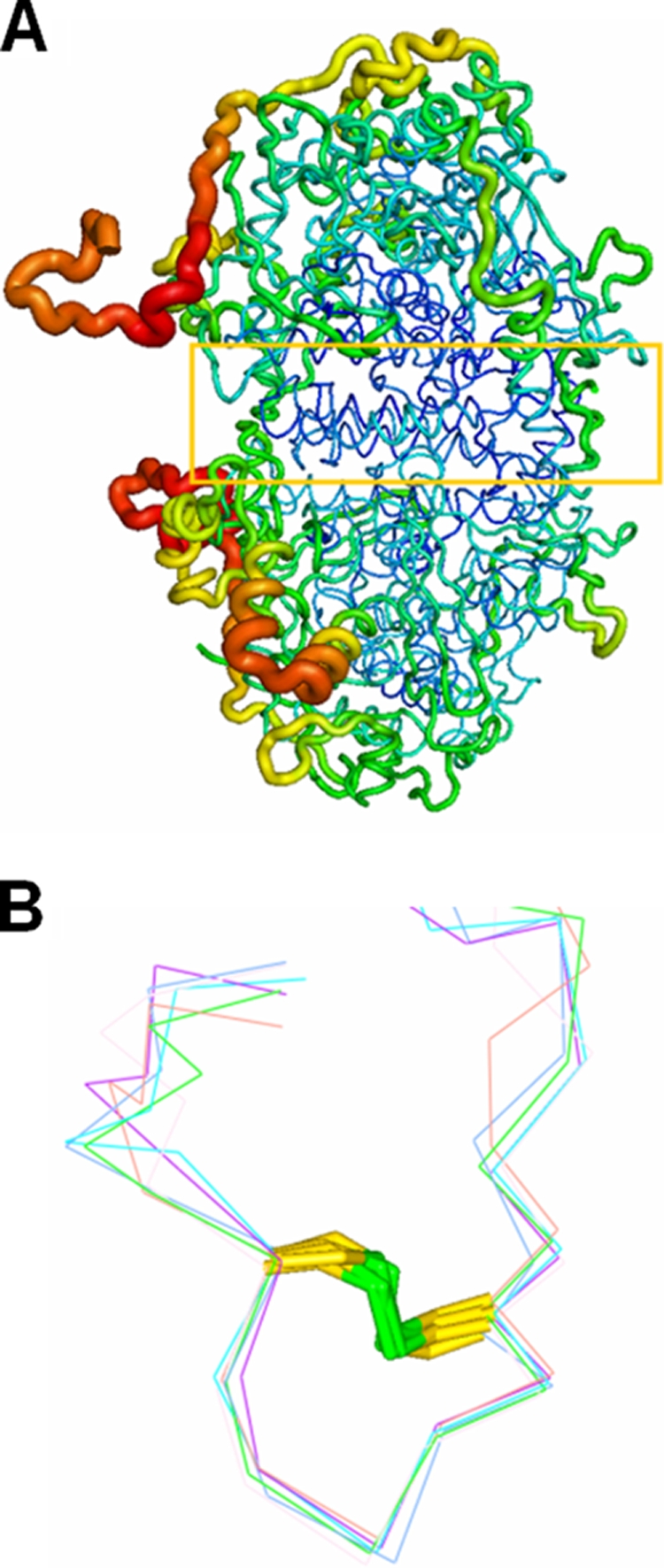
(A) B-factor-labeled structure of the-heterotetramer. The contact area (yellow box) between two αβ-heterodimers shows lower B-factors (blue). (B) Superimposition of six C-terminal loops of the β-subunits. The disulfide bond fixes the structure of the C-terminal loop of the β-subunit to block the channel entrance.
To evaluate the stability of the C-terminal loop in the various β-subunits, we undertook a superimposition of the C-terminal loops (aa 152 to 167) in the β-subunit among the separate αβ-heterodimers. This analysis indicates that the formation of a disulfide bond between Cys156 and Cys162 stabilizes the structure of the C-terminal loop (Fig. 7B). The temperature B-factors of the main chains in the β-subunit increase from residues Val130 to Glu152 and decrease after Glu152. The electron density map at the C terminus of the β-subunit exhibits an indistinct region from residues Glu147 to Ala151. The increased B-factors and the blurred electron density at the C terminus of the β-subunit imply instability in this region and suggest that the C terminus in the β-subunit is flexible, whereas the C-terminal loop in the β-subunit (aa 156 to 162) exhibits much less flexibility. We propose that the flexible C terminus of the β-subunit may possess a stable terminal loop as an anchor to allow the conformation search needed for it to interact with an accessible α-subunit from an adjoining αβ-heterodimer.
