

Abstract
Regardless of genome polarity, intermediaries of complementary sense must be synthesized and used as templates for the production of new genomic strands. Depending on whether these new genomic strands become themselves templates for producing extra antigenomic ones, thus giving rise to geometric growth, or only the firstly synthesized antigenomic strands can be used to this end, thus following Luria's stamping machine model, the abundances and distributions of mutant genomes will be different. Here we propose mathematical and bit string models that allow distinguishing between stamping machine and geometric replication. We have observed that, regardless the topology of the fitness landscape, the critical mutation rate at which the master sequence disappears increases as the mechanism of replication switches from purely geometric to stamping machine. We also found that, for a wide range of mutation rates, large-effect mutations do not accumulate regardless the scheme of replication. However, mild mutations accumulate more in the geometric model. Furthermore, at high mutation rates, geometric growth leads to a population collapse for intermediate values of mutational effects at which the stamping machine still produces master genomes. We observed that the critical mutation rate was weakly dependent on the strength of antagonistic epistasis but strongly dependent on synergistic epistasis. In conclusion, we have shown that RNA viruses may increase their robustness against the accumulation of deleterious mutations by replicating as stamping machines and that the magnitude of this benefit depends on the topology of the fitness landscape assumed.
The mode of RNA virus replication has important consequences for understanding the rates at which deleterious mutations accumulate and the statistical properties of the cloud of mutants around the master sequence (4, 17). For the sake of illustration, let's assume that the infecting virus has an mRNA sense genome (positive strand), such as, for example, picornaviruslike viruses. The different steps of the infectious cycle are schematically illustrated in Fig. 1a. The first step of infection is the uncoating of the RNA molecule, followed by its translation into structural and nonstructural proteins, the latter including the replicase. The replicase then copies the genomic strand to make antigenomic (negative-sense) strands. These are used as templates to produce the genomic progeny that will accumulate in the cell, serve as templates for translation, and, following encapsidation by coat proteins, form new virions. If the antigenomic strands produced at the first round are the only templates for producing the entire progeny of genomic strands, the distribution of mutations per genome within an infected cell is expected to be Poisson because mutants do not replicate (25). Consequently, the fraction of mutation-free genomes produced is given by the Poisson null class e−μL, where μ is the per site mutation rate and L the genome length. This scheme of replication corresponds to the linear stamping machine replication (SMR) model first proposed by Luria (25). However, if all genomic strand progeny can also immediately serve as templates for additional rounds of antigenomic strand synthesis, the replication model is effectively geometric (GR) and the distribution of mutant genomes per cell increases in variance because mutant progeny are themselves producing more mutant viruses. In this case, the distribution of mutant genomes conforms to the Luria-Delbrück distribution (9). The fraction of mutation-free genomes produced would depend on the number of replication rounds experienced, τ, according to e−τμL. Therefore, it is straightforward to see that GR will produce f more mutant genomes than SMR according to the equation f = (1 − e−τμL)/(1 − e−μL). If only a fraction of the genomic strand progeny replicates, then the replication model will be a mixture of GR and SMR that deviates from the Poisson expectation as much as the GR contribution. The effect of replication mode in virus mutational load can be better understood with the following example. The genomic mutation rate of tobacco mosaic virus was estimated to be in the range 0.043 ≤ μL ≤ 0.063 per replication round, and there were about 40 viral particles produced per infected tobacco cell, which is equivalent to 5.322 generations of GR (26). Therefore, the excess production of mutant genomes for GR over SMR will lie in the range of 16.38 ≤ f ≤ 23.76.
FIG. 1.

