

Abstract
Mammalian sterile 20-like kinases 1 and 2 (Mst1 and Mst2, respectively) are potent serine/threonine kinases that are involved in cell proliferation and cell death. To investigate the physiological functions of Mst1 and Mst2, we generated Mst1 and Mst2 mutant mice. Mst1−/− and Mst2−/− mice were viable and fertile and developed normally, suggesting possible functional overlaps between the two genes. A characterization of heterozygous and homozygous combinations of Mst1 and Mst2 mutant mice showed that mice containing a single copy of either gene underwent normal organ development; however, Mst1−/−; Mst2−/− mice lacking both Mst1 and Mst2 genes started dying in utero at approximately embryonic day 8.5. Mst1−/−; Mst2−/− mice exhibited severe growth retardation, failed placental development, impaired yolk sac/embryo vascular patterning and primitive hematopoiesis, increased apoptosis in placentas and embryos, and disorganized proliferating cells in the embryo proper. These findings indicate that both Mst1 and Mst2 kinases play essential roles in early mouse development, regulating placental development, vascular patterning, primitive hematopoiesis, and cell proliferation and survival.
Mammalian sterile 20-like kinases 1 and 2 (Mst1 and Mst2, respectively) belong to the germinal center kinase group II family of mitogen-activated protein kinase-related kinases (4). The amino acid sequences of Mst1 and Mst2 are highly conserved in humans and mice; both kinases contain an N-terminal kinase domain and a C-terminal SARAH domain (17). Mst1/2 play a role in mediating the apoptosis induced by various types of apoptotic stress in vitro (1, 6, 17, 22). The kinase domain of Mst1, when activated by caspase-mediated cleavage, phosphorylates histone H2B, promoting the chromatin condensation that precedes apoptotic cell death (2, 32). Mst1/2 promote neuronal cell death by phosphorylating and activating forkhead box O (FOXO) transcription factors in response to oxidative stress (18, 36). AKT and Mst1 are mutually antagonistic in mammalian cells and in the zebrafish (3, 8).
In addition to their roles in mammalian apoptosis, Mst1/2 are also key components in the Hippo signaling pathway, which is a key developmental pathway involved in restricting cell proliferation and promoting apoptosis in Drosophila melanogaster (7, 9, 24, 31, 33). This pathway involves Hippo (Hpo) kinase (Drosophila homolog of Mst1 and Mst2) and its scaffold, Salvador (Sav) (Drosophila homolog of WW45); Warts kinase (Drosophila homolog of LATS kinases) and its scaffold, Mats (Drosophila homolog of Mob); and the transcriptional coactivator, Yorkie (Yki) (Drosophila homolog of YAP). In flies, deletion of any Hippo pathway component or overexpression of Yki leads to increased cell proliferation and decreased cell death in developing epithelial tissues (5, 7, 9, 11, 12, 24, 26, 30, 31, 33, 34). In contrast to these fly phenotypes, mice with a deletion of one of these Hippo components show more complicated phenotypes during embryogenesis. Lats2-null embryos die at approximately embryonic day 12.5 (E12.5) with pleiotropic defects, including neural tube and pericardium defects (19). Yap knockout embryos die at about E8.5 with defects in chorioallantoic fusion and yolk sac vasculogenesis (20). WW45-null embryos also die at about E18.5 in utero due to placental defects (16).
Several studies of flies have supported the importance of the Hippo pathway in mammalian tumorigenesis. Consistent with the overexpression of Yorkie in flies, overexpression of YAP also causes increased cell proliferation, expansion of progenitor cells, and reduced cell death (16, 23, 38). In fact, amplification of Yap has frequently been observed in liver and breast tumor models (23, 37), and mice with a homozygous deletion of LATS1 develop tumors (27). We have recently shown that loss of WW45 affects developing epithelial tissues, producing precancerous characteristics (16). Consistent with these findings, loss of LATS1 and LATS2 expression and mutations of WW45 and MATS1 are found in cancer cell lines (10, 14, 28, 30).
Two recent studies have showed that mice lacking Mst1 display reduced numbers of naïve T cells in secondary lymphoid organs and peripheral blood (13, 39). However, the physiological functions of Mst1 and Mst2 kinases in mice are not fully understood. To reveal the functions of these two proteins, we disrupted Mst1 and Mst2 genes by gene targeting in mice. Mst1-Mst2 double-knockout embryos die early in gestation and show defects in placental development, vascular patterning, primitive hematopoiesis, and regulation of cell proliferation and survival. These data suggest that Mst kinases are essential for the early developmental program in mice.
MATERIALS AND METHODS
Generation and genotyping of Mst1 and Mst2 knockout mice.
We isolated the mouse Mst1 genomic locus encompassing exons 2 to 5 and the Mst2 locus encompassing exon 1 from a 129/SvJ mouse bacterial artificial chromosome library. The murine Mst1-targeting vector was constructed by sequentially subcloning a 2.3-kb BamHI fragment (right arm) obtained by PCR (forward primer, 5′-AAT TGG ATC CCT TCT TCT GGC CTT TTG TGC-3′; reverse primer, 5′-AAT TGG ATC CCA AAA TAT TTC CCG CCT TGA-3′) into the BamHI site of a PGK-puro targeting vector (PGK stands for phosphoglycerokinase, and puro stands for the 1.5-kb puromycin resistance gene) and then a 3.7-kb HindIII fragment (left arm) into the HindIII site of the vector. An EcoRI site at the 5′ end of the PGK-puro cassette was retained in this targeting vector to facilitate genotyping of the targeted allele. The murine Mst2-targeting vector was constructed by subcloning a 4.4-kb BamHI-SpeI fragment into a BamHI-XbaI site of the PGK-puro targeting vector, and a 4.0-kb EcoRV fragment into an EcoRI site (blunted). The diphtheria toxin A chain gene was then inserted into a unique NotI site at the 3′ end of the right arm in each targeting vector for negative selection. In this strategy, an Mst1 genomic region spanning exons 3 and 4 (3.3 kb) and an Mst2 genomic region coding exon 1 (3.8 kb) were replaced with a 1.5-kb puromycin resistance gene (puro), introduced as a positive-selection marker.
After electroporation, embryonic stem (ES) cell clones were grown in the presence of 3 μg/ml puromycin and isolated after culturing for 8 days. Homologous recombination was confirmed by Southern blot analysis of genomic DNA using 3′ external probes (Fig. 1B). Embryos and mice were genotyped by PCR using primers M-8 (5′-AGC ATG TTT GGG AAA TTT AAA AGA-3′), M-9 (5′-AAT CTG GCC AGT CTC TTT ATG AAT-3′), and PGK-3 (5′-GCA CGA GAC TAG TGA GAC GTG CTA C-3′) for Mst1, and M2gF-01 (5′-AAA GCT GGT ACT GGG GTT CA-3′), M2gR-04 (5′-CCC CAA GAA TAC ATG CCA AT-3′), and PGK-3 (5′-GCA CGA GAC TAG TGA GAC GTG CTA C-3) for Mst2. The resulting PCR products were 679 bp (wild-type allele) and 556 bp (puro allele) for Mst1 and 1,027 bp (wild-type allele) and 675 bp (puro allele) for Mst2 (Fig. 1C). To generate Mst1−/−; Mst2−/− embryos, Mst1+/−; Mst2−/− males were mated with Mst1−/−; Mst2+/− females (Fig. 1D). The embryos were dissected from the yolk sacs on E8.0 to E9.5 and genotyped using genomic DNA from yolk sacs. Embryo morphology was analyzed after dissection, and digital photographs were obtained using stereomicroscopy (Olympus).
