

Abstract
Clathrin is involved in the endocytosis and exocytosis of cellular proteins and the process of virus infection. We have previously demonstrated that large hepatitis delta antigen (HDAg-L) functions as a clathrin adaptor, but the detailed mechanisms of clathrin involvement in the morphogenesis of hepatitis delta virus (HDV) are not clear. In this study, we found that clathrin heavy chain (CHC) is a key determinant in the morphogenesis of HDV. HDAg-L with a single amino acid substitution at the clathrin box retained nuclear export activity but failed to interact with CHC and to assemble into virus-like particles. Downregulation of CHC function by a dominant-negative mutant or by short hairpin RNA reduced the efficiency of HDV assembly, but not the secretion of hepatitis B virus subviral particles. In addition, the coexistence of a cell-permeable peptide derived from the C terminus of HDAg-L significantly interfered with the intracellular transport of HDAg-L. HDAg-L, small HBsAg, and CHC were found to colocalize with the trans-Golgi network and were highly enriched on clathrin-coated vesicles. Furthermore, genotype II HDV, which assembles less efficiently than genotype I HDV does, has a putative clathrin box in its HDAg-L but interacted only weakly with CHC. The assembly efficiency of the various HDV genotypes correlates well with the CHC-binding activity of their HDAg-Ls and coincides with the severity of disease outcome. Thus, the clathrin box and the nuclear export signal at the C terminus of HDAg-L are potential new molecular targets for HDV therapy.
Pathogens often take advantage of intracellular pathways involved in the trafficking of cellular macromolecules in order to carry out their life cycle, which consists of virus entry, translation, genome replication, assembly, and release. The clathrin-mediated endocytic route is a pathway commonly used for virus entry (29). Following clathrin-mediated endocytosis, incoming viruses are transported together with their receptors from the plasma membrane into early and late endosomes. Several links between clathrin adaptor complexes and viral biogenesis, including those of influenza virus (37), reovirus (13), and vesicular stomatitis virus (33), have been demonstrated.
Clathrin and its adaptor proteins (APs), which constitute the major components of clathrin-coated vesicles (CCVs), are often the carriers of proteins and lipids that are transported from the trans-Golgi network (TGN) to the endosome (20, 35). Clathrin-mediated exocytosis has been found to participate in viral multiplication. The envelope protein of vesicular stomatitis virus, glycoprotein 1, recruits clathrin adaptor complex adaptor protein 1 (AP1) onto Golgi membranes and possibly leaves the TGN in CCVs for subsequent transport to endosomes (1). It is also known that interaction of AP1 with the matrix domain of human immunodeficiency virus type 1 Gag protein promotes viral release (5). In addition, Vpu inhibits the endosomal accumulation of the human immunodeficiency virus type 1 structural proteins Env and Gag, which is known to enhance viral assembly and release at the plasma membrane (39). Furthermore, large hepatitis delta antigen (HDAg-L) encoded by the hepatitis delta virus (HDV) has recently been identified as a novel clathrin adaptor-like protein (18). HDAg-L specifically interacts with clathrin heavy chain (CHC) at the TGN and inhibits clathrin-mediated protein transport. However, the role of CHC in the life cycle of HDV remains unclear.
HDV is a highly pathogenic virus. The virion is coated with the envelope proteins of hepatitis B virus (HBV), the hepatitis B virus surface antigens (HBsAgs) (24). Superinfection or coinfection with HBV may result in fulminant hepatitis and progressive chronic liver cirrhosis (3, 36). The small HDAg (HDAg-S) lacks the unique C-terminal 19-amino-acid sequence of HDAg-L (6, 41, 43) and functions as a transactivator of HDV genome replication in the nucleus (23, 24). Both HDAg-S and HDAg-L possess nuclear localization signals (NLSs) spanning amino acid residues 35 to 88 and are mainly localized in the nuclei of transfected cells in the absence of HBsAg (7, 8). However, HDAg-L has been demonstrated to be a nucleocytoplasmic shuttling protein with a nuclear export signal (NES) at its unique C terminus, and this is important for HDV assembly (27). In the presence of HBsAg, HDAg-L relocalizes to the cytoplasm (29). In addition, a NES-interacting protein of HDAg-L, NESI, has been identified to be essential for the HDAg-L-mediated nuclear export of HDV RNA (42). Furthermore, the proline-rich motif within the unique 19-amino-acid extension together with isoprenylation of the CXXX motif (15) are essential for HDAg-L to form delta virus-like particles (VLPs) with HBsAg (19, 22). Taken together, these results imply that an intracellular association between HDAg-L and HBsAg in the cytoplasm is the driving force of HDV assembly. The interaction of HDAg-L with HBsAg facilitates the assembly and secretion of HDV particles. Nevertheless, the cellular proteins and pathways involved in the transport, packaging, and secretion of HDV are poorly understood.
In this study, the involvement of clathrin-mediated trafficking in the propagation of HDV is biochemically characterized. Downregulation of functional CHC significantly reduced the efficiency of the CCV-mediated HDV assembly. However, CHC is not essential for the assembly of HBV subviral particles (SVPs). These results indicate that, although HBV and HDV share common surface antigens, different mechanisms are involved in their viral assembly and release. In addition, the assembly efficiency of the various HDV genotypes correlates well with the ability of HDAg-L to interact with CHC. This may reflect the fact that there is lower pathogenicity among patients infected with HDV genotype II than among those infected with genotype I.
MATERIALS AND METHODS
Plasmids. (i) pGEX-HDAg-L(198-210) and pGEX-HDAg-L(198-210)-II.
Plasmid pGEX-HDAg-L(198-210) has been described previously (18). For construction of plasmid pGEX-HDAg-L(198-210)-II, a cDNA fragment encompassing the C terminus of the genotype II HDAg-L from amino acid residues 198 to 210 was generated by annealing two synthetic oligonucleotides representing the two strands of HDAg-L(198-210) with additional recognition sequences of EcoRI/SalI at the ends. The cDNA fragment was subsequently cloned into the EcoRI/SalI sites of plasmid pGEX-6P-1 (GE Healthcare Bio-Sciences) to generate plasmid pGEX-HDAg-L(198-210)-II.
(ii) pEGFP-N1-CHC1-107.
For construction of plasmid pEGFP-N1-CHC1-107 encoding a fusion protein of green fluorescent protein (GFP) and CHC1-107, a cDNA fragment representing the N-terminal 107 amino acid residues of the CHC with additional HindIII/SacII recognition sequences at the ends was generated by PCR from pET15b-CHC1-107 (18) and subcloned into the HindIII/SacII sites of plasmid pEGFP-N1 (Clontech).
(iii) pECE-d-BE, pECE-d-SM, pECE-HDAgL-L199A, pECE-HDAgL-D203A, pECE-d-BE(P205A), and pECE-C-ES.