(a) Schematic representation of the virus infectious cycle. During infection, the viral particle enters into the host cell and, after uncoating, the genomic RNA (acting as mRNA) forms the translational complex, Tc, by binding with ribosomes. This directs the synthesis of the viral polyprotein, p, which is converted to both structural and nonstructural proteins at rates of 1 − β and β, respectively. The nonstructural proteins are involved in the synthesis of genomic and antigenomic RNAs. We distinguish between master and mutant genomic (+) and antigenomic (−) strands. We simulate SMR with Γ+ ≪ Γ−, where the initial genomic RNA directs the synthesis of one or very few negative copies, which are used as templates for the synthesis of new genomic strands. To model GR, we use Γ+ = Γ−, where all the synthesized strands replicate at the same rate (see Table 1 for a description of the variables and the parameters used in the model). ssRNA, single-stranded RNA; RdRp, RNA-dependent RNA polymerase. (b) Sequence spaces for a population of genomic and antigenomic binary strands of length (L = 3) bits. The consideration of genomic and antigenomic senses can be interpreted as two coupled hypercubes (ℋL±). Each node of the hypercube generates by replication of the complementary strand, SM± (blue line), or a mutant one, SM± (yellow line), in the coupled sequence space. (c) Diagram of the replication, mutation, and degradation rules implemented for the simulations. (d) Fitness landscapes analyzed in this study. The plot shows the effect of increasing numbers of mutations on the fitness of the genotype: the Swetina-Schuster (black), additive (blue), synergistic (green), and antagonistic (red) fitness landscapes. The parameters defining the landscapes shown in panel d are arbitrary.
Experimental data support different models of replication for different viruses. For example, bacteriophage T2 is thought to replicate mostly by GR because the number of mutants per infected cell fails to fit a Poisson distribution (25). However, phage φX174 data fit well the Poisson distribution, and hence the phage is thought to replicate according to an SMR model (7). Lying within these two extremes, phage φ6 slightly deviated from the Poisson expectation, an observation interpreted as a result of a mixed model in which some progeny were also able to replicate (4). Plant positive strand RNA viruses are also thought to reproduce mostly according to an SMR model (15, 18). Despite the apparent importance of the model of RNA virus replication for the accumulation of mutant genomes, most mathematical models proposed for the study of the dynamics of RNA virus populations rely on the assumption of GR. For example, the most commonly used theoretical paradigm for the study of virus evolution, Eigen's quasispecies picture (10, 13), assumes geometric growth and the Swetina-Schuster fitness landscape (41). This approach assumes that all mutations have the same deleterious fitness effect and that fitness does not depend on the number of mutations carried by a genome (i.e., the fitnesses of genomes having 1, 10, or 100 mutations would be identical) (Fig. 1d).
How to model viral replication is a timely research topic in virology. The growth of a virus in its host cell is a complex process. In seeking to understand this process and the effect of the interactions between the macromolecules involved in viral growth, the crossing of the disciplines of biochemistry, molecular biology, population genetics, and nonlinear dynamical systems might provide a powerful way to study the overall behavior of virus dynamics. In this sense, models play a crucial role in a qualitative and quantitative study of virus replication, as well as also being useful for predicting the system's time evolution and analyzing its sensitivity with respect to parameter changes. Furthermore, insights into interactions of viruses with host cells might help us to improve our understanding of virus-mediated diseases and to develop antiviral strategies (37). Several models of intracellular viral growth kinetics can be found in the literature, ranging from simple (unstructured) models capturing the basic replication processes (12, 19, 39, 46) to the so-called structured models that consider replication in different cellular compartments such as membranes, endosomes, cytoplasm, or the nucleus. Some examples of structured models have been developed for bacteriophage T7 (17, 45), human immunodeficiency virus type 1 (29), subgenomic hepatitis C virus (6), influenza A virus (37), and vesicular stomatitis virus (VSV) (24). However, to the best of our knowledge and despite its relevance, none has considered the effect that nongeometric modes of replication in combination with different fitness landscapes may have on the accumulation of mutant genomes.