FIG. 1.

Targeted disruption of the Mst1 and Mst2 genes. (A) Structures of the targeting vector for the mouse Mst1 (mMst1) (top) and Mst2 (mMst2) (bottom) loci and the targeted allele after homologous recombination. Black boxes denote exons. The PGK-puromycin resistance cassette (puro?) replaced exons 3 and 4 in Mst1 and exon 1 in Mst2. The DT-A box represents a polymerase II promoter-diphtheria toxin A chain (DT-A) used for negative selection of ES cells. WT, wild type; Mut, mutant; E, EcoRI site. (B) Southern blot analyses of Mst1 ES cells and Mst2 ES cells and mice. Homologous recombination was verified using external digests and 3′ external probes. (C) Genotyping of Mst1 mice and Mst2 ES cells and mice by PCR analysis. The specific primers were indicated by arrowheads in panel A (M-8, M-9, and PGK-3 primers for the Mst1 locus and M2gF-01, M2gR-04, and PGK-3 primers for the Mst2 locus). (D) Genotyping of E8.5 embryos from mating of Mst1+/−; Mst2−/− mice and Mst1−/−; Mst2+/− mice. The genotypes of the embryos were determined by PCR amplification of yolk sac genomic DNA. (E) Protein extracts derived from MEFs with the indicated genotypes were analyzed by immunoblotting using antibodies against Mst1 (Cell Signaling) and Mst2 (Cell Signaling [N-terminal region], Santa Cruz [C-terminal region], and GP 2 [N-terminal region]). N-term, N-terminal region; C-term, C-terminal region.
BrdU labeling, terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) analysis, and immunohistochemistry.
For 5-bromo-2-deoxyuridine (BrdU; Sigma) treatment of embryos, pregnant mice from Mst1+/−; Mst2−/− and Mst1−/−; Mst2+/− matings at specific times were injected intraperitoneally with BrdU (50 μg/g of body weight) on E8.5 to E9.5. Pregnant mice were sacrificed 30 min after injection, and the uteri were removed. For histological analysis, embryos and tissues were fixed in 4% paraformaldehyde overnight at 4°C and embedded in paraffin wax for sectioning. Generally, 5-μm sections were cut and stained with hematoxylin and eosin (H&E).
The embryos were immunostained as described previously (16). Briefly, the embryos were fixed in 4% paraformaldehyde, bleached with 3% H2O2, blocked in 5% bovine serum albumin, incubated sequentially with primary and second antibodies, and stained with the EnVision dual-link system-horseradish peroxidase (DAB+) (DakoCytomation). Immunohistochemical analyses were performed using standard protocols with primary antibodies against PECAM-1 (BD), BrdU (BD), and Ki67 (Novocastra) and secondary horseradish peroxidase-conjugated goat anti-rat immunoglobulin G (Jackson ImmunoResearch), Alexa Fluor 488- or Alexa Fluor 594-conjugated antibodies (Molecular Probes), respectively. TUNEL assays were performed on sections using an in situ cell death detection kit (Roche). TUNEL/BrdU/Ki67/phosphorylated histone H3-positive cells were counted, and the mean number was used to obtain the relative ratio to Mst1+/−; Mst2+/− embryos.
Generation of antibodies.
Guinea pig polyclonal antibodies to Mst2 (GP 2) were generated by injecting guinea pigs with a keyhole-limpet-hemocyanin-conjugated peptide corresponding to the N-terminal amino acids of Mst2 (EQPPAPKSKLKKLSCys). Specific antibodies were affinity purified with the appropriate antigens.
Western blotting analysis.
Mst1 and Mst2 knockout mouse embryonic fibroblasts (MEFs) and embryos were lysed in lysis buffer (25 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1 mM MgCl2, 0.2% Triton X-100, 0.3% NP-40, leupeptin, pepstatin A, phenylmethylsulfonyl fluoride, NaF, Na3OVO3, and beta-glycerophosphate). Successful ablation of Mst1 and Mst2 was confirmed by Western blot analysis using antibodies against Mst1 (Cell Signaling) and Mst2 (Cell Signaling [N-terminal region], Santa Cruz [C-terminal region], and GP 2 [N-terminal region]). Western blot analysis from E8.5 embryos were performed using antibodies against YAP, phosphorylated YAP, Mst1, Mst2, FOXO3A (Cell Signaling), WW45 (16), LATS1, LATS2 (Bethyl Laboratories Inc.), and glyceraldehyde-3-phosphate dehydrogenase (Abcam). Densitometry was performed using the software Multi Gauge V3 (Fujifilm).
Passive transport of rhodamine 123.
Pregnant mice at specific times were injected intraperitoneally on E8.5 with rhodamine 123 (1 μg/g of body weight; Sigma) (29). The mice were sacrificed 2 h after injection, and the embryos were removed from the uteri and analyzed with a fluorescence microscope.
Reverse transcription-PCR (RT-PCR) and quantitative real-time PCR.
RNA preparation and cDNA synthesis were previously described (15). Briefly, total RNAs were extracted from Mst1 and Mst2 MEFs, E8.5 Mst1+/−; Mst2+/− and Mst1−/−; Mst2−/− embryos and placentas using Trizol reagent (Intron) according to the manufacturer's instructions. Aliquots of 1 μg of RNA were used as template for reverse transcription with oligo(dT) primers (Intron). Real-time PCR was performed with iQ SYBR green reagent kits (Genet Bio) on an iQ5 PCR machine (Bio-Rad). For quantitative PCR, SYBR fluorescence was utilized to generate a threshold cycle (CT) value for each transcript. These values were normalized for absolute quantity against Gapdh. The normalized CT values for Mst1+/−; Mst2+/− embryos were compared to the CT values for Mst1−/−; Mst2−/− embryos using the 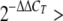 calculation method. The sequences of specific primers are available upon request.
calculation method. The sequences of specific primers are available upon request.
Hematopoietic colony assay.
Hematopoietic colonies were generated as described previously (15). Briefly, E8.5 yolk sacs were isolated and dissociated in collagenase with 20% fetal bovine serum, and the resulting cells were replated in 1% methylcellulose containing hematopoietic cytokines. Hematopoietic colonies were counted 4 to 7 days later.
Whole-mount in situ hybridization.
Whole-mount in situ hybridization was carried out as previously described (15). Briefly, antisense digoxigenin-labeled riboprobes were generated from the pBluescript(+) KS vector containing amplified Mst1 and Mst2 cDNA fragments (Mst1 locus, positions 889 to 1118 in cDNA, and Mst2 locus, positions 1 to 314 in cDNA) using the digoxigenin RNA labeling kit (Roche) according to the manufacturer's instructions. The sequences of specific primers are listed in Table 1.
TABLE 1.