The oligomerization of APSR from D. vulgaris has been observed in the presence of various substrates and in different buffers (1, 37). In a phosphate buffer, Tris-HCl or other buffers at low concentrations, a large aggregate of molecular mass of about 440 kDa is observed in D. vulgaris, but altering the buffer to Tris-maleate or adding 10 mM AMP decreases the apparent molecular mass of APSR to 220 kDa. In the present study, oligomerization of APSR from D. gigas is also observed by DLS. The average size of APSR multimers decreases from 12.8 nm to 8.1 nm upon the addition of 3.5 mM AMP (Fig. 8). From the crystal structures of APSR from D. gigas, the sizes of the APSR hexamer and dimer can be estimated to be 135 by 120 by 100 Å3 and 100 by 70 by 65 Å3, respectively. Moreover, the analytical ultracentrifugation experiment using the sedimentation velocity method showed a peak corresponding to a molecular mass of about 600 kDa for APSR in the Tris-HCl buffer. This peak was shifted to another peak position corresponding to the molecular mass of about 220 kDa in the presence of AMP at the molar ratio of APSR to added AMP of 1:1. Thus, the DLS and analytical ultracentrifugation data suggest the existence of multiple forms of APSR, ranging from the hexamer to the dimer in the presence of AMP. Furthermore, our DLS data showed that the average size of APSR also decreases from 12.8 nm (hexamer) to 8.3 nm (dimer) upon the presence of 186 mM β-mercaptoethanol (Fig. 8). This result confirms our previous hypothesis that the formation of a disulfide bond between Cys156 and Cys162 stabilizes the structure of the C-terminal loop and is crucial for oligomerization.
FIG. 8.

Oligomerization of APSR observed by DLS analysis. The average size of oxidized APSR (0.25 mg/ml) was decreased from 12.8 nm (black line) to 8.1 nm (gray line) and to 8.3 nm (dashed line) upon the addition of 3.5 mM AMP and 186 mM β-mercaptoethanol, respectively.
From the sequence alignments of APSRs from D. gigas and A. fulgidus, it is clear that there must be a major difference in the C termini of the β-subunits between the two enzymes. The structure of APSR from D. gigas shows that the C terminus of the β-subunit interacts with and obstructs the activity channel in the α-subunit of a contiguous αβ-heterodimer to form the α2β2-heterotetramer. In the crystal structure, the unit cell is constructed from three α2β2-heteroteramers that are connected via interactions between the rigid C-terminal loops of the β-subunits to form the hexamer structure. It is clear that APSR in the hexameric form is inactive to prevent other comparable substrate analogs from diffusing into the active site when AMP is limiting. In an environment where AMP is no so limiting or abundant, upon the C terminus of β-subunit switching away from the active site, hexameric APSR dissociates spontaneously partially or fully into the functional dimeric form to disclose the active site for allowing the AMP entrance and subsequently triggering the catalytic reaction. It is possible that in APSR from D. gigas, the catalytic efficiency of the enzyme is regulated through oligomerization, exploiting the loop of the β-subunit C terminus. Moreover, the concentration of AMP might play a prominent role in initiating catalysis by competition with the C terminus of the β-subunit for binding to the activity channel and the active site in the α-subunit. Attempts to obtain the structure of APSR complexed with AMP have been unsuccessful so far, suggesting that the catalysis and regulation are more complicated than a simple substrate competition.
Supplementary Material
Acknowledgments
We are indebted to Yuch-Cheng Jean and the supporting staffs at beamlines BL13B1 and BL13C1 at the NSRRC and Hirofumi Ishii at the Taiwan-contracted beamline BL12B2 at SPring-8 for technical assistance. We are grateful to Go Ueno and his staffs for assistance in the experiments carried out at BL26-I and -II of SPring-8 at RIKEN. We thank Wen-guey Wu and Rong-Long Pan for the valuable discussion and suggestion.
This work was supported in part by National Science Council (NSC) grants 94-2313-B-213-001 and 95-2313-B-009-001-MY and NSRRC grants 963RSB02 and 973RSB02 to C.-J.C.