In the first part of this work we analyze a mathematical model describing the single-cell reproductive cycle of positive-sense RNA viruses that make no subgenomic mRNAs and encode a single polyprotein that is self-processed into structural and nonstructural proteins, sensu picornaviruslike viruses (Fig. 1a). This model describes the main features of cytoplasmatic intracellular amplification of a viral genome (see reference 1 and references therein) without getting deep into the peculiarities of any particular system. By doing so, the model remains as general as possible and would be applicable to a wide variety of RNA viruses. By focusing on intracellular replicative dynamics, we are not explicitly considering cell lysis and transmission among cells. The main goal of this first part is to analyze the effect of SMR and GR under the simplistic but mathematically convenient Swetina-Schuster fitness landscape (41). For each replication scheme, we compare the dynamics of nonmutated and mutated strains of both polarities, the value of the critical mutation rate for which the nonmutated strain disappears from the population, and the sensitivity to mutations. The model considers explicitly both genomic and antigenomic master and mutant strands, the viral polyprotein, the translational complexes, and the mature virions. Whenever possible, model parameter values were chosen to be in the same ranges as experimental determinations taken from the literature (Table 1). For those parameters for which no experimental values were found, we ran a sensitivity analysis with over a million parameter combinations and selected values providing the most consistent outcome. In the second part of our study, we confirm the validity of the results by using a stochastic model involving digital genomes and incorporating more-complex and more-realistic fitness landscapes to be representative of RNA viruses.
TABLE 1.
Notations used in the differential equations model and the values assigned to each parametera
| Notation | Description | Value |
|---|---|---|
| β | Fraction of the viral polyprotein used as replicase (1 − β is the fraction used as structural proteins) | 0.9b |
| ɛ | Strand degradation rate | 0.005c |
| ɛΤ | Degradation rate of the translational complex | 10−5d |
| ɛp | Degradation rate of the viral polyprotein | 1.5 × 10−3e |
| Γ± | Probabilities of replication for the genomic (+) and antigenomic (−) master strands | 0-1 |
| Λ± | Probabilities of replication for the genomic and antigenomic mutant strands | 0-0.1 |
| kR | Effective rate of interaction between the master genomic strands and the available ribosomes | 0.04d |
| k1 | Rate of dissociation of the master genomic strand from the translational complex | 0.02d |
| k2 | Encapsidation probability of genomic strands | 0.75 |
| m | No. of monomers of structural protein necessary for building up a virion | 200f |
| μ | Avg mutation rate | 0-1 |
| Rtot | Constant concentration of ribosomes inside the cell | 1 |
| σ | Rate of elimination of mature virions (either by degradation or by leaking out of the cell) | 3.5 × 10−3g |
Initial conditions: x0+(0) = 0.01 and x0−(0) = x1+(0) = x1−(0) = Tc(0) = p(0) = V(0) = 0 if not otherwise specified.
The nonstructural genomic components represent ∼90% of the genome in picornaviruslike viruses (1).
RNA stability depends on intrinsic properties of the molecule, such as sequence structure and the presence of CAP, VPg, or a poly(A) tail, as much as the action of cellular RNases, and thus it must be independent of the replication model. We assume that mutations have negligible effects on RNA stability. Since we found no useful values in the literature, we performed a sensitivity analysis for ɛ in the range 0.002 to 0.01 by exploring more than a million parameter combinations. We obtained a good qualitative convergence of equilibrium concentrations inside this range.
These values were chosen in such a way that the ratio of formation rate to degradation rate plus dissociation rate lies in the same range as in reference 6.
Once the polyprotein has been produced, we assume that either it can be degraded at rate given by ɛp or is cleaved with absolute efficiency into the mature peptides, which is equivalent to a cleavage rate of 1, as in the range reported in reference 6. The exact value employed in our simulations was obtained from the same sensitivity analyses described for ɛ.
For picornaviruslike viruses, the number of monomers of structural proteins required for encapsidation is in the range of hundreds to thousands. The value has been fixed to make it similar to that for the potyviruses (36).
The virion degradation rate has been fixed at 2 orders of magnitude smaller than the formation rate (k2) as reported for VSV (14).
MATERIALS AND METHODS
Mathematical model.