Plasmids pECE-d-BE and pECE-d-SM contain cDNAs encoding the HDAg-L and HDAg-S, respectively, and pECE-C-ES encodes the small form of HBsAg (9). Plasmid pECE-d-BE(P205A) encodes HDAg-L with the P205A amino acid substitution as previously described (27). For construction of plasmids pECE-HDAgL-L199A and pECE-HDAgL-D203A, cDNA fragments encompassing the C terminus of HDAg-L from amino acid residues 195 to 212 with the single amino acid substitution L199A and D203A, respectively, and additional recognition sequences of NcoI/SalI at the ends were generated by annealing two synthetic oligonucleotides. The cDNA fragments were cloned into the NcoI/SalI sites of plasmid pX9 (10) to generate plasmids pX9-L199A and pX9-D203A. Subsequently, 1-kb BamHI/HindIII fragments were obtained from pX9-L199A and pX9-D203A and cloned into the BamHI/HindIII sites of plasmid pECE to generate plasmids pECE-HDAgL-L199A and pECE-HDAgL-D203A, respectively.
(iv) pCMV-Tag2C-HDAgL, pCMV-Tag2C-HDAgS, pCMV-Tag2C-HDAgL-d35/88, pCMV-Tag2C-HDAgS-d35/88, pCMV-Tag2C-HDAgL-INIIC, pCMV-Tag2C-HDAgL-II, and pCMV-Tag2C-HDAgL-IINIC.
Plasmids pCMV-Tag2C-HDAgL (CMV stands for cytomegalovirus), pCMV-Tag2C-HDAgS, pCMV-Tag2C-HDAgL-d35/88, and pCMV-Tag2C-HDAgS-d35/88 have been described previously (18). For construction of plasmid pCMV-Tag2C-HDAgL-INIIC, a cDNA fragment encompassing the C terminus of genotype II HDAg-L from amino acid residues 195 to 214 was generated by annealing two synthetic oligonucleotides with additional recognition sequences of NcoI/HindIII at the ends and subsequently cloned into the NcoI/HindIII sites of plasmid pX9 to generate pX9-INIIC. The 1-kb BamHI/HindIII cDNA fragment from plasmid pX9-INIIC was then cloned into the BamHI/HindIII sites of plasmid pCMV-Tag2C (Stratagen) to generate pCMV-Tag2C-HDAgL-INIIC. For construction of pCMV-Tag2C-HDAgL-II, the full-length cDNA of genotype II HDAg-L (44) with additional recognition sequences of BamHI/HindIII at the ends was synthesized and cloned into the BamHI/HindIII sites of plasmid pBH (Genomics, Taiwan) to generate pBH-HDAg-L-II. The 0.7-kb BamHI/HindIII cDNA fragment from plasmid pBH-HDAg-L-II was then cloned into the BamHI/HindIII sites of plasmid pCMV-Tag2C to generate pCMV-Tag2C-HDAgL-II. For generation of plasmid pCMV-Tag2C-HDAgL-IINIC, a 0.6-kb BamHI/NcoI cDNA fragment from plasmid pBH-HDAg-L-II was cloned into the BamHI/NcoI sites of plasmid pX9 to generate pX9-IINIC. The 1-kb BamHI/HindIII cDNA fragment from plasmid pX9-IINIC was then cloned into the BamHI/HindIII sites of plasmid pCMV-Tag2C to generate pCMV-Tag2C-HDAgL-IINIC.
(v) pLKO.1-shCHC-1, pLKO.1-shCHC-2, and pLKO.1-shLuc.
Plasmids pLKO.1-shCHC-1, pLKO.1-shCHC-2, and pLKO.1-shLuc were obtained from the National RNAi Core Facility (Academia Sinica, Taiwan). pLKO.1-shCHC-1 and pLKO.1-shCHC-2 transcribed short hairpin RNAs (shRNAs) CHC-1 (5′-CCUGUGUAGAUGGGAAAGAAU-3′) and CHC-2 (5′-GCCAAUGUGAUCUGGAACUUA-3′) for CHC RNA interference, whereas pLKO.1-shLuc transcribed shRNA 5′-CAAAUCACAGAAUCGUCGUAU-3′ for luciferase RNA interference. All expression constructs were verified by DNA sequencing.
Synthetic peptides TAT-HDAg-L(198-210) and TAT-NS4A(35-54).
Cell-permeable peptide TAT-HDAg-L(198-210) consists of a 10-amino-acid carrier peptide derived from HIV-TAT48-57 sequence (GRKKRRQRRR) (31, 40) N-terminally linked to the 13 amino acid residues of HDAg-L(198-210) with two proline residues inserted as spacer between the TAT and HDAg-L(198-210) sequences for maximal flexibility (4). TAT-NS4A(35-54) consists of the HIV-TAT48-57 sequence and spacer linked to the NS4A protein of hepatitis C virus from amino acid residues 35 to 54.
Antibodies and reagents.
Rabbit polyclonal antibodies specific to HDAg were generated as described previously (7). Mouse monoclonal and rabbit polyclonal antibodies against HBsAg were provided by Mi-Hua Tao (Academia Sinica, Taipei, Taiwan). Goat polyclonal antibodies specific to HBsAg were from Dako. Mouse monoclonal antibodies against CHC were purchased from Transduction Laboratories. Rabbit polyclonal antibodies against Golgin-245 were generously provided by Fang-Jen Lee (National Taiwan University, Taipei, Taiwan). Mouse monoclonal antibodies to calnexin were from Affinity BioReagents. Mouse monoclonal antibodies against GFP and His epitope were purchased from Clontech. Horseradish peroxidase-conjugated secondary antibodies were purchased from Jackson Laboratory. Alexa Fluor 488-, and Alexa Fluor 594-conjugated secondary antibodies were from Invitrogen.
Cell lines and DNA transfection.
COS7 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Huh7 cells were maintained in the same medium but with additional nonessential amino acids. DNA transfection was performed with cationic liposomes (Invitrogen) according to the manufacturer's instructions. For establishment of the stable cell line LB-15 that constitutively expresses HDAg-L and HBsAg, COS7 cells were cotransfected with plasmids pCMV-Tag2C-HDAgL and pECE-C-ES. The transfected cells were selected with 0.5 mg G418/ml for 5 weeks. Individual clones were picked, expanded, and screened. In addition, cotransfection was performed with pCMV-Tag2C and pECE-C-ES to establish HBsAg-expressing control stable cell line B-15, and with pCMV-Tag2C-HDAgL and pECE vector to establish HDAg-L-expressing stable cell line L-1.
GST pulldown assay.
Expression and purification of recombinant fusion proteins and glutathione S-transferase (GST) pulldown assay were carried out as described previously (18). For GST pulldown competition assay, GST fusion proteins were incubated with glutathione-Sepharose 4B beads in the presence of increasing amounts of HDAg-L(198-210) peptide (ILFPADPPFSPQS) at 4°C overnight before incubation with His-tagged CHC from residues 1 to 107 (His-CHC1-107). The protein-bound beads were then precipitated, washed extensively with phosphate-buffered saline containing 1% Triton X-100, and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Immunofluorescence staining, coimmunoprecipitation, and Western blot analysis.
Immunofluorescence staining (26), coimmunoprecipitation (26), and Western blot analysis (27) were performed as previously described. The immunoblots were developed with the ECL reagent (GE Healthcare Bio-Sciences).
Harvest of VLPs from culture media and purification of CCVs from cultured cells.