Footnotes
Published ahead of print on 9 October 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Bramlett, R. N., and H. D. Peck, Jr. 1975. Some physical and kinetic properties of adenylyl sulfate reductase from Desulfovibrio vulgaris. J. Biol. Chem. 250:2979-2986. [PubMed] [Google Scholar]
- 2.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, G.-R. W. Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography and NMR system (CNS), a new software suite for macromolecular structure determination. Acta Crystallogr. D 54:905-921. [DOI] [PubMed] [Google Scholar]
- 3.Chen, K., G. J. Tilley, V. Sridhar, G. S. Prasad, C. D. Stout, F. A. Armstrong, and B. K. Burgess. 1999. Alteration of the reduction potential of the [4Fe-4S](2+/+) cluster of Azotobacter vinelandii ferredoxin I. J. Biol. Chem. 274:36479-36487. [DOI] [PubMed] [Google Scholar]
- 4.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50:760-763. [DOI] [PubMed] [Google Scholar]
- 5.Denke, E., T. Merbitz-Zahradnik, O. M. Hatzfeld, C. H. Snyder, T. A. Link, and B. L. Trumpower. 1998. Alteration of the midpoint potential and catalytic activity of the rieske iron-sulfur protein by changes of amino acids forming hydrogen bonds to the iron-sulfur cluster. J. Biol. Chem. 273:9085-9093. [DOI] [PubMed] [Google Scholar]
- 6.Fritz, G., T. Buchert, H. Huber, K. O. Stetter, and P. M. H. Kroneck. 2000. Adenylylsulfate reductases from archaea and bacteria are 1:1 alpha beta-heterodimeric iron-sulfur flavoenzymes—high similarity of molecular properties emphasizes their central role in sulfur metabolism. FEBS Lett. 473:63-66. [DOI] [PubMed] [Google Scholar]
- 7.Fritz, G., A. Roth, A. Schiffer, T. Buchert, G. Bourenkov, H. D. Bartunik, H. Huber, K. O. Stetter, P. M. H. Kroneck, and U. Ermler. 2002. Structure of adenylylsulfate reductase from the hyperthermophilic Archaeoglobus fulgidus at 1.6-Å resolution. Proc. Natl. Acad. Sci. USA 99:1836-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fritz, G., T. Buchert, and P. M. H. Kroneck. 2002. The function of the [4Fe-4S] clusters and FAD in bacterial and archaeal adenylylsulfate reductases—evidence for flavin-catalyzed reduction of adenosine 5′-phosphosulfate. J. Biol. Chem. 277:26066-26073. [DOI] [PubMed] [Google Scholar]
- 9.Fritz, G., O. Einsle, M. Rudolf, A. Schiffer, and P. M. H. Kroneck. 2005. Key bacterial multi-centered metal enzymes involved in nitrate and sulfate respiration. J. Mol. Microbiol. Biotechnol. 10:223-233. [DOI] [PubMed] [Google Scholar]
- 10.Gibson, G. R., J. H. Cummings, and G. T. Macfarlane. 1991. Growth and activities of sulfate-reducing bacteria in gut contents of healthy subjects and patients with ulcerative colitis. FEMS Microbiol. Ecol. 86:103-111. [Google Scholar]
- 11.Hamilton, W. A. 1998. Bioenergetics of sulphate-reducing bacteria in relation to their environmental impact. Biodegradation 9:201-212. [DOI] [PubMed] [Google Scholar]
- 12.Hammack, R. W., and H. M. Edenborn. 1992. The removal of nickel from mine waters using bacterial sulfate reduction. Appl. Microbiol. Biotechnol. 37:674-678. [Google Scholar]
- 13.Hipp, W. M., A. S. Pott, N. Thum-Schmitz, I. Faath, C. Dahl, and H. G. Trüper. 1997. Towards the phylogeny of APS reductases and sirohaem sulfite reductases in sulfate-reducing and sulfur-oxidizing prokaryotes. Microbiology 143:2891-2902. [DOI] [PubMed] [Google Scholar]
- 14.Iverson, T. M., C. Luna-Chavez, L. R. Croal, G. Cecchini, and D. C. Rees. 2002. Crystallographic studies of the Escherichia coli quinol-fumarate reductase with inhibitors bound to the quinol-binding site. J. Biol. Chem. 277:16124-16130. [DOI] [PubMed] [Google Scholar]
- 15.Jones, T. A., J.-Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47:110-119. [DOI] [PubMed] [Google Scholar]
- 16.Kissinger, C. R., L. C. Sieker, E. T. Adman, and L. H. Jensen. 1991. Refined crystal structure of ferredoxin II from Desulfovibrio gigas at 1.7 Å. J. Mol. Biol. 219:693-715. [DOI] [PubMed] [Google Scholar]
- 17.Kopriva, S., and A. Koprivova. 2004. Plant adenosine 5′-phosphosulphate reductase: the past, the present, and the future. J. Exp. Bot. 55:1775-1783. [DOI] [PubMed] [Google Scholar]
- 18.Kremer, D. R., G. Fauque, H. D. Peck, Jr., J. LeGall, J. Lampreia, J. J. G. Moura, and T. A. Hansen. 1988. Immunocytochemical localization of APS reductase and bisulfite reductase in three Desulfovibrio species. Arch. Microbiol. 150:296-301. [Google Scholar]