Our quasispecies mathematical model of intracellular viral replication is based on the scheme shown in Fig. 1a (see Table 1 for details on the notation used hereafter). The model is used to analyze the dynamics of replication of positive-sense RNA viruses that make no subgenomic mRNAs. We explicitly define the polarity of the strands constituting the quasispecies, studying its dynamics using the Swetina-Schuster fitness landscape (41) (Fig. 1d). In this landscape, the wild-type sequence has fitness 1, while all mutant genotypes (regardless the number and nature of mutations) have equal fitnesses that are <1 (41) and, thus, represent a single sharp peak emerging from a flat surface. In such a scenario we may divide the population in either master or mutant genomic and antigenomic viral strands. The pool of mutant strands of each polarity is thus grouped in an average sequence different from the master one. Therefore, the variables of this dynamical system include the genomic (superscript +) and antigenomic (superscript −) viral strands, designated x, with the master ones indicated by subscript 0 and the mutants by subscript 1. Moreover we also consider as variables the viral polyprotein, p, the translation complex formed by genomic RNAs and ribosomes, Tc, and the mature viral particles, V.
Our model assumes that all the interacting macromolecules are homogeneously mixed and that mutant genomic RNAs (x1+) fail to be translated into the polyprotein and thus they do not compete for the available ribosomes. This assumption is reasonable because, as confirmed below, for moderately large mutational effects (Λ+ ≪ Γ+, where Λ and Γ are as defined in Table 1), the concentration of mutant genomic RNAs should be low compared to that of the wild-type RNAs, thus making the amount of ribosome-RNA complexes for mutant strains quickly drop to zero. For instance, one can envision these mutations as introducing stop codons or inducing conformational changes in the ribosomal entry sites, hindering the interaction of the RNA with the ribosomes. More importantly, with this assumption, we eliminate the mathematical complications of complementation, keeping the model more simple and focused. Next we proceed to give a detailed explanation of the processes described by the model. Four steps, which correspond to the main phases of viral replication inside the host cell, are considered in the following subsections (Fig. 1a).
(i) Translation complex kinetics.
Upon entry and uncoating, the genomic strand binds with the cellular ribosomes, forming the translational complexes. Following Dahari et al. (6), the amount of free available ribosomes, Rav, is used as an upper bound to the formation of the translation complexes, Tc, and is given by Rav = Rtot − Tc. Note that here we assume that the total number of ribosomes, Rtot, is constant and the number of available ribosomes decreases due to the formation of the translational complexes. The dynamics of the translational complex is then defined by dTc/dt = kRx0+Rav − k1Tc − ɛTTc, where t is time and the parameter kR is the effective rate of interaction between the genomic RNA and the available ribosomes. The second term denotes the dissociation of the translation complex, after which the ribosomes and the genomic RNA again become available. We consider that the translation complex is degraded at a rate (ɛT) that is ≪k1.
(ii) Viral polyprotein dynamics.
The dynamics of the viral polyprotein, p, depends on the presence of the translation complexes, the formation of mature virions, its intrinsic degradation, and the replication of viral strands. An appropriate description is given by the equation dp/dt = k1Tc − 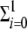 ϕ(xi+) − ɛpp − ℛ(X). Here ℛ(X), given by βp[ψ(X−)Γ+ℛ0+ + Γ−ℛ0− + ψ(X−)Λ+x1+ + Λ−x1−], determines the amount of nonstructural protein involved in replication [see below for the definition of the terms involved in function ℛ(X)]. The function ϕ(xi+), which corresponds to the formation of virions by encapsidation of the genomic strands, is defined as k2[(1 − β)p]m. Here k2 is the encapsidation rate and m is the number of monomers of structural protein required for building up a mature virion. This formula assumes that virion assembly follows an m-order mass action kinetics. The polyprotein is proteolytically self-processed, resulting in the formation of both structural (capsid) and nonstructural (replicase) proteins, which represent, respectively, 1 − β and β of the entire polyprotein (19). The polyprotein degrades at rate ɛp.
ϕ(xi+) − ɛpp − ℛ(X). Here ℛ(X), given by βp[ψ(X−)Γ+ℛ0+ + Γ−ℛ0− + ψ(X−)Λ+x1+ + Λ−x1−], determines the amount of nonstructural protein involved in replication [see below for the definition of the terms involved in function ℛ(X)]. The function ϕ(xi+), which corresponds to the formation of virions by encapsidation of the genomic strands, is defined as k2[(1 − β)p]m. Here k2 is the encapsidation rate and m is the number of monomers of structural protein required for building up a mature virion. This formula assumes that virion assembly follows an m-order mass action kinetics. The polyprotein is proteolytically self-processed, resulting in the formation of both structural (capsid) and nonstructural (replicase) proteins, which represent, respectively, 1 − β and β of the entire polyprotein (19). The polyprotein degrades at rate ɛp.