Summary of primer sequences
| Use | Gene | Direction | Sequence (5′-3′) | Positions in cDNA |
|---|---|---|---|---|
| RT-PCR probe | Mst1 | Forward | CTG TGG GGC TGG TTC TGT AT | 349-766 |
| Reverse | TGT GGG AGG AGG GTT TGT AG | |||
| Mst2 | Forward | TGG TCC CTT GGC ATTACT TC | 746-1275 | |
| Reverse | TCC TCC TCC TCC TCT TCC TC | |||
| Whole-mount in situ hybridization probe | Mst1 | Forward | AATTGGATCCTTGAGCACGGTGACAC | 889-1118 |
| Reverse | AATTGGATCCCGGATCTCTTCCATTTC | |||
| Mst2 | Forward | AATTGGTCCATGGAGCAGCCGCCGGC | 1-314 | |
| Reverse | AATTGGATCCCCCGCTCCACAGTACTC |
RESULTS
Generation of Mst1−/− and Mst2−/− mutant mice.
Mst kinases are key regulators of the apoptotic response to various cell death signals. In addition, loss of Hippo kinase (Mst1/2 homolog) in flies results in uncontrolled cell proliferation and reduced cell death. To further explore the physiological functions of Mst kinases in mice, we targeted the murine Mst1 and Mst2 genes by homologous recombination in ES cells, generating Mst1 and Mst2 mutant mice (Fig. 1A, B, and C), as described in Materials and Methods. Western blot analysis (Fig. 1E) and RT-PCR analysis (data not shown) confirmed the absence of Mst1 and Mst2 protein in mutant MEFs, demonstrating that each mutant was null for the respective allele. Mice homozygous null for the Mst1 allele (Mst1−/−) developed normally and were viable but had markedly reduced numbers of peripheral naïve T cells and exhibited dysregulated lymphocyte trafficking as described previously (13, 39; S. Oh and D.-S. Lim, personal observations). In contrast, mice homozygous for the mutant Mst2 allele (Mst2−/−) were viable, fertile, and phenotypically indistinguishable from wild-type littermates over a 24-month observation period (data not shown). These observations were in apparent contradiction to in vitro data suggesting their prominent role in apoptosis.
Spatiotemporal and functional overlaps in expression of Mst1 and Mst2.
The lack of obvious developmental defects in Mst1−/− or Mst2−/− mice prompted us to suggest that Mst1 and Mst2 can functionally compensate for each other's deficiency. Thus, we first examined whether spatiotemporal regulation of Mst1 and Mst2 expression overlap by analyzing Mst1 and Mst2 protein levels in various adult tissues and at different developmental stages (Fig. 2A). Both Mst1 and Mst2 proteins were expressed ubiquitously at particularly high levels in the lymphoid organs for Mst1 and in the brain for Mst2. During development, Mst1 and Mst2 were detected as early as E8.5, and their expression appeared to increase to a maximum on E12.5 and decrease thereafter, suggesting their possibly important roles in early embryonic development. Next, we performed a whole-mount in situ hybridization on E8.5 to determine the expression profile of Mst1 and Mst2 transcripts. The embryos had ubiquitous and largely overlapping expression of Mst1 and Mst2 with particularly high expression along the neural fold (Fig. 2B). Given the spatiotemporal overlap in embryonic and adult tissues and extensive homology of Mst1 and Mst2 proteins, it is likely that functional redundancy may account for the lack of an overt phenotype in Mst1−/− and Mst2−/− single-knockout mice.
FIG. 2.

Expression profile of Mst1 and Mst2 in adult tissues and during embryonic development. (A) Western blot analysis for Mst1 and Mst2 proteins from wild-type adult tissues and embryos on E8.5 to E14.5. (B) Whole-mount in situ hybridization of E8.5 Mst1+/−; Mst2−/− embryos (Mst1 antisense probe [a] and Mst1 sense probe [b]) and Mst1−/−; Mst2+/− embryos (Mst2 antisense probe [c] and Mst2 sense probe [d]). Bars, 200 μm.
Next, we crossed Mst1−/− and Mst2−/− mice to generate Mst1+/− Mst2+/− mice, and these mice are consequently intercrossed to generate Mst1+/−; Mst2−/− mice, Mst1−/−; Mst2+/− mice, and Mst1−/−; Mst2−/− mice (Fig. 1D). We first found that Mst1+/−; Mst2+/− mice Mst1+/−; Mst2−/− mice, and Mst1−/−; Mst2+/− mice were viable and fertile. We then crossed Mst1+/−; Mst2−/− animals with Mst1−/−; Mst2+/− animals to obtain Mst1−/−; Mst2−/− double mutant mice. Notably, we did not observe any Mst1−/−; Mst2−/− mice at birth, indicating that these double-null mice were embryonic lethal. Next, we determined the developmental stage at which lethality occurs and found viable Mst1−/−; Mst2−/− embryos on E8.0 to E9.5 with distorted Mendelian frequency (Table 2). This is consistent with our suggestion that Mst1 and Mst2 functionally overlap at least during early development.
TABLE 2.
Summary of embryo genotypinga
| Age (dpcb) | No. (%) of mice with the following genotype: |
No. (%) of embryos resorbed | Total no. of embryos | |||
|---|---|---|---|---|---|---|
| Mst1+/−; Mst2+/− | Mst1+/−; Mst2−/− | Mst1−/−; Mst2+/− | Mst1−/−; Mst2−/− | |||
| E8.0-9.5 | 41 (22.4) | 52 (28.4) | 50 (27.3) | 32 (17.5) | 8 (4.4) | 183 |
| E9.5 | 6 (42.9) | 5 (35.7) | 3 (21.4) | 0 (0) | NAc | 14 |
| P21d | 17 (28.8) | 28 (47.5) | 14 (23.7) | 0 (0) | NA | 59 |
| Total | 64 (25.0) | 85 (33.2) | 67 (26.2) | 32 (12.5) | 8 (3.1) | 256 |
Mst1+/−; Mst2−/− mice and Mst1−/−; Mst2+/− mice were mated. The expected Mendelian frequency was 25% each for the Mst1+/−; Mst2+/− genotype, Mst1+/−; Mst2−/− genotype, Mst1−/−; Mst2+/− genotype, and Mst1−/−; Mst2−/− genotype.
dpc, days postcoitum.
NA, not applicable.
P21, neonate.
Growth retardation and subsequent lethality on approximately E8.5 in Mst1−/−; Mst2−/− double mutant mice.
By E8.5, the Mst1−/−; Mst2−/− embryos were deformed and were significantly smaller in size than Mst1+/−; Mst2+/− littermates (Fig. 3A to D). Although some Mst1−/−; Mst2−/− embryos were occasionally recovered on E9.0 or E9.5, many were undergoing resorption by E9.5 (Fig. 3H and L). Mst1−/−; Mst2−/− embryos on E9.5 showed overt developmental delay, as evidenced by incomplete embryo turning, fusion of cranial neural plates, and growth retardation (Fig. 3L). No Mst1−/−; Mst2−/− embryos were recovered beyond E9.5 (data not shown).
FIG. 3.

Photomicrographs of Mst1+/−; Mst2+/− embryos (A, E, and I), Mst1+/−; Mst2−/− embryos (B, F, and J), Mst1−/−; Mst2+/− embryos (C, G, and K), and Mst1−/−; Mst2−/− (D, H, and L) embryos. Panels A to D were all taken at one magnification, and panels E to L were taken at a different, higher magnification. Bars, 200 μm (A) and 400 μm (E and I).