VLPs were harvested from media of cultured cells previously transfected with HDAg- and HBsAg-expressing plasmids and subjected to Western blot analysis as described previously (27) in order to determine the package activities of HDAgs and HBsAg. In addition, CCVs were purified from cultured cells as previously described (30) with modifications. Briefly, cultured cells were homogenized in buffer A containing 0.1 M morpholineethanesulfonic acid (MES) (pH 6.5), 1 mM EGTA, 0.5 mM MgCl2, and protease inhibitor cocktail (1 mM phenylmethylsulfonyl fluoride, 10 μg aprotinin/ml, 1 μg pepstatin A/ml, and 1 μg leupeptin/ml) using a glass homogenizer. The homogenate fraction (designated H) was subjected to centrifugation in a RA-48J rotor for 20 min at 18,000 × g. The pellet was designated P1. The supernatant was collected and subjected to a second centrifugation in an SW 55 Ti rotor (Beckman) for 1 h at 53,500 × g. The supernatant was designated S2. The pellet was resuspended in buffer A, homogenized, and dispersed through a 26-gauge needle. The resuspended pellet was then loaded onto the top of a solution containing 8% sucrose in buffer A made in D2O and centrifuged in an SW 55 Ti rotor for 2 h at 110,000 × g. The pellet containing CCVs was collected.
RESULTS
The clathrin box of HDAg-L is critical for HDV morphogenesis.
We have previously identified a clathrin box (199-LFPAD-203) that is conserved among clathrin adaptors at the C terminus of HDAg-L and demonstrated that HDAg-L can function as a clathrin adaptor-like protein (18, 20). To study the role of the clathrin box in the assembly of HDV, a plasmid encoding HDAg-L with either a L199A or a D203A mutation was cotransfected into Huh7 cells together with an HBsAg-encoding plasmid. In addition, HDAg-L with a mutation at Pro-205, which has been shown to be critical for the nuclear export of HDAg-L and the assembly of HDV (27), was analyzed in parallel. As shown in Fig. 1A, wild-type HDAg-L was coimmunoprecipitated with endogenous CHC in the presence of small HBsAg. This contrasted with the situation with HDAg-L-L199A and HDAg-L-D203A, where only a small portion of these two proteins formed complexes with CHC. Furthermore, neither HDAg-S, which lacks the unique C-terminal sequence of HDAg-L, nor HDAg-L-P205A, which has lost nuclear export, formed complexes with CHC. Similar results were observed in COS7 cells (Fig. 1B). The specific interaction between CHC and HDAg-L was further demonstrated in COS7 cells that were coexpressing HDAg-L, HBsAg, and GFP fused to CHC1-107 (GFP-CHC1-107) (Fig. 2C). In addition, VLPs in the culture medium were harvested and analyzed for the presence of small HBsAg and HDAg-L. As shown in Fig. 1C and D, both forms of the small HBsAg, p24 and gp27, were detected in all the conditions examined. Nonetheless, the VLP packaging activities of the various mutants were significantly different from that of the wild-type HDAg-L. Specifically, HDAg-L-L199A and HDAg-L-D203A showed reduced package activities of 9% and 23% in Huh7 cells and 5% and 15% in COS7 cells, respectively, relative to wild-type HDAg-L (Fig. 1E). In addition, HDAg-S and HDAg-L-P205A failed to form VLPs with the small HBsAg as we have previously reported (27). These results indicated that Leu-199 and Asp-203 in the clathrin box of HDAg-L are critical for the interaction of HDAg-L with CHC and for the assembly of HDV.
FIG. 1.

The conserved amino acid residues of the clathrin box of HDAg-L are critical for HDV assembly. Huh7 cells (A and C) and COS7 cells (B and D) were cotransfected with plasmid pECE-C-ES encoding the small HBsAg and a plasmid encoding HDAg as indicated. Two days posttransfection, cells were harvested and subjected to immunoprecipitation with antibodies specific to CHC, followed by Western blot analysis with antibodies against CHC and HDAgs as indicated (A and B). HDAgs present in cell lysates (10% input) and small HBsAg secreted into culture medium in the form of HDV VLPs or HBV SVPs were detected by Western blotting as controls. On the other hand, HDV VLPs were collected 4 days posttransfection from the culture medium and subjected to Western blot analysis with antibodies against HDAg and HBsAg (C and D). NT represents untransfected cells. The VLP packaging activities of the various mutant HDAgs were calculated and normalized to the VLP packaging activity of the wild-type HDAg-L (E). The results shown represent the means plus standard deviations (error bars) from three independent experiments.
FIG. 2.

Subcellular localization and CHC-binding activity of HDAg-L mutants. (A and B) Cellular distribution of HDAgs in the presence of small HBsAg. HDAgs as indicated were coexpressed with small HBsAg in COS7 cells. At 72 h posttransfection, immunofluorescence staining was performed with HDAg-specific antibodies. Three representative staining patterns of the HDAgs are shown in panel A. Type I, nucleolus distribution; type II, both nucleolus and nucleoplasm distribution; type III, nucleolus, nucleoplasm, and cytoplasm distribution. For statistical analysis, fields (each containing at least 200 HDAg-positive cells) were randomly selected. Cell numbers bearing each type of the defined staining patterns of HDAg were counted and plotted as the percentage of the total number of the HDAg-positive cells in the same field (B). (C) CHC-binding activity of HDAg-L mutants. HBsAg and GFP-CHC1-107 were coexpressed with HDAgs as indicated in COS7 cells. Two days posttransfection, cell lysates were harvested and subjected to immunoprecipitation (IP) and Western blot analysis (WB) with antibodies as shown to the right of the blots. HDAgs present in the cell lysates (10% input) and HBsAg secreted into culture medium were used as controls. The positions of molecular mass markers (in kilodaltons) are shown to the left of the blots.
Mutations at Leu-199 and Asp-203 had little effect on the cytoplasmic relocalization of HDAg-L.
We have previously demonstrated that HDAg-L is a nuclear protein that relocalizes to the cytoplasm in the presence of small HBsAg (27). The staining patterns of HDAg-L in the presence of HBsAg were classified into three types as shown in Fig. 2A, namely, type I (nucleolus staining), type II (both nucleolus and nucleoplasm staining), and type III (nucleolus, nucleoplasm, and cytoplasm staining). To understand the molecular effects of the amino acid substitutions at Leu-199 and Asp-203 in terms of inhibition of HDV assembly, the nuclear export activities of the HDAg-L mutants were examined by immunofluorescence staining. The results of statistical analysis indicated that the localization of wild-type HDAg-L was restricted to the nucleolus and nucleoplasm, giving the type I and type II patterns in up to 98% of the cells in the absence of HBsAg, but this changed to a nucleus and cytoplasm distribution, the type III pattern, in about 41% of transfected cells in the presence of small HBsAg (Fig. 2B). As expected, both HDAg-S and the package-defective HDAg-L-P205A mutant were confined to the nuclei even in the presence of small HBsAg. Interestingly, the distribution patterns of the HDAg-L-L199A and HDAg-L-D203A mutants were similar to that of the wild-type HDAg-L in the presence of HBsAg. These results indicated that Leu-199 and Asp-203 in the clathrin box of HDAg-L are not the major determinants of the nuclear export of HDAg-L, although they are critical for HDV assembly. The wild-type HDAg and the cytoplasm-localized HDAg-L-d35/88 and HDAg-S-d35/88, which represent NLS deletion mutants of HDAg-L and HDAg-S, respectively, were then coexpressed with HBsAg and GFP-CHC1-107 in cultured cells. The cells were harvested 2 days posttransfection and subjected to an immunoprecipitation assay. As shown in Fig. 2C, the cytoplasm-localized HDAg-L-d35/88 coimmunoprecipitated with GFP-CHC1-107 similar to wild-type HDAg-L (lanes 1 and 3), whereas HDAg-S and HDAg-S-d35/88 failed to form complexes with GFP-CHC1-107 (lanes 2 and 4). These results indicated that both the clathrin box and a cytoplasmic localization are essential for HDAg-L to form a complex with CHC.