(iii) RNA synthesis and degradation.
Four different classes of RNA sequences (xj ∈ {x0+, x0−, x1+, x1−} = X) are considered, and their growth is limited by a logisticlike function, ℒ, that imposes finite cellular resources (i.e., carrying capacity [𝒦 = 1]) according to the equation ℒ(X) = 1 − (1/𝒦)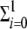 (xi+ + xi−). The replicase uses a given strand as a template to synthesize its perfectly complementary sequence at rate Γ±(1 − μ), where μ the average mutation rate and Γ± is the replication rate of the master strands. Hence, master sequences will generate mutant sequences at rate Γ±μ. Mutant strands replicate at rates (Λ±) that are ≪Γ± because we have assumed that mutations are deleterious. Note that backward mutations are assumed to be so infrequent that their effect may be neglected. The concentration of free positive-sense strands will grow according to the equation dx0+/dt = r+ℒ(X) − ɛx0+ + k1Tc − ϕ(x0+) − kRRavx0+, where ɛx0+ represents the decay of RNA molecules as a consequence of inherent degradation or the action of cellular RNases, which is assumed to be the same for all strands. The three last terms on the right side correspond to the dissociation from the translation machinery, the sequestration rate due to the formation of new viral particles, and the capture by free ribosomes to produce new translational complexes, respectively. The growth rate, r+, incorporates the presence of x0− templates, the fraction of the polyprotein used as replicase (βp), and the fidelity of replication, 1 − μ, according to the equation r+ = Γ−(1 − μ)x0−βp. For the x0− strands, the dynamics is now given by dx0−/dt = r−ℒ(X) − ɛx0−, where now r− = ψ(X−)Γ+(1 − μ)x0+βp [see below for the definition of the function ψ(X−)]. Similarly, we can build the equations for the mutant populations as dx1+/dt = r+′ℒ(X) − ϕ(x1+) − ɛx1+ and dx1−/dt = r−′ℒ(X) − ɛx1−, with their replication rates now given by r+′ = βp(Γ−μx0− + Λ−x1−) and r−′ = ψ(X−)βp(Γ+μx0+ + Λ+x1+), consistent with the reactions outlined in Fig. 1.
(xi+ + xi−). The replicase uses a given strand as a template to synthesize its perfectly complementary sequence at rate Γ±(1 − μ), where μ the average mutation rate and Γ± is the replication rate of the master strands. Hence, master sequences will generate mutant sequences at rate Γ±μ. Mutant strands replicate at rates (Λ±) that are ≪Γ± because we have assumed that mutations are deleterious. Note that backward mutations are assumed to be so infrequent that their effect may be neglected. The concentration of free positive-sense strands will grow according to the equation dx0+/dt = r+ℒ(X) − ɛx0+ + k1Tc − ϕ(x0+) − kRRavx0+, where ɛx0+ represents the decay of RNA molecules as a consequence of inherent degradation or the action of cellular RNases, which is assumed to be the same for all strands. The three last terms on the right side correspond to the dissociation from the translation machinery, the sequestration rate due to the formation of new viral particles, and the capture by free ribosomes to produce new translational complexes, respectively. The growth rate, r+, incorporates the presence of x0− templates, the fraction of the polyprotein used as replicase (βp), and the fidelity of replication, 1 − μ, according to the equation r+ = Γ−(1 − μ)x0−βp. For the x0− strands, the dynamics is now given by dx0−/dt = r−ℒ(X) − ɛx0−, where now r− = ψ(X−)Γ+(1 − μ)x0+βp [see below for the definition of the function ψ(X−)]. Similarly, we can build the equations for the mutant populations as dx1+/dt = r+′ℒ(X) − ϕ(x1+) − ɛx1+ and dx1−/dt = r−′ℒ(X) − ɛx1−, with their replication rates now given by r+′ = βp(Γ−μx0− + Λ−x1−) and r−′ = ψ(X−)βp(Γ+μx0+ + Λ+x1+), consistent with the reactions outlined in Fig. 1.