Since the growth retardation characteristic of Mst1−/−; Mst2−/− embryos manifested on approximately E8.5, we further confirmed these phenotypes in serial histological sections of embryos on E8.0 to E9.5. Histological sections revealed that E8.0 Mst1−/−; Mst2−/− embryos were in the process of gastrulation and had normally formed all three germ layers and extraembryonic tissues (Fig. 4A, panels a and b). We also analyzed the levels of expression of endodermal genes (Foxa2, Sox17, Afp, and Cerl), ectodermal genes (Fgf5, NeuroD, Sox2, and Otx2), and mesodermal genes (Tpbp, Gsc, Fgf8, and Tbx6) by quantitative RT-PCR (Fig. 4B). Expression levels of these marker genes expressed in the endoderm, ectoderm, and mesoderm had no significant difference between Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos, indicating normal germ layer formation in Mst1−/−; Mst2−/− embryos. Of interest, transverse sections from E8.5 to 9.0 showed that the Mst1−/−; Mst2−/− neural plate failed to fuse to form a neural tube and was kinked dorsoventrally (Fig. 4A, panels c to f). These observations indicate that even Mst1 and Mst2 are required for mouse development in the early postimplantation period, but not for axial or germ layer patterning.
FIG. 4.

Histological analyses of Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos on E8.0 to E9.5. (A) H&E-stained Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos on E8.0 to E9.5. Abbreviations: Ys, yolk sac; Al, allantois; Am, amnion; Da, dorsal aorta; Nt, neural tube; Ht, heart; Ne, neural ectoderm. Bar, 200 μm. (B) Representative quantitative PCR data showing the levels of genes expressed in endoderm (Foxa2, Sox17, Afp, and Cerl), ectoderm (Fgf5, NeuroD, Sox2, and Otx2), and mesoderm (Tpbp [T], Gsc, Fgf8, and Tbx6) from Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos. (C) Sagittal sections of Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos. (a and b) H&E-stained hearts from embryos on E9.5. (c and d) Immunofluorescence staining of hearts from embryos on E9.5 for smooth muscle α-actin. (e and f) H&E-stained brains from embryos on E9.5. The boxed areas are shown at a higher magnification. Abbreviations: Mc, myocardium; Ec, endocardium; Ht, heart; SM, smooth muscle; DAPI, 4′,6′-diamidino-2-phenylindole. Panels a, b, e, and f were all taken at one magnification, and panels c and d were taken at another, higher magnification. Bars, 100 μm (a, e, and f) and 200 μm (c and d). (D) H&E-stained Mst1+/−; Mst2+/− placentas and Mst1−/−; Mst2−/− placentas on E8.0 to E9.5. Dotted lines indicate the border between the ectoplacental cone/labyrinth layer and giant cells. The boxed areas are shown at a higher magnification. Abbreviations: Epc, ectoplacental cone; Ch, chorionic plate; La, labyrinth layer. Bar, 200 μm. (E) (a and b) TUNEL analysis of sections of Mst1+/−; Mst2+/− placentas and Mst1−/−; Mst2−/− placentas on E8.5. (c and d) BrdU incorporation and immunohistochemical analysis of Ki-67 labeling in placentas on E9.0. Thick dotted lines indicate the border of the chorionic plate. Thin dotted lines indicate the border between the ectoplacental cone and giant cells. Abbreviations: Epc, ectoplacental cone; Ch, chorionic plate. Bar, 200 μm. (e) The graph shows the mean relative ratios plus standard errors of the means (error bars) (SEMs) of TUNEL-positive cells from placentas on E8.5 and BrdU- and Ki67-positive cells from placentas on E9.0. (F) Representative quantitative PCR data showing the levels of genes expressed in trophoblasts for the placenta (Gcm1 [for labyrinthine trophoblasts], Mash2 [for ectoplacental cone and the chorion], Tpbp [for spongiotrophoblasts], and Pl1 [for trophoblast giant cells]) from Mst1+/−; Mst2+/− and Mst1−/−; Mst2−/− placentas. The sequences of specific primers are available upon request. (G) Rhodamine 123 transport from mother to fetus. (a and b) Two hours after intraperitoneal injection, transport of rhodamine 123 was impaired in Mst1−/−; Mst2−/− embryos. Dotted lines indicate the head regions in Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos. Panels a and b were taken at the same magnification. Bar, 200 μm. (c) The graph shows relative ratios plus SEMs of rhodamine 123 intensities.
Severe disorganization of the heart and neuroectoderm in Mst1−/−; Mst2−/− embryos.
Examination of organogenesis in Mst1−/−; Mst2−/− embryos on E9.5 revealed a severe developmental retardation of early endocardium (Fig. 4C, panel b). Because Mst1 is a key kinase in cardiomyocyte apoptosis (35), we stained for smooth muscle α-actin, a marker for cardiomyocytes, in Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos. Unexpectedly, the smooth muscle α-actin immunohistochemical staining pattern was similar in the Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos (Fig. 4C, panels c and d), suggesting that cardiomyocytes developed normally. Interestingly, Mst1−/−; Mst2−/− embryos also exhibited marked disorganization of the neuroectoderm: they lacked any obvious boundary of the neuroectoderm in the forebrain region and had a disorganized accumulation of neuroepithelial cells (Fig. 4C, panel f). These findings indicate that Mst1 and Mst2 are dispensable for differentiation of progenitor cells that constitute vital organs, such as the heart and brain, but are actively involved in organogenesis.
Mst1−/−; Mst2−/− embryos exhibit placental defects.
The initial step in placenta development involves attachment of the allantois to the chorionic plate on about E8.5, followed by vascular invasion of the labyrinth layer. On E8.0, we observed normal fusion of the chorionic plate and the allantois in both Mst1+/−; Mst2+/− and Mst1−/−; Mst2−/− embryos (Fig. 4A, panel b). From E8.5 to E9.5, Mst1+/−; Mst2+/− embryos showed normal morphogenesis of the labyrinth layer, including invasion of fetal blood vessels from the allantois into the chorionic plate, and differentiation of trophoblast cells. In contrast, formation of the labyrinth layer and all other subsequent steps in the development of the placenta were severely disrupted in Mst1−/−; Mst2−/− embryos (Fig. 4D). Notably, whereas the labyrinth layer of Mst1+/−; Mst2+/− placentas contained blood vessels with embryo-derived nucleated erythrocytes, that of the Mst1−/−; Mst2−/− placenta contained sparse blood vessels with few nucleated erythrocytes (boxed areas in Fig. 4D).
To further characterize placental defects, we analyzed cell proliferation and cell death in the placenta. TUNEL staining of E8.5 placentas revealed a significant increase in the number of dying cells in Mst1−/−; Mst2−/− placentas (Fig. 4E, panels a and b). Immunostaining of Mst1−/−; Mst2−/− placentas with anti-BrdU and anti-Ki-67 antibodies revealed that Ki-67 nuclear staining was apparently reduced in the ectoplacental cone and was restricted to the chorion (Fig. 4E, panels c and d). However, we observed no detectable difference in BrdU incorporation between Mst1+/−; Mst2+/− placentas and Mst1−/−; Mst2−/− placentas (Fig. 4E, panels c and d). To further examine the origin of placental defect, we isolated RNA from E8.5 placentas and performed quantitative RT-PCR for transcriptional factor Gcm1 (for labyrinthine trophoblasts), Mash2 (for ectoplacental cone and the chorion), Tpbp (for spongiotrophoblast), and Pl1 (for trophoblast giant cell) (29). In Mst1−/−; Mst2−/− placentas, the Pl1 and Mash2 mRNA levels were significantly decreased, whereas the mRNA levels of other markers, such as Gcm1 and Tpbp, showed no alteration (Fig. 4F). On the basis of these results, ablation of Mst1 and Mst2 leads to disruption of the generation of specific placental lineages.