HDV morphogenesis is diminished by a dominant-negative mutant of CHC and by CHC shRNA.
Since GFP-CHC1-107 has been shown to interact specifically with HDAg-L and HDAg-L-d35/88 (Fig. 2C), it was expected to function as a dominant-negative mutant and compete for the endogenous clathrin. To functionally examine whether HDV assembly is indeed a clathrin-dependent pathway, transfection experiments were performed with plasmids encoding HBsAg, GFP-CHC1-107, and HDAg-L or HDAg-L-d35/88 followed by a coimmunoprecipitation assay. The results demonstrated that, in the absence of GFP-CHC1-107 or in the presence of the GFP control, both the cytoplasm-localized wild-type HDAg-L and HDAg-L-d35/88 coimmunoprecipitated with the endogenous CHC (Fig. 3A, lanes 1, 2, 4, and 5) and the formation of HDV VLPs could be detected (Fig. 3B). However, in the presence of GFP-CHC1-107, neither HDAg-L nor HDAg-L-d35/88 formed complexes with the endogenous CHC (Fig. 3A, lanes 3 and 6), and no HDV VLPs could be detected in the culture medium (Fig. 3B, lanes 3 and 6). When a transferrin internalization assay was performed as previously described (18), a decreased level of Alexa Fluor 594-conjugated transferrin uptake was detected in the presence of GFP-CHC1-107 (data not shown), indicating that GFP-CHC1-107 interfered with the transport function of CHC. These results indicated that GFP-CHC1-107 acted as a dominant-negative mutant of CHC and competed with endogenous CHC during the formation of complexes with HDAg-L, which, in turn, interfered with the secretion of HDV VLPs. The critical role of CHC in HDV assembly was further confirmed by shRNA knockdown experiments. As shown in Fig. 4A and B, CHC-2, a shRNA against CHC that was capable of downregulating the expression of endogenous CHC, significantly reduced the level of HDV VLPs in the culture medium but had little effect on the production of HBV SVPs. On the other hand, a shRNA against luciferase and the nonfunctional CHC shRNA, CHC-1, which had little effect on the expression of endogenous CHC, had no effect on the secretion of HDV VLPs. Taken together, these results demonstrated a critical role of CHC in HDV assembly.
FIG. 3.

Inhibition of HDV assembly in cells expressing the CHC dominant-negative mutant, GFP-CHC1-107. (A) GFP-CHC1-107 competes against the endogenous clathrin for interacting with the cytoplasm localized HDAg-L. COS7 cells were transfected with plasmids encoding wild-type or mutant HDAg-L, small HBsAg, and GFP-CHC1-107 or control GFP as indicated. Two days posttransfection, the cells were harvested and subjected to immunoprecipitation assay (IP) and Western blot analysis (WB) with antibodies as shown to the right of the blots. The positions of molecular mass markers (in kilodaltons) are shown to the left of the blots. (B) Analysis of HDV VLPs. COS7 cells were transfected as described above for panel A. Four days posttransfection, VLPs were collected from culture medium and examined for the presence of HDAgs and HBsAg by Western blot analysis.
FIG. 4.
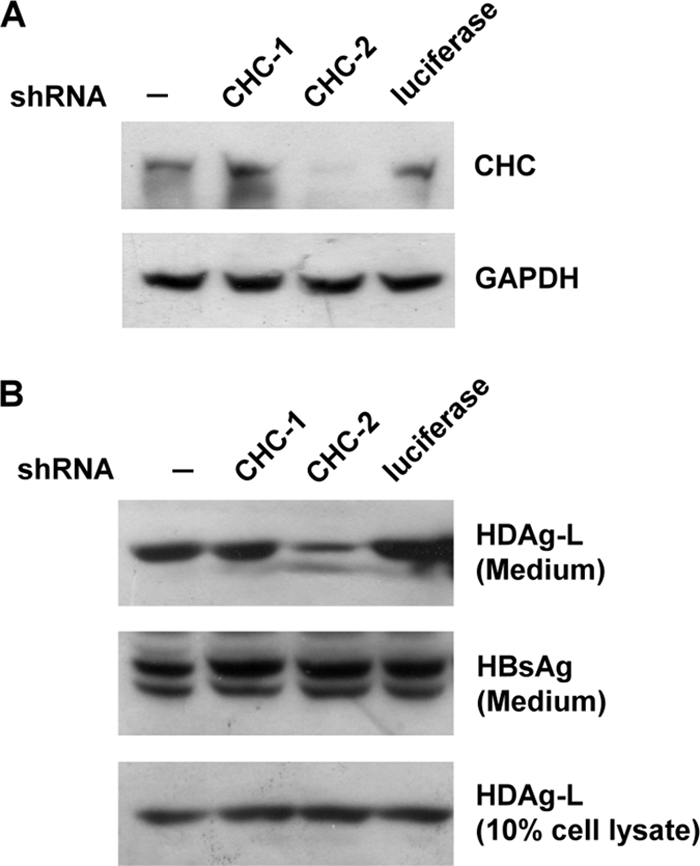
Interference of HDV assembly in cells expressing CHC-targeted shRNA. COS7 cells were transfected with plasmid expressing shRNAs as indicated (A) or cotransfected with plasmids expressing HDAg-L, small HBsAg, and the shRNAs (B). Cells were harvested, and VLPs in the culture medium were collected for Western blot analysis with antibodies as indicated. Cells not expressing shRNA (−) and cells expressing shRNA against luciferase serve as controls. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
The cell-permeable peptide TAT-HDAg-L(198-210) interferes with the transport of HDAg-L.