Note that the differences in the replication rates of both genomic and antigenomic strands allow us to analyze both SMR and GR kinetics. To model GR, we set Γ+ = Γ−, i.e., all the synthesized strands are allowed to replicate. For SMR, however, we use Γ+ ≪ Γ−, that is, the infectious genomic RNA entering into the host cell synthesizes one or very few negative copies, which are then used as the only templates for the synthesis of new genomic strands in a Luria's stamping machine strategy. Indeed, to further stress the assumption that at the beginning of the infectious cycle only the antigenomic strands need to be produced but that, as infectious progresses, this production has to be shut off to favor production of genomic strands, we assume a negative feedback of antigenomic strand concentration on its own rate of production. In mathematical terms, this constraint can be incorporated by setting ψ(X−) equal to 1/(1 + 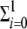 xi−) in the production of antigenomic strands from genomic ones. For GR, ψ(X−) is 1.
xi−) in the production of antigenomic strands from genomic ones. For GR, ψ(X−) is 1.
(iv) Formation of viral particles.
The new virions are produced both from master and mutant genomic strands combined with the structural proteins. From the previous steps, we can see that the formation of mature virions, V, will follow the equation dV/dt = 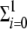 ϕ(xi+) − σV, where ϕ(xi+) represents the encapsidation of positive strands and σV is introduced to control the amount of virions (e.g., degradation of viral particles and elimination of mature particles that may leak out the cell).
ϕ(xi+) − σV, where ϕ(xi+) represents the encapsidation of positive strands and σV is introduced to control the amount of virions (e.g., degradation of viral particles and elimination of mature particles that may leak out the cell).
For the sake of simplicity we hereafter use (by default) Γ− = 1 and Λ± = Γ±/10 (i.e., assuming that mutations are largely deleterious and that the replication rate of mutants is reduced by a factor of 10). The 10% average fitness effect was chosen to be of the same order of magnitude as that for experimental data on the effect of single point mutations gathered for VSV (33) and tobacco etch virus (3). The other parameters will be either explored in this work or remain fixed at the biologically meaningful values provided in Table 1.
Stochastic dynamics of digital genomes.
During the first stages of the infection and due to the stochastic nature of transmission events, cells are usually invaded by one or a few viral particles. Therefore, a stochastic description of the replication process would better capture the fluctuations due to small population sizes (see reference 39 and references therein). We use an unstructured discrete model of in silico genome evolution considering a bit string description of the population structure (35, 38), which allows us to explicitly simulate the complex and heterogeneous structure of populations of replicators. Although real RNA is composed of a four-letter alphabet, we use Leuthäusser's approach by considering that each bit would represent either purines or pyrimidines (22, 23). Digital strands will thus be represented as chains of bits. Each chain will have 32 bits, and a maximum population size (N) of 1,000 chains will be allowed.
We define a population of digital genomes. We indicate as Si+ and Si− positive and negative strands, respectively. A given string will be defined as Si± = (Si1± Si2±… SiL±), with Sik± ∈ {0, 1}. The genomic and antigenomic master sequences in our model (labeled Sm) are defined as Sm+ = (11… 1) and Sm− = (00… 0), respectively. We initially “inoculate” our system with N(0) replicating strings. These strings can now replicate (generating complementary strands) and mutate. For instance, each bit in Si+ can mutate according to the equation Sik+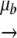 1 − Sik+ = Sik− with a given mutation probability, μb, per bit and replication cycle. They also degrade with probability ɛ (Fig. 1c). We first investigate the Swetina-Schuster fitness landscape (41) used in the previous mathematical model (Fig. 1d). In this case, the master sequences have the highest fitness, i.e., their replication probabilities are set to 1, whereas all other strings replicate with an arbitrary probability of 0.1. Then, we move to more complex and realistic fitness landscapes that are described by an equation of the general form Γ± = 1 − dimξ/L, where dim is the Hamming distance between string Si± and the master sequence Sm± and ξ measures the sign and strength of epistasis. If ξ is 1, then we have an additive fitness landscape in which all mutations have the same effect and thus fitness declines linearly with the number of mutations (Fig. 1d). Values of ξ that are >1 or between 0 and 1 correspond to synergistic and antagonistic epistasis, respectively (Fig. 1d). The latter case is in good agreement with the results of recent experiments showing that, on average, antagonistic epistasis predominates in RNA viruses (2, 30, 34, 42).