- 19.Kuang, F., J. Wang, L. Yan, and D. Zhang. 2007. Effects of sulfate-reducing bacteria on the corrosion behavior of carbon steel. Electrochim. Acta 52:6084-6088. [Google Scholar]
- 20.Lampreia, J., A. S. Pereira, and J. J. G. Moura. 1994. Adenylylsulfate reductases from sulfate-reducing bacteria. Methods Enzymol. 243:241-260. [Google Scholar]
- 21.Lampreia, J., I. Moura, M. Teixeira, H. D. Peck, J. LeGall, B. H. Huynh, and J. J. G. Moura. 1990. The active-centers of adenylylsulfate reductase from Desulfovibrio gigas—characterization and spectroscopic studies. Eur. J. Biochem. 188:653-664. [DOI] [PubMed] [Google Scholar]
- 22.Lebowitz, J., M. S. Lewis, and P. Schuck. 2002. Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11:2067-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeGall, J. 1963. A new species of Desulfovibrio. J. Bacteriol. 86:1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, J., M. R. Nelson, C. Y. Peng, D. Bashford, and L. Noodleman. 1998. Incorporating protein environments in density functional theory: a self-consistent reaction field calculation of redox potentials of [2Fe2S] clusters in ferredoxin and phthalate dioxygenase reductase. J. Phys. Chem. A 102:6311-6324. [Google Scholar]
- 25.Matias, P. M., I. A. Pereira, C. M. Soares, and M. A. Carrondo. 2005. Sulphate respiration from hydrogen in Desulfovibrio bacteria: a structural biology overview. Prog. Biophys. Mol. Biol. 89:292-329. [DOI] [PubMed] [Google Scholar]
- 26.Mattevi, A., G. Tedeschi, L. Bacchella, A. Coda, A. Negri, and S. Ronchi. 1999. Structure of L-aspartate oxidase: implications for the succinate dehydrogenase/fumarate reductase oxidoreductase family. Structure 7:745-756. [DOI] [PubMed] [Google Scholar]
- 27.Meyer, B., and J. Kuever. 2007. Phylogeny of the alpha and beta subunits of the dissimilatory adenosine-5′-phosphosulfate (APS) reductase from sulfate-reducing prokaryotes—origin and evolution of the dissimilatory sulfate-reduction pathway. Microbiology 153:2026-2044. [DOI] [PubMed] [Google Scholar]
- 28.Meyer, B., and J. Kuever. 2007. Molecular analysis of the distribution and phylogeny of dissimilatory adenosine-5′-phosphosulfate reductase-encoding genes (aprBA) among sulfur-oxidizing prokaryotes. Microbiology 153:3478-3498. [DOI] [PubMed] [Google Scholar]
- 29.Moulis, J. M., L. C. Sieker, K. S. Wilson, and Z. Dauter. 1996. Crystal structure of the 2[4Fe-4S] ferredoxin from Chromatium vinosum: evolutionary and mechanistic inferences for [3/4Fe-4S] ferredoxins. Protein Sci. 5:1765-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53:240-255. [DOI] [PubMed] [Google Scholar]
- 31.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307-326. [DOI] [PubMed] [Google Scholar]
- 32.Peck, H. D., Jr., T. E. Deacon, and J. T. Davidson. 1965. Studies on adenosine 5′-phosphosulfate reductase from Desulfovibrio desulfuricans and Thiobacillus thioparus. I. The assay and purification. Biochim. Biophys. Acta 96:429-446. [DOI] [PubMed] [Google Scholar]
- 33.Pochart, P., J. Dore, F. Lemann, I. Goderel, and J. C. Rambaud. 1992. Interrelations between populations of methanogenic Archaea and sulfate-reducing bacteria in the human colon. FEMS Microbiol. Lett. 98:225-228. [DOI] [PubMed] [Google Scholar]
- 34.Schiffer, A., G. Fritz, P. M. H. Kroneck, and U. Ermler. 2006. Reaction mechanism of the iron-sulfur flavoenzyme adenosine-5′-phosphosulfate reductase based on the structural characterization of different enzymatic states. Biochemistry 45:2960-2967. [DOI] [PubMed] [Google Scholar]
- 35.Traore, A. S., C. E. Hatchikian, J. LeGall, and J. P. Belaich. 1982. Microcalorimetric studies of the growth of sulfate-reducing bacteria: comparison of the growth parameters of some Desulfovibrio species. J. Bacteriol. 149:606-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vagin, A., and A. Teplyakov. 1997. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30:1022-1025. [Google Scholar]
- 37.Verhagen, M., I. M. Kooter, R. B. G. Wolbert, and W. R. Hagen. 1994. On the iron-sulfur cluster of adenosine phosphosulfate reductase from Desulfovibrio vulgaris (Hildenborough). Eur. J. Biochem. 221:831-837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.