Next, we assessed the functional capacity of the placenta by injecting rhodamine 123 and measuring the efficiency of transport from the placenta to the fetus (29). Rhodamine 123 was readily detectable within 2 h after intraperitoneal injection into the pregnant female in Mst1+/−; Mst2+/− embryos, but significantly less rhodamine 123 was detected in Mst1−/−; Mst2−/− littermate embryos (Fig. 4G). Therefore, placental vascular defects, disruption in specific placental lineages, and the resulting deficit in placental function contribute to the death of Mst1−/−; Mst2−/− embryos.
Analysis of cell proliferation and apoptotic cell death in Mst1−/−; Mst2−/− embryos.
Since deletion of Mst kinases decreased cell proliferation and increased cell death in the placenta, we evaluated Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos for similar effects. We first pulse-labeled E9.0 embryos with BrdU for 30 min to examine embryonic cell proliferation (Fig. 5A, panels a and b). However, whereas BrdU-positive cells were readily detected in Mst1+/−; Mst2+/− embryos, BrdU-positive cells were largely absent in Mst1−/−; Mst2−/− embryos, likely reflecting limited availability of BrdU due to defects in placental function. Therefore, we measured embryonic cell proliferation by staining for Ki-67 (Fig. 5A, panels c and d). Ki-67-positive cells were densely distributed in highly proliferative regions of Mst1+/−; Mst2+/− embryos, particularly in the neural tube. The numbers of proliferating cells in E9.0 and E9.5 Mst1−/−; Mst2−/− embryos, as detected by anti-Ki-67 staining, were not higher than those in Mst1+/−; Mst2+/− embryos. Of interest, these anti-Ki-67-positive proliferating cells were restricted to the proliferative neural tube but were rather disorganized (Fig. 5A). This indicates that ablation of Mst1 and Mst2 does not significantly increase cell proliferation in the embryo proper. However, consistent with observations related to the organogenesis of heart and neuroectoderm, ablation of Mst1 and Mst2 severely disrupted the spatiotemporal patterning of proliferating cells within the embryo.
FIG. 5.

Cell proliferation and apoptosis in Mst1−/−; Mst2−/− embryos. (A) (a and b) Immunofluorescence staining of BrdU and Ki-67 in embryos on E9.0. Dotted lines indicate the border of the neural tube and neural ectoderm. (c and d) Immunohistochemical study of Ki-67 labeling in embryos on E9.5. Dotted lines indicate the forebrain regions in embryos. Abbreviations: Nt, neural tube; Ne, neural ectoderm; Fb, forebrain; DAPI, 4′,6′-diamidino-2-phenylindole. Panels a to d were taken at the same magnification. Bar, 200 μm. (e) The graph shows the relative ratios plus standard errors of the means (error bars) (SEMs) of Ki67- and BrdU-positive cells from embryos on E9.0 and Ki67-positive cells from embryos on E9.5. (B) TUNEL staining of transverse sections from embryos on E8.5 and sagittal sections from embryos on E9.5. Dotted lines indicate the border of neural tube and neural ectoderm (a and b), forebrain (c and d), and somite (e and f) regions in embryos. Abbreviations: Nt, neural tube; Ne, neural ectoderm; Fb, forebrain; Sm, somites. Panels a to f were all taken at one magnification. Bar, 200 μm. (g) The graph shows the relative ratios plus SEMs of TUNEL-positive cells from embryos on E8.5 and E9.5. (C) (a and b) Phosphorylated histone H3 (P-H3) and TUNEL costaining in sagittal sections from embryos on E9.5. The boxed area is shown at a higher magnification. Dotted lines indicate the forebrain regions of embryos. Panels a and b were both taken at one magnification. Bar, 200 μm. (c) The graph shows the relative ratios plus SEMs of phosphorylated histone H3 (PH3)- and TUNEL-positive cells from embryos on E9.5.
We next analyzed cell death in Mst1−/−; Mst2−/− embryos by TUNEL staining of E8.5 and E9.5 embryo sections (Fig. 5B). Mst1−/−; Mst2−/− embryos had greater numbers of apoptotic cells on both E8.5 and E9.5than Mst1+/−; Mst2+/− embryos did. Since Mst kinases are involved in cell cycle progression (25), we further examined the number and distribution of mitotic cells in double mutant embryos. Although we have observed the severe disorganization in distribution of mitotic cells due to overall disorganization of neural tissue, we found no difference in the number of mitotic cells in Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos (Fig. 5C). Collectively, these data suggest that the loss of Mst kinases increases cell death during embryonic development. This is unlike the case in cancer cells, where loss of Mst function disrupts the normal apoptotic process, consistent with the role of these kinases as tumor suppressors. The increased apoptosis, growth and developmental retardation, and eventual death of Mst1−/−; Mst2−/− embryos could be a secondary consequence of the observed placental functional defects.
Mst1 and Mst2 are required for normal developmental vascularization and primitive hematopoiesis.
Even though placental functional defects in Mst1−/−; Mst2−/− embryos could cause the embryonic defects, including increased apoptosis and growth and developmental retardation, there is a possibility that Mst1−/−; Mst2−/− embryos also have the intrinsic embryonic functional defects. In fact, whereas histological examination of Mst1−/−; Mst2−/− embryos from E8.0 to E9.0 revealed apparently normal yolk sac development (Fig. 6A), by E9.5 we observed a collapse in the yolk sac vasculature. Thus, we further examined the vascular architecture of embryos by immunostaining whole-mounted E9.5 Mst1−/−; Mst2−/− embryos with anti-PECAM-1. As shown in Fig. 6B, panels b and c, differentiated endothelial cells were strikingly abnormal in the Mst1−/−; Mst2−/− embryos, and total vascularity was decreased. On E9.5, PECAM-1 staining readily detected well-organized blood vessels in the dorsal aorta and intersomitic regions of Mst1+/−; Mst2+/− embryos. However, the dorsal aorta of Mst1−/−; Mst2−/− embryos had only a primitive vasculature and the somites of Mst1−/−; Mst2−/− embryos did not have small sprouting blood vessels. These results indicate that disruption of Mst1 and Mst2 genes affects vascular patterning and vascular branching in the yolk sac and embryonic tissues.
FIG. 6.

Abnormal primitive hematopoiesis and vascular development with impaired vascular patterning in mice deficient for Mst1 and Mst2. (A) H&E-stained Mst1+/−; Mst2+/− yolk sacs and Mst1−/−; Mst2−/− yolk sacs on E8.0 to E9.5. Abbreviations: Me, mesoderm; Ve, visceral endoderm; Ec, endothelial cells. All the sections shown in panel A were taken at the same magnification. Bar, 100 μm. (B) Whole-mount PECAM-1-stained embryos on E9.5. The boxed areas are shown at a higher magnification. Arrowheads indicate the somites (Sm) (a) and dorsal aorta (Da) (b and c). Panels a to c were taken at the same magnification. Bar, 400 μm. (C) Hematopoietic progenitor assay. Yolk sac cells (YS) from embryos on E8.5 were replated into methylcellulose medium supplemented with hematopoietic cytokines.