A peptide competition GST pulldown assay was carried out to further demonstrate a specific interaction in vitro between His-CHC1-107 and HDAg-L(198-210), which contains the clathrin box. As shown in Fig. 5, the HDAg-L(198-210) peptide significantly reduced the amount of purified His-CHC1-107 protein captured by GST-HDAg-L(198-210) (Fig. 5A), whereas the nonsense control peptide NS4A(35-54) had no effect at all (Fig. 5B). Correspondingly, when the cell-permeable peptide TAT-HDAg-L(198-210) was added to cultured cells transiently expressing HDAg-L and HBsAg, the specific association of HDAg-L and CHC was disrupted (Fig. 5C), and at the same time, no HDV VLPs could be detected (Fig. 5D). On the other hand, with the nonsense cell-permeable control peptide TAT-NS4A(35-54) and the nonpermeable peptide HDAg-L(198-210), no inhibitory effects were observed. The effect of the TAT-HDAg-L(198-210) peptide, whereby it inhibited the interaction between CHC and HDAg-L and reduced the assembly of HDV, was also observed in cells coexpressing cytoplasmic HDAg-L-d35/88 and HBsAg (Fig. 5C and D). Interestingly, when immunofluorescence staining was carried out, HDAg-L was detected only in the nucleus when TAT-HDAg-L(198-210) was introduced into the cells coexpressing HDAg-L and HBsAg (Fig. 5E). This indicated that the TAT-HDAg-L(198-210) peptide was able to block the nuclear export of HDAg-L that normally occurs in the presence of small HBsAg. Moreover, HDAg-L-d35/88 normally colocalized with CHC mainly in the perinuclear region, but it dissociated from CHC and was found distributed throughout the cytoplasm in the presence of TAT-HDAg-L(198-210) peptide (Fig. 5F). These observations strongly suggest that the TAT-HDAg-L(198-210) interferes with the interaction between HDAg-L and CHC and may inhibit the assembly and secretion of HDV.
FIG. 5.

Effect of the cell-permeable peptide TAT-HDAg-L(198-210) on the nuclear transport of HDAg-L and the assembly of HDV. (A and B) Competition of HDAg-L(198-210) peptide with GST-HDAg-L(198-210) for interacting with His-CHC1-107. GST pulldown competition assay was performed with GST-HDAg-L(198-210), purified His-CHC1-107, and various amounts of HDAg-L(198-210) peptide (A) or NS4A(35-54) peptide (B) as indicated. Coomassie blue staining (top) and Western blot analysis with antibodies against the His tag of CHC1-107 (bottom) are shown. The positions of molecular mass markers (in kilodaltons) are shown to the left of the blots. (C and D) Effects of TAT-HDAg-L(198-210) peptide on the interaction of HDAg-L with CHC and the assembly of HDV. COS7 cells were transfected with plasmids encoding small HBsAg and HDAg-L or HDAg-L-d35/88. Eight hours posttransfection, the cell-permeable peptides TAT-HDAg-L(198-210) and TAT-NS4A(35-54) and the nonpermeable control peptide HDAg-L(198-210) as indicated were added at a concentration of 20 μg/ml into the culture medium. Cells harvested 2 days posttransfection and VLPs collected 4 days posttransfection were subjected to analysis as described in the legend to Fig. 1. (E and F) Effects of TAT-HDAg-L(198-210) peptide on the subcellular distribution of HDAgs. COS7 cells were cotransfected with plasmids encoding HDAg-L and small HBsAg (E) or transfected with plasmid encoding HDAg-L-d35/88 (F). Eight hours posttransfection, cells were incubated with TAT-HDAg-L(198-210) peptide (+) at a concentration of 20 μg/ml for 40 h, followed by immunofluorescence staining with antibodies against HDAg or CHC as indicated.
CHC colocalizes with HDAg-L and small HBsAg at the TGN.
In order to better understand the localization of HDAg-L and HBsAg in association with CHC, stable cell lines were established. B-15, L-1, and LB-15 represent COS7 cell lines constitutively expressing small HBsAg alone, HDAg-L alone, and both HBsAg and HDAg-L, respectively (Fig. 6A). Immunofluorescence staining showed that HDAg-L localized to the nucleus in L-1 cells, while HDAg-L colocalized with CHC in the cytoplasm of LB-15 cells (Fig. 6B). In contrast, the small HBsAg was distributed throughout the cytoplasm of B-15 cells (Fig. 6C, panel i) and consistently colocalized with the distribution of the endoplasmic reticulum (ER) marker calnexin (Fig. 6E, panel i), but not with CHC at the TGN (Fig. 6D, panel i). Interestingly, in LB-15 cells, HDAg-L that was relocalized to the cytoplasm was enriched in a perinuclear pool with a punctate staining pattern and was colocalized with CHC (Fig. 6B to D). In addition, the coexistence of HDAg-L in LB-15 cells restricted a portion of the small HBsAg to a perinuclear compartment that is different from ER (Fig. 6E, panel ii). Double labeling with antibodies against HBsAg and the Golgi apparatus-specific protein Golgin-245 identified the distribution pattern of small HBsAg in LB-15 cells being related to the Golgi apparatus (Fig. 6F). These observations suggest that HDV assembly occurs in association with CHC and takes place at the TGN, whereas the assembly of HBV SVPs occurs at the ER and is CHC independent.
FIG. 6.

Subcellular localizations of HDAg-L, small HBsAg, and CHC in stable cell lines. (A) Western blot analysis. HDAg-L and small HBsAg in stable cell lines L1, B-15, and LB-15 and the culture media were examined by Western blot analysis. (B to F) Immunofluorescence staining. Subcellular localizations of HDAg-L, small HBsAg, and CHC in the stable cell lines were examined by immunofluorescence staining with antibodies against HDAg, CHC, and organelle markers calnexin and Golgin-245 using confocal microscopy. Bars, 20 μm.
Targeting of HBsAg to CCVs by cytoplasm-localized HDAg-L.
To learn whether HDV particles leave cells through a clathrin-mediated post-Golgi membrane trafficking pathway, CCVs from COS7 cells transiently or stably expressing HDAg with or without HBsAg were purified by subcellular fractionation. Endogenous AP2 that is highly enriched in the CCV fractions was used as an internal control in the Western blot analysis. As shown in Fig. 7, although both HDAg-L-d35/88 and HDAg-S-d35/88 that lack NLSs were localized exclusively to the cytoplasm, only HDAg-L-d35/88, which possesses the clathrin box, was detected in the CCV fractions (Fig. 7A and B). The profile of HDAg-L-d35/88 enrichment in the CCV fractions was comparable with those of CHC and AP2. In addition, neither the nucleus-localized HDAg-L in the L-1 cells nor the cytoplasm-localized small HBsAg in the B-15 cells could be detected in the CCV fractions (Fig. 7D and E). Interestingly, coexpression of HDAg-L and HBsAg in LB-15 cells drove relocalization and enrichment of both proteins on the CCVs (Fig. 7C). These results indicate that both cytoplasm localization and the clathrin box of HDAg-L are essential for targeting of HDAg-L to CCVs. Thus, HDAg-L functions as a clathrin adaptor protein through which HDV particles may be released by a CCV-mediated pathway.
FIG. 7.

Targeting of HDAg-L and HBsAg to CCVs. CCVs were partially purified by subcellular fractionation from COS7 cells transiently transfected with plasmids encoding HDAg-L-d35/88 (A) or HDAg-S-d35/88 (B) or from cells stably expressing HDAg-L alone (D), HBsAg alone (E), or HDAg-L/HBsAg together (C). Aliquots (20 μg) of each fraction as described in Materials and Methods were analyzed by Western blot analysis with antibodies against CHC, AP2, HDAgs, and HBsAg as shown to the right of the blots. The positions of molecular mass markers (in kilodaltons) are shown to the left of the blots. H, homogenate fraction; P1, pellet from first centrifugation; S2, supernatant from second centrifugation.