Mst1 protein is highly expressed in lymphoid tissues (Fig. 2A) and Mst1−/−; Mst2−/− yolk sacs on E9.5 appeared to have reduced numbers of primitive hematopoietic cells (Fig. 6A). Therefore, we determined primitive hematopoietic capacity of Mst1−/−; Mst2−/− yolk sacs by replating the E8.5 yolk sacs into methylcellulose containing hematopoietic cytokines (Fig. 6C). The numbers of hematopoietic colonies that were generated from Mst1+/−; Mst2+/− yolks sacs, Mst1+/−; Mst2−/− yolk sacs, and Mst1−/−; Mst2+/− yolk sacs were not significantly different (Fig. 6C). In contrast, E8.5 Mst1−/−; Mst2−/− yolk sacs had significantly decreased number of colonies formed when subjected to hematopoietic replating. These results suggest decreased numbers and/or differentiation capacities of intrinsic hematopoietic progenitors in Mst1−/−; Mst2−/− yolk sacs.
Normal embryonic development in Mst1+/−; Mst2−/− embryos and Mst1−/−; Mst2+/− embryos.
Because the phenotypic differences between Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos during the course of development were very dramatic, we examined the development of Mst1+/−; Mst2−/− embryos and Mst1−/−; Mst2+/− embryos. As noted above, Mst1+/−; Mst2−/− mice and Mst1−/−; Mst2+/− mice were viable and fertile. On E8.0 to E9.0, Mst1+/−; Mst2−/− embryos and Mst1−/−; Mst2+/− embryos had phenotypically normal yolk sacs and placentas (Fig. 7A for Mst1+/−; Mst2−/− embryos and 7B for Mst1−/−; Mst2+/− embryos). PECAM-1 staining showed that neither Mst1+/−; Mst2−/− embryos nor Mst1−/−; Mst2+/− embryos had vascular defects (Fig. 7C). Even at the later E12.5 stage, Mst1+/−; Mst2−/− embryos were indistinguishable from the normally developing Mst1+/−; Mst2+/− embryos (Fig. 7D, panel a for Mst1+/−; Mst2+/− embryos and panel b for Mst1+/−; Mst2−/− embryos). Collectively, these data indicate that a haplosufficiency of either Mst1 or Mst2 can compensate for the loss of the other's function; only complete ablation of both Mst1 and Mst2 leads to developmental defects.
FIG. 7.

Histological analysis of Mst1+/−; Mst2−/− embryos and placentas and Mst1−/−; Mst2+/− embryos and placentas. (A and B) H&E-stained Mst1+/−; Mst2−/− embryos and placentas and Mst1−/−; Mst2+/− embryos and placentas on E8.0 to E9.0. Dotted lines indicate the border between the ectoplacental cone/labyrinth layer and giant cells. Abbreviations: Ys, yolk sacs; Am, amnion; Nt, neural tube; Ht, heart. Panels a to f were all taken at one magnification. Bars, 200 μm (a and b) and 100 μm (g). (C) Whole-mount PECAM-1-stained E9.5 Mst1+/−; Mst2−/− embryos and Mst1−/−; Mst2+/− embryos. The boxed areas are shown at a higher magnification. Abbreviations: Da, dorsal aorta; Sm, somites. Panels a and b were taken at the same magnification. Bar, 400 μm. (D) Gross phenotypes and H&E staining of Mst1+/−; Mst2+/− embryos and Mst1+/−; Mst2−/− embryos on E12.5. Panels and d were taken at one magnification, and panels b, c, e, and f were taken at another, higher magnification. Bars, 1 mm (a) and 200 μm (b).
Expression levels of components in the Mst signaling pathway during early embryonic development in Mst1−/−; Mst2−/− embryos.
Finally, we determined whether components in Mst signaling pathway were dysregulated in Mst1−/−; Mst2−/− embryos on E8.5 by Western blot analysis (Fig. 8A). Phosphorylation of FOXO3A on serine 207 (18) and LATS1/2, known substrates of Mst1/2, as judged by phospho-specific antibody or shift in electrophoretic mobility, were not observed in both Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos. We observed unaltered phosphorylation of YAP by LATS1/2 in both Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos. Since YAP is the principal regulator of TEAD4 transcription factor for trophectoderm lineage development (21), we determined the levels of expression of Cdx2, a major transcription target of TEAD4 (Fig. 8C). However, expression of Cdx2 was not significantly changed in Mst1−/−; Mst2−/− embryos. Of interest, ablation of Mst1 and Mst2 significantly decreased the protein level of WW45, suggesting that Mst kinases are required for maintaining WW45 stability. Overall, Mst1−/−; Mst2−/− embryos did not exhibit any dysregulation in phosphorylation or levels of components in the Mst signaling pathway compared to Mst1+/−; Mst2+/− embryos on E8.5 (Fig. 8B). WW45 is required for Mst1 activation and consequent phosphorylation of LATS1/2 and YAP to sequester YAP in the cytoplasm during epithelial differentiation (16). Comparing this previous report with these data in present study, the effects of ablation of the Mst signaling pathway likely vary between early embryonic development and epithelial differentiation.
FIG. 8.

Ablation of Mst1 and Mst2 did not affect the levels of components in the Mst signaling pathway but significantly decreased the level of WW45 protein. (A) Whole-cell extracts from embryos on E8.5 [Mst1+/−; Mst2+/− embryos (n = 10), Mst1+/−; Mst2−/− embryos (n = 17), Mst1−/−; Mst2+/− embryos (n = 18), and Mst1−/−; Mst2−/− embryos (n = 9) were analyzed by Western blotting using antibodies against YAP, phosphorylated YAP (p-YAP), WW45, LATS1, LATS2, FOXO3A, Mst1, and Mst2. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B) Relative protein levels of genes of interest in Mst1−/−; Mst2−/− embryos compared to those in Mst1+/−; Mst2+/− embryos. (C) Representative quantitative PCR data showing the levels of cdx2 gene from Mst1+/−; Mst2+/− embryos and Mst1−/−; Mst2−/− embryos relative to the level of the GAPDH gene. The sequences of specific primers are available upon request.
DISCUSSION
Mst1 and Mst2 are best known for their originally identified function as key apoptotic kinases that can be activated by various cell death signals in in vitro culture systems. Hippo (Hpo), the Drosophila homolog of Mst kinases, is involved in restricting cell proliferation and promoting apoptosis during development of the fly. However, the roles of Mst kinases in mammalian development have not been fully investigated.
This study reveals that Mst kinases are essential for normal mouse development. Although both Mst1−/− and Mst2−/− single-knockout mice were outwardly normal and lacked severe morphological or histological defects, Mst1−/−; Mst2−/− double mutant embryos died in utero with multiple developmental defects. First, Mst1−/−; Mst2−/− embryos showed functional placental defects that could be expected to restrict the availability of nutrients to the embryo. These placental defects could account for the observed growth retardation and decreased rate of cell proliferation in Mst1−/−; Mst2−/− embryos, and ultimately for inducing cell death in the embryo proper. Second, Mst1−/−; Mst2−/− embryos showed circulation defects, including abnormal vascular patterning in the yolk sac and embryo proper, as well as turning failure, cellular disorganization, and failure of neural ectodermal cells to fuse to form a neural tube. In particular, Mst1−/−; Mst2−/− embryos showed decreased numbers and/or differentiation capacities of hematopoietic progenitors in the yolk sacs. These data suggest that Mst1 and Mst2 are essential for regulating primitive hematopoiesis intrinsically. As previously noted, defects of many tissues in double mutant embryos, including placental and/or intrinsic embryonic defects could be secondary to the observed developmental retardation. Taken together with the high sequence homology between Mst kinases, the phenotypic differences between single and double knockouts suggest that Mst1 and Mst2 kinases are functionally redundant. Collectively, these results indicate that Mst1 and Mst2 are both essential for early developmental processes, including placental development, vascular patterning, and primitive hematopoiesis.
In Drosophila and mammals, the Hippo pathway restricts cell proliferation and promotes apoptosis in differentiating epithelia (7, 9, 11, 12, 16, 24, 30, 31, 33, 34). We have recently shown that Mst1 kinase phosphorylates LATS1/2 and consequent phosphorylation of YAP to sequester YAP in cytoplasm (16) during epithelial differentiation in mammals. In this study, however, we could not observe any phosphorylation of known substrate proteins even in Mst1+/−; Mst2+/− embryos on E8.5. Moreover, transcriptional coactivation by YAP was not significantly altered as judged by Cdx2 transcription. Instead, loss of Mst1/2 decreased the level of the WW45 protein, suggesting possible regulation of stability. This finding suggests that although the Hippo pathway conserved from Drosophila is important for epithelial proliferation and differentiation, Mst kinases may be involved in other pathways crucial for early mammalian development. It could also be possible that spatiotemporal regulation affects either the composition of Mst signaling components or substrate specificities during embryogenesis.
In mammals, dysfunctions in the Hippo pathway lead to inappropriate proliferation and expansion of cell compartment and subsequent induction of tumor formation. Unexpectedly, we found no evidence for tumor development over a 24-month observation period in mice lacking either Mst1 or Mst2 (data not shown), a result that may indicate that that Mst1 and Mst2 functionally compensate for each other to prevent tumorigenesis. Instead, we observed that ablation of Mst kinases increased cell death and decreased proliferation. This observation is not in accord with in vitro data from mammalian cells. Thus, it is possible that these defects are not direct consequences of ablation of Mst kinases in the embryo proper but are secondary to placental defects and insufficient nutrient transport. To exclude this possibility and investigate a possible role for Mst kinases in cell proliferation and cell death, we attempted to isolate and culture MEFs from Mst1−/−; Mst2−/− embryos on E8.5 to E9.5. However, we were unable to propagate fibroblasts from embryos on E8.5 or E9.5 under standard growth conditions. Interestingly, in contrast to previous reports that depletion of Mst1 reduced the incidence of Fas- and oxidative stress-induced apoptosis in cancer cell lines (18, 22), we saw no significant decrease in the incidence of Fas- or H2O2-induced apoptosis in Mst1 or Mst2 single-null MEFs (Oh and Lim, personal observations), again suggesting the functional redundancy of Mst1 and Mst2. The reason for the discrepancy between our results and previous reports is not clear but may reflect differences in cellular context between primary MEFs and cancer cells.
We also examined possible haploinsufficiency effects of Mst1 in an Mst2-deficient background and vice versa. However, Mst1+/−; Mst2−/− mice and Mst1−/−; Mst2+/− mice were developmentally normal and viable and did not develop tumors over a 10-month observation period (data not shown). Further investigation of tumorigenesis in Mst1+/−; Mst2−/− mice and Mst1−/−; Mst2+/− mice will be required to understand the gene dosage-dependent functions of Mst genes. Because Mst1−/−; Mst2−/− embryos have severe developmental defects at an early embryonic stage, the use of conditional knockout mice to exclude developmental defects is warranted in future studies of the molecular mechanisms of Mst proteins in cell proliferation and apoptosis.
Acknowledgments
This work was supported by the National Research Laboratory Program, the South Korea National Cancer Center Control Program, and the Nuclear Research Program of South Korea. This work was also partly supported by a grant from the KOSEF (M10503010002) to K.-S.K.
Footnotes
Published ahead of print on 28 September 2009.
REFERENCES
- 1.Ahn, S. H., W. L. Cheung, J. Y. Hsu, R. L. Diaz, M. M. Smith, and C. D. Allis. 2005. Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell 120:25-36. [DOI] [PubMed] [Google Scholar]
- 2.Cheung, W. L., K. Ajiro, K. Samejima, M. Kloc, P. Cheung, C. A. Mizzen, A. Beeser, L. D. Etkin, J. Chernoff, W. C. Earnshaw, and C. D. Allis. 2003. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 113:507-517. [DOI] [PubMed] [Google Scholar]
- 3.Cinar, B., P. K. Fang, M. Lutchman, D. Di Vizio, R. M. Adam, N. Pavlova, M. A. Rubin, P. C. Yelick, and M. R. Freeman. 2007. The pro-apoptotic kinase Mst1 and its caspase cleavage products are direct inhibitors of Akt1. EMBO J. 26:4523-4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dan, I., N. M. Watanabe, and A. Kusumi. 2001. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 11:220-230. [DOI] [PubMed] [Google Scholar]
- 5.Edgar, B. A. 2006. From cell structure to transcription: Hippo forges a new path. Cell 124:267-273. [DOI] [PubMed] [Google Scholar]
- 6.Graves, J. D., K. E. Draves, Y. Gotoh, E. G. Krebs, and E. A. Clark. 2001. Both phosphorylation and caspase-mediated cleavage contribute to regulation of the Ste20-like protein kinase Mst1 during CD95/Fas-induced apoptosis. J. Biol. Chem. 276:14909-14915. [DOI] [PubMed] [Google Scholar]
- 7.Harvey, K. F., C. M. Pfleger, and I. K. Hariharan. 2003. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114:457-467. [DOI] [PubMed] [Google Scholar]
- 8.Jang, S. W., S. J. Yang, S. Srinivasan, and K. Ye. 2007. Akt phosphorylates MstI and prevents its proteolytic activation, blocking FOXO3 phosphorylation and nuclear translocation. J. Biol. Chem. 282:30836-30844. [DOI] [PubMed] [Google Scholar]
- 9.Jia, J. H., W. S. Zhang, B. Wang, R. Trinko, and J. Jiang. 2003. The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev. 17:2514-2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang, Z., X. Li, J. Hu, W. Zhou, Y. Jiang, G. Li, and D. Lu. 2006. Promoter hypermethylation-mediated down-regulation of LATS1 and LATS2 in human astrocytoma. Neurosci. Res. 56:450-458. [DOI] [PubMed] [Google Scholar]
- 11.Justice, R. W., O. Zilian, D. F. Woods, M. Noll, and P. J. Bryant. 1995. The Drosophila tumor-suppressor gene Warts encodes a homolog of human myotonic-dystrophy kinase and is required for the control of cell-shape and proliferation. Genes Dev. 9:534-546. [DOI] [PubMed] [Google Scholar]
- 12.Kango-Singh, M., R. Nolo, C. Tao, P. Verstreken, P. R. Hiesinger, H. J. Bellen, and G. Halder. 2002. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development 129:5719-5730. [DOI] [PubMed] [Google Scholar]