Functional correlation of the CHC-binding activity of HDAg-L with HDV assembly in different genotypes.
HDV genotype I is widespread in Asia, North America, and Europe, but genotype II has been isolated only in Japan and Taiwan (28, 45). Both the NLSs and RNA-binding motifs in the HDAg-L are highly conserved between genotypes I and II, but a sequence comparison has revealed divergence within the C termini (17). Genotype II HDAg-L possesses a putative clathrin box, but it differs from the one in genotype I HDAg-L in terms of both location and sequence (Fig. 8A). To learn whether the putative clathrin box of genotype II HDAg-L can function in a manner similar to that of genotype I, a GST pulldown assay was performed to examine the interaction between CHC and the genotype II HDAg-L. The results demonstrated that neither the cellular CHC (Fig. 8B) nor the partially purified His-tagged recombinant CHC1-107 (Fig. 8C) could be pulled down by the GST-genotypeII-HDAg-L(197-209) as efficiently as by the GST-genotypeI-HDAg-L(198-210). The key player that made the difference in interaction strength between CHC and genotype I/II HDAg-Ls was further examined using the HDV assembly system. In addition, two HDAg-L chimeras, INIIC and IINIC, in which the unique C-terminal 19 amino acid residues of genotype I and II HDAg-Ls were replaced by those of genotypes II and I, respectively (Fig. 8A), were generated by domain swapping. As shown in Fig. 8D, when expressed in cultured cells, both genotype II HDAg-L and the HDAg-L(INIIC) chimera could coimmunoprecipitate with CHC, but this was to a lesser extent than for genotype I HDAg-L and the HDAg-L(IINIC) chimera. In agreement with this observation, both genotype II HDAg-L and HDAg-L(INIIC) demonstrated a reduced efficiency when forming HDV VLPs with small HBsAg (Fig. 8E). The statistical analysis showed that the assembly efficiencies of genotype II HDAg-L and HDAg-L(INIIC) are about 22% and 27%, respectively, that of genotype I HDAg-L (Fig. 8F). Nevertheless, immunofluorescence staining demonstrated that genotype II HDAg-L and HDAg-L(INIIC) had cellular distribution patterns similar to those of genotype I HDAg-L and HDAg-L(IINIC) (Fig. 8G), indicating that the nuclear export activities carried by the C-terminal domain of genotype I and II HDAg-Ls are equivalent. In conclusion, the assembly efficiency correlated well with the ability of the unique C-terminal domains of the two HDAg-Ls to interact with CHC, implying a critical role of the clathrin box of HDAg-L in the morphogenesis of HDV genotypes I and II.
FIG. 8.

Correlation of the CHC-interacting activity of HDAg-L with HDV assembly in genotypes I and II. (A) Schematic representations and amino acid sequences at the C termini of genotypes I, II, and chimeras of HDAg-L. The putative clathrin boxes are underlined. (B and C) Specific interaction between HDAg-L(198-210) and CHC. GST pulldown assay was performed with GST-genotypeI-HDAg-L(198-210) or GST-genotypeII-HDAg-L(197-209) precoupled to glutathione-Sepharose beads and lysates prepared from COS7 cells (B) or purified His-CHC1-107 protein (C) as indicated. Cellular CHC and purified His-CHC1-107 that interacted with the GST fusion proteins were detected by Western blot analysis with antibodies against CHC and the His-tag of CHC1-107, respectively. GST fusion proteins that serve as loading controls were detected by Coomassie blue staining. The positions of molecular mass markers (in kilodaltons) are shown to the left of the blots. (D to G) Critical roles of the clathrin boxes of the genotype I and genotype II HDAg-L in HDV assembly. COS7 cells were cotransfected with plasmids encoding HBsAg and various HDAg-Ls as indicated. Cell lysates harvested 2 days posttransfection (D) and VLPs collected 4 days posttransfection (E and F) were subjected to analysis as described in the legend to Fig. 1. The VLP packaging activities of the various HDAg-Ls were calculated and normalized to the VLP packaging activity of genotype I HDAg-L. The results shown in panel F represent the means plus standard deviations (error bars) from three independent experiments. In addition, subcellular distributions of the HDAg-Ls in the presence of small HBsAg were analyzed by immunofluorescence staining (G). Cell numbers bearing each type of the defined staining patterns of HDAg as described in the legend to Fig. 2A were counted and plotted as the percentage of the total number of the HDAg-positive cells in the same field.
DISCUSSION
Our previous studies have demonstrated that the relocalization of HDAg-L from nucleus to the cytoplasm in the presence of small HBsAg is facilitated by the unique C terminus of the HDAg-L (27). During HDV morphogenesis, the cytoplasm-localized HDAg-L functions as a cargo and a clathrin adaptor protein that interacts through its C-terminal clathrin box with CHC at TGN (18). In this study, we further demonstrated that the morphogenesis of HDV is mediated by a clathrin-mediated post-Golgi membrane trafficking and that CHC is essential for the formation of HDV VLPs by HDAg-L. Conceivably, this correlation between the relocalization and package activities suggests that both NES and clathrin box in the unique C terminus of HDAg-L are functionally involved in the trafficking of HDAg-L and the assembly of HDV. We conclude that nuclear HDAg-L is transported to the TGN where it interacts with HBsAg and CHC for HDV morphogenesis.
The cellular transport pathways utilized by HBV and HDV for package and secretion are poorly understood. Our results suggest that HDV assembly is mediated by a clathrin-dependent pathway, whereas the assembly of HBV SVPs is clathrin independent. When coexpressed, the distribution of HDAg-L and small HBsAg parallel the CHC subcellular fraction, whereas HDAg-L or small HBsAg alone do not (Fig. 7). In addition, functional inhibition of the CHC protein has little effect on the secretion of HBV SVPs (Fig. 3 and 4). Previous reports have demonstrated a coat-mediated concentration of cargo proteins to within a relatively small TGN domain. The AP1 adaptor protein drives the concentration of the mannose-6-phosphate receptor, and as a result, more than 50% of the total Golgi apparatus mannose-6-phosphate receptors are associated with AP1-positive clathrin-coated membranes (21, 34). In our earlier studies, we found that both brefeldin A, which inhibits recruitment of clathrin adaptors onto the Golgi membrane, and wortmannin, which reduces the number of internal endosome vesicles, decreased the level of HDV VLPs in the culture medium (18). In this study, immunofluorescence staining with subcellular markers suggests that HDAg-L may direct ER-localized HBsAg to the TGN. Alternatively, HDAg-L may catch HBsAg as it undergoes endosome trafficking to the TGN. Subsequently, HDV particles are packaged into the clathrin-coated vesicles (or partially clathrin-coated vesicles); these leave the TGN and enter into late endosome or secretory vesicles before release. In contrast, without forming a complex with HDAg-L, the small HBsAg seems to aggregate in the ER, form spherical and filamentous HBV SVPs, and directly undergo ER export (2, 11).
Our current study is the first report to distinguish independent routes for the morphogenesis of HDV VLPs and HBV SVPs. The function of HDAg-L in clathrin-mediated transport may be to ensure an efficient release of HDV. However, virus infection and assembly can occur via a number of different pathways and through a variety of compartments. In this study, inhibiting the expression and function of CHC did not completely abolish the assembly and release of HDV VLPs, which suggests that alternative pathways other than the clathrin-mediated exocytosis of HDV may exist. Identification of the cellular factors capable of interacting with HDAg and HBsAg would facilitate further our understanding on these events.