- 13.Katagiri, K., T. Katakai, Y. Ebisuno, Y. Ueda, T. Okada, and T. Kinashi. 2009. Mst1 controls lymphocyte trafficking and interstitial motility within lymph nodes. EMBO J. 28:1319-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai, Z. C., X. Wei, T. Shimizu, E. Ramos, M. Rohrbaugh, N. Nikolaidis, L. L. Ho, and Y. Li. 2005. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 120:675-685. [DOI] [PubMed] [Google Scholar]
- 15.Lee, D., C. Park, H. Lee, J. J. Lugus, S. H. Kim, E. Arentson, Y. S. Chung, G. Gomez, M. Kyba, S. Lin, R. Janknecht, D. S. Lim, and K. Choi. 2008. ER71 acts downstream of BMP, Notch, and Wnt signaling in blood and vessel progenitor specification. Cell Stem Cell 2:497-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee, J. H., T. S. Kim, T. H. Yang, B. K. Koo, S. P. Oh, K. P. Lee, H. J. Oh, S. H. Lee, Y. Y. Kong, J. M. Kim, and D. S. Lim. 2008. A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J. 27:1231-1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee, K. K., T. Ohyama, N. Yajima, S. Tsubuki, and S. Yonehara. 2001. MST, a physiological caspase substrate, highly sensitizes apoptosis both upstream and downstream of caspase activation. J. Biol. Chem. 276:19276-19285. [DOI] [PubMed] [Google Scholar]
- 18.Lehtinen, M. K., Z. Yuan, P. R. Boag, Y. Yang, J. Villen, E. B. Becker, S. DiBacco, N. de la Iglesia, S. Gygi, T. K. Blackwell, and A. Bonni. 2006. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125:987-1001. [DOI] [PubMed] [Google Scholar]
- 19.McPherson, J. P., L. Tamblyn, A. Elia, E. Migon, A. Shehabeldin, E. Matysiak-Zablocki, B. Lemmers, L. Salmena, A. Hakem, J. Fish, F. Kassam, J. Squire, B. G. Bruneau, M. P. Hande, and R. Hakem. 2004. Lats2/Kpm is required for embryonic development, proliferation control and genomic integrity. EMBO J. 23:3677-3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morin-Kensicki, E. M., B. N. Boone, M. Howell, J. R. Stonebraker, J. Teed, J. G. Alb, T. R. Magnuson, W. O'Neal, and S. L. Milgram. 2006. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol. Cell. Biol. 26:77-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishioka, N., K. Inoue, K. Adachi, H. Kiyonari, M. Ota, A. Ralston, N. Yabuta, S. Hirahara, R. O. Stephenson, N. Ogonuki, R. Makita, H. Kurihara, E. M. Morin-Kensicki, H. Nojima, J. Rossant, K. Nakao, H. Niwa, and H. Sasaki. 2009. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev. Cell 16:398-410. [DOI] [PubMed] [Google Scholar]
- 22.Oh, H. J., K. K. Lee, S. J. Song, N. S. Jin, M. S. Song, J. H. Lee, C. R. Im, J. O. Lee, S. Yonehara, and D. S. Lim. 2006. Role of the tumor suppressor RASSF1A in Mst1-mediated apoptosis. Cancer Res. 66:2562-2569. [DOI] [PubMed] [Google Scholar]
- 23.Overholtzer, M., J. Zhang, G. A. Smolen, B. Muir, W. Li, D. C. Sgroi, C. X. Deng, J. S. Brugge, and D. A. Haber. 2006. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 103:12405-12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pantalacci, S., N. Tapon, and P. Leopold. 2003. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 5:921-927. [DOI] [PubMed] [Google Scholar]
- 25.Praskova, M., F. Xia, and J. Avruch. 2008. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr. Biol. 18:311-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saucedo, L. J., and B. A. Edgar. 2007. Filling out the Hippo pathway. Nat. Rev. Mol. Cell Biol. 8:613-621. [DOI] [PubMed] [Google Scholar]
- 27.St John, M. A., W. Tao, X. Fei, R. Fukumoto, M. L. Carcangiu, D. G. Brownstein, A. F. Parlow, J. McGrath, and T. Xu. 1999. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat. Genet. 21:182-186. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi, Y., Y. Miyoshi, C. Takahata, N. Irahara, T. Taguchi, Y. Tamaki, and S. Noguchi. 2005. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin. Cancer Res. 11:1380-1385. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka, H., K. Nagaike, N. Takeda, H. Itoh, K. Kohama, T. Fukushima, S. Miyata, S. Uchiyama, S. Uchinokura, T. Shimomura, K. Miyazawa, N. Kitamura, G. Yamada, and H. Kataoka. 2005. Hepatocyte growth factor activator inhibitor type 1 (HAI-1) is required for branching morphogenesis in the chorioallantoic placenta. Mol. Cell. Biol. 25:5687-5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tapon, N., K. F. Harvey, D. W. Bell, D. C. R. Wahrer, T. A. Schiripo, D. A. Haber, and I. K. Hariharan. 2002. salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 110:467-478. [DOI] [PubMed] [Google Scholar]
- 31.Udan, R. S., M. Kango-Singh, R. Nolo, C. Y. Tao, and G. Halder. 2003. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 5:914-920. [DOI] [PubMed] [Google Scholar]
- 32.Ura, S., N. Masuyama, J. D. Graves, and Y. Gotoh. 2001. Caspase cleavage of MST1 promotes nuclear translocation and chromatin condensation. Proc. Natl. Acad. Sci. USA 98:10148-10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu, S., J. B. Huang, J. X. Dong, and D. J. Pan. 2003. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114:445-456. [DOI] [PubMed] [Google Scholar]
- 34.Xu, T., W. Wang, S. Zhang, R. A. Stewart, and W. Yu. 1995. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 121:1053-1063. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto, S., G. P. Yang, D. Zablocki, J. Liu, C. Hong, S. J. Kim, S. Soler, M. Odashima, J. Thaisz, G. Yehia, C. A. Molina, A. Yatani, D. E. Vatner, S. F. Vatner, and J. Sadoshima. 2003. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J. Clin. Investig. 111:1463-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan, Z., M. K. Lehtinen, P. Merlo, J. Villen, S. Gygi, and A. Bonni. 2009. Regulation of neuronal cell death by MST1-FOXO1 signaling. J. Biol. Chem. 284:11285-11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zender, L., M. S. Spector, W. Xue, P. Flemming, C. Cordon-Cardo, J. Silke, S. T. Fan, J. M. Luk, M. Wigler, G. J. Hannon, D. Mu, R. Lucito, S. Powers, and S. W. Lowe. 2006. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125:1253-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao, B., J. Kim, X. Ye, Z. C. Lai, and K. L. Guan. 2009. Both TEAD-binding and WW domains are required for the growth stimulation and oncogenic transformation activity of Yes-associated protein. Cancer Res. 69:1089-1098. [DOI] [PubMed] [Google Scholar]
- 39.Zhou, D., B. D. Medoff, L. Chen, L. Li, X. F. Zhang, M. Praskova, M. Liu, A. Landry, R. S. Blumberg, V. A. Boussiotis, R. Xavier, and J. Avruch. 2008. The Nore1B/Mst1 complex restrains antigen receptor-induced proliferation of naive T cells. Proc. Natl. Acad. Sci. USA 105:20321-20326. [DOI] [PMC free article] [PubMed] [Google Scholar]