The reasons for HDV genotype I infection being associated with a more severe outcome than the genotype II infection (45) remain unclear. Previous studies have demonstrated that the packaging activity of genotype II is much lower than that of the genotype I (16), but mechanisms that contribute to this difference are also not clear. It is known that prenylation on the CXXX motif at the C terminus of HDAg-L is essential to allowing HDAg-L to form HDV VLPs with HBsAg (19, 22), but there is little difference between genotypes I and II HDAg-L in terms of the level of farnesylation (32). Our current study has revealed that genotype II HDAg-L and the HDAg-L(INIIC) chimera exhibit a decreased efficiency of binding to CHC and a lower level of HDV assembly compared to genotype I HDAg-L and the HDAg-L(IINIC) chimera (Fig. 8). Since the assembly of HDV genotype I is clathrin dependent, the inefficient binding of genotype II HDAg-L to CHC may at least partially explain its relatively low efficiency in terms of virus assembly, which coincides with its milder disease phenotype. Furthermore, genotype III HDAg-L is quite divergent from genotypes I and II (44). The lack of a C-terminal clathrin box-like domain in genotype III HDAg-L implies that a different molecular mechanism(s) is involved in the assembly of HDV genotype III.
Despite considerable geographical variation, more than 350 million individuals worldwide are chronically infected with HBV. As estimated, 5% of HBV carriers are coinfected with HDV (14). HDV coinfection causes more severe liver disease than HBV infection alone (12, 38). Unfortunately, no specific therapy is currently available for HDV infection and patients with HDV coinfection respond differently to the therapies used for patients with HBV infection alone (25). We have previously reported that the inhibitory effects of HDAg-L on clathrin-mediated protein transport, including endocytosis of transferrin and trafficking of epidermal growth factor receptor, could be one possible mechanism by which HDV is responsible for progressive chronic liver disease (18). In this study, we have further demonstrated an inhibitory effect of the cell-permeable HDAg-L(198-210) peptide on HDV morphogenesis (Fig. 5). While details of the functional roles of the CHC-HDAg-L interaction in natural HDV infection await further clarification, our results suggest that chemicals capable of inhibiting the interaction between HDAg-L and host factors, including CHC and NESI (42), may be potential therapeutic agents capable of fighting HDV.
Acknowledgments
We thank Fang-Jen Lee (Institute of Molecular Medicine, National Taiwan University, Taipei, Taiwan), Mi-Hua Tao (Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan) and the National RNAi Core Facility (Institute of Molecular Biology/Genomic Research Center, Academia Sinica) for providing reagents.
This work was supported by research grants NSC95-2320-B-002-107-MY3 and NSC96-2752-B-002-009-PAE from the National Science Council of the Republic of China. The National RNAi Core Facility is supported by the National Research Program for Genomic Medicine grant NSC97-3112-B-001-016.
Footnotes
Published ahead of print on 30 September 2009.
REFERENCES
- 1.Alconada, A., U. Bauer, and B. Hoflack. 1996. A tyrosine-based motif and a casein kinase II phosphorylation site regulate the intracellular trafficking of the varicella-zoster virus glycoprotein I, a protein localized in the trans-Golgi network. EMBO J. 15:6096-6110. [PMC free article] [PubMed] [Google Scholar]
- 2.Blanchet, M., and C. Sureau. 2006. Analysis of the cytosolic domains of the hepatitis B virus envelope proteins for their function in viral particle assembly and infectivity. J. Virol. 80:11935-11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonino, F., K. H. Heermann, M. Rizzetto, and W. H. Gerlich. 1986. Hepatitis delta virus: protein composition of delta antigen and its hepatitis B virus-derived envelope. J. Virol. 58:945-950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonny, C., A. Oberson, S. Negri, C. Sauser, and D. F. Schorderet. 2001. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50:77-82. [DOI] [PubMed] [Google Scholar]
- 5.Camus, G., C. Segura-Morales, D. Molle, S. Lopez-Verges, C. Begon-Pescia, C. Cazevieille, P. Schu, E. Bertrand, C. Berlioz-Torrent, and E. Basyuk. 2007. The clathrin adaptor complex AP-1 binds HIV-1 and MLV Gag and facilitates their budding. Mol. Biol. Cell 18:3193-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang, M.-F., S. C. Baker, L. H. Soe, T. Kamahora, J. G. Keck, S. Makino, S. Govindarajan, and M. M. C. Lai. 1988. Human hepatitis delta antigen is a nuclear phosphoprotein with RNA-binding activity. J. Virol. 62:2403-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang, M. F., S. C. Chang, C. I. Chang, K. Wu, and H. Y. Kang. 1992. Nuclear localization signals, but not putative leucine zipper motifs, are essential for nuclear transport of hepatitis delta antigen. J. Virol. 66:6019-6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, M. F., C. H. Chen, S. L. Lin, C. J. Chen, and S. C. Chang. 1995. Functional domains of delta antigens and viral RNA required for RNA packaging of hepatitis delta virus. J. Virol. 69:2508-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang, M. F., C. J. Chen, and S. C. Chang. 1994. Mutational analysis of delta antigen: effect on assembly and replication of hepatitis delta virus. J. Virol. 68:646-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang, M. F., C. Y. Sun, C. J. Chen, and S. C. Chang. 1993. Functional motifs of delta antigen essential for RNA binding and replication of hepatitis delta virus. J. Virol. 67:2529-2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chua, P. K., R. Y. Wang, M. H. Lin, T. Masuda, F. M. Suk, and C. Shih. 2005. Reduced secretion of virions and hepatitis B virus (HBV) surface antigen of a naturally occurring HBV variant correlates with the accumulation of the small S envelope protein in the endoplasmic reticulum and Golgi apparatus. J. Virol. 79:13483-13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colombo, M., R. Cambieri, M. G. Rumi, G. Ronchi, E. Del Ninno, and R. De Franchis. 1983. Long-term delta superinfection in hepatitis B surface antigen carriers and its relationship to the course of chronic hepatitis. Gastroenterology 85:235-239. [PubMed] [Google Scholar]
- 13.Ehrlich, M., W. Boll, A. Van Oijen, R. Hariharan, K. Chandran, M. L. Nibert, and T. Kirchhausen. 2004. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 118:591-605. [DOI] [PubMed] [Google Scholar]
- 14.Farci, P., L. Chessa, C. Balestrieri, G. Serra, and M. E. Lai. 2007. Treatment of chronic hepatitis D. J. Viral Hepat. 14(Suppl. 1):58-63. [DOI] [PubMed] [Google Scholar]
- 15.Glenn, J. S., J. A. Watson, C. M. Havel, and J. M. White. 1992. Identification of a prenylation site in delta virus large antigen. Science 256:1331-1333. [DOI] [PubMed] [Google Scholar]
- 16.Hsu, S. C., W. J. Syu, I. J. Sheen, H. T. Liu, K. S. Jeng, and J. C. Wu. 2002. Varied assembly and RNA editing efficiencies between genotypes I and II hepatitis D virus and their implications. Hepatology 35:665-672. [DOI] [PubMed] [Google Scholar]
- 17.Hsu, S. C., J. C. Wu, I. J. Sheen, and W. J. Syu. 2004. Interaction and replication activation of genotype I and II hepatitis delta antigens. J. Virol. 78:2693-2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang, C., S. C. Chang, I. C. Yu, Y. G. Tsay, and M. F. Chang. 2007. Large hepatitis delta antigen is a novel clathrin adaptor-like protein. J. Virol. 81:5985-5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang, S. B., and M. M. C. Lai. 1993. Isoprenylation mediates direct protein-protein interactions between hepatitis large delta antigen and hepatitis B virus surface antigen. J. Virol. 67:7659-7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirchhausen, T. 2000. Clathrin. Annu. Rev. Biochem. 69:699-727. [DOI] [PubMed] [Google Scholar]
- 21.Klumperman, J., A. Hille, T. Veenendaal, V. Oorschot, W. Stoorvogel, K. von Figura, and H. J. Geuze. 1993. Differences in the endosomal distributions of the two mannose 6-phosphate receptors. J. Cell Biol. 121:997-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komla-Soukha, I., and C. Sureau. 2006. A tryptophan-rich motif in the carboxyl terminus of the small envelope protein of hepatitis B virus is central to the assembly of hepatitis delta virus particles. J. Virol. 80:4648-4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuo, M. Y., M. Chao, and J. Taylor. 1989. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: role of delta antigen. J. Virol. 63:1945-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai, M. M. C. 1995. The molecular biology of hepatitis delta virus. Annu. Rev. Biochem. 64:259-286. [DOI] [PubMed] [Google Scholar]
- 25.Lau, D. T., E. Doo, Y. Park, D. E. Kleiner, P. Schmid, M. C. Kuhns, and J. H. Hoofnagle. 1999. Lamivudine for chronic delta hepatitis. Hepatology 30:546-549. [DOI] [PubMed] [Google Scholar]
- 26.Lee, C. H., S. C. Chang, C. J. Chen, and M. F. Chang. 1998. The nucleolin binding activity of hepatitis delta antigen is associated with nucleolus targeting. J. Biol. Chem. 273:7650-7656. [DOI] [PubMed] [Google Scholar]
- 27.Lee, C. H., S. C. Chang, C. H. Wu, and M. F. Chang. 2001. A novel chromosome region maintenance 1-independent nuclear export signal of the large form of hepatitis delta antigen that is required for the viral assembly. J. Biol. Chem. 276:8142-8148. [DOI] [PubMed] [Google Scholar]
- 28.Lee, C. M., C. S. Changchien, J. C. Chung, and Y. F. Liaw. 1996. Characterization of a new genotype II hepatitis delta virus from Taiwan. J. Med. Virol. 49:145-154. [DOI] [PubMed] [Google Scholar]
- 29.Marsh, M., and A. Helenius. 1989. Virus entry into animal cells. Adv. Virus Res. 36:107-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metzler, M., V. Legendre-Guillemin, L. Gan, V. Chopra, A. Kwok, P. S. McPherson, and M. R. Hayden. 2001. HIP1 functions in clathrin-mediated endocytosis through binding to clathrin and adaptor protein 2. J. Biol. Chem. 276:39271-39276. [DOI] [PubMed] [Google Scholar]
- 31.Nagahara, H., A. M. Vocero-Akbani, E. L. Snyder, A. Ho, D. G. Latham, N. A. Lissy, M. Becker-Hapak, S. A. Ezhevsky, and S. F. Dowdy. 1998. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 4:1449-1452. [DOI] [PubMed] [Google Scholar]
- 32.O'Malley, B., and D. W. Lazinski. 2005. Roles of carboxyl-terminal and farnesylated residues in the functions of the large hepatitis delta antigen. J. Virol. 79:1142-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pelkmans, L., E. Fava, H. Grabner, M. Hannus, B. Habermann, E. Krausz, and M. Zerial. 2005. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature 436:78-86. [DOI] [PubMed] [Google Scholar]
- 34.Polishchuk, E. V., A. Di Pentima, A. Luini, and R. S. Polishchuk. 2003. Mechanism of constitutive export from the Golgi: bulk flow via the formation, protrusion, and en bloc cleavage of large trans-Golgi network tubular domains. Mol. Biol. Cell 14:4470-4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puertollano, R. 2004. Clathrin-mediated transport: assembly required. EMBO Rep. 5:942-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rizzetto, M., M. G. Canese, S. Arico, O. Crivelli, C. Trepo, F. Bonino, and G. Verme. 1977. Immunofluorescence detection of new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut 18:997-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rust, M. J., M. Lakadamyali, F. Zhang, and X. Zhuang. 2004. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 11:567-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saracco, G., S. Macagno, F. Rosina, and M. Rizzetto. 1988. Serologic markers with fulminant hepatitis in persons positive for hepatitis B surface antigen. A worldwide epidemiologic and clinical survey. Ann. Intern. Med. 108:380-383. [DOI] [PubMed] [Google Scholar]
- 39.Van Damme, N., and J. Guatelli. 2008. HIV-1 Vpu inhibits accumulation of the envelope glycoprotein within clathrin-coated, Gag-containing endosomes. Cell. Microbiol. 10:1040-1057. [DOI] [PubMed] [Google Scholar]
- 40.Vives, E., P. Brodin, and B. Lebleu. 1997. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 272:16010-16017. [DOI] [PubMed] [Google Scholar]
- 41.Wang, K. S., Q. L. Choo, A. J. Weiner, J. H. Ou, R. C. Najarian, R. M. Thayer, G. T. Mullenbach, K. J. Denniston, J. L. Gerin, and M. Houghton. 1986. Structure, sequence and expression of the hepatitis delta (δ) viral genome. Nature 323:508-514. [DOI] [PubMed] [Google Scholar]
- 42.Wang, Y. H., S. C. Chang, C. Huang, Y. P. Li, C. H. Lee, and M. F. Chang. 2005. Novel nuclear export signal-interacting protein, NESI, critical for the assembly of hepatitis delta virus. J. Virol. 79:8113-8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weiner, A. J., Q. L. Choo, K. S. Wang, S. Govindarajan, A. G. Redeker, J. L. Gerin, and M. Houghton. 1988. A single antigenomic open reading frame of the hepatitis delta virus encodes the epitope(s) of both hepatitis delta antigen polypeptides p24δ and p27δ. J. Virol. 62:594-599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu, J. C., T. Y. Chiang, and I. J. Sheen. 1998. Characterization and phylogenetic analysis of a novel hepatitis D virus strain discovered by restriction fragment length polymorphism analysis. J. Gen. Virol. 79:1105-1113. [DOI] [PubMed] [Google Scholar]
- 45.Wu, J. C., K. B. Choo, C. M. Chen, T. Z. Chen, T. I. Huo, and S. D. Lee. 1995. Genotyping of hepatitis D virus by restriction-fragment length polymorphism and relation to outcome of hepatitis D. Lancet 346:939-941. [DOI] [PubMed] [Google Scholar]