1 − Sik+ = Sik− with a given mutation probability, μb, per bit and replication cycle. They also degrade with probability ɛ (Fig. 1c). We first investigate the Swetina-Schuster fitness landscape (41) used in the previous mathematical model (Fig. 1d). In this case, the master sequences have the highest fitness, i.e., their replication probabilities are set to 1, whereas all other strings replicate with an arbitrary probability of 0.1. Then, we move to more complex and realistic fitness landscapes that are described by an equation of the general form Γ± = 1 − dimξ/L, where dim is the Hamming distance between string Si± and the master sequence Sm± and ξ measures the sign and strength of epistasis. If ξ is 1, then we have an additive fitness landscape in which all mutations have the same effect and thus fitness declines linearly with the number of mutations (Fig. 1d). Values of ξ that are >1 or between 0 and 1 correspond to synergistic and antagonistic epistasis, respectively (Fig. 1d). The latter case is in good agreement with the results of recent experiments showing that, on average, antagonistic epistasis predominates in RNA viruses (2, 30, 34, 42).
The simulation algorithm repeats, at every generation τ, the replication and degradation rules 1,000 times. This is a standard Metropolis importance sampling Monte Carlo updating scheme, which ensures that, on average, the rules are applied to all the population of strings per generation and defines the time scale (21). To differentiate between both types of replication, we implemented the following strategy. When a genomic strand replicates, producing an antigenomic one, the latter will always keep replicating (unless degraded). On the other hand, when an antigenomic strand replicates, the synthesized genomic strand will become a replicator with probability ρ. Note that, whenever ρ is 1, all the progeny strands copied from the negative templates will replicate in the following generations and replication will be purely geometric. However, if ρ is ≪1, the negative strands will be mainly used as templates while the positive ones will not replicate. With this second strategy the kinetics will be closer to the SMR. Indeed, to potentiate the effect of SMR, the nonreplicating genomic strands are not degraded and the degradation probability for the replicating sequences is kept very low. We also consider differential replication rates for each strategy of replication by using δΓ+ and δΛ+, where δ is the constant of asymmetry in replication used to differentiate between both replication modes. For SMR we set δ at 0.1 and the genomic strands will synthesize few antigenomic ones. For GR we use a δ of 1 and all the synthesized strands will always replicate proportionally to their fitness.
All numerical analyses of the deterministic mathematical model were done using a C program implemented to solve the differential equations by the standard fourth-order Runge-Kutta method (40) using a constant time step size (Δt) of 0.1. The stochastic bit string model was also implemented in a C program (available upon request).
RESULTS
Mathematical model. (i) Quantitative differences in the accumulation of master and mutant genomes of both polarities.
The first important analysis for any replication model involving antigenomic intermediates of replication should be to explore the kinetics of accumulation of both genomic and antigenomic strands and to understand how mutations accumulate in both strands. The effects of mutation rate in each replicating strategy are illustrated in Fig. 2. We show the temporal dynamics of all the viral strands for each replication strategy using three different mutation rates. The genomic strands achieve higher equilibrium concentrations than the antigenomic ones for the SMR. For GR, however, the genomic and antigenomic strands always asymptotically achieve identical equilibria. With a μ of 0.1 (Fig. 2a) the master strands achieve population equilibria higher than the mutant strands regardless of the replication kinetics. If the mutation rate is increased (μ = 0.3) (Fig. 2b), then the concentration of the mutant strands grows while the master strand concentration decreases, although for SMR the concentration of mutant genomic strands achieves a higher value than that of antigenomic master strands. For either replication mechanism, both master strands achieve higher population densities than the mutant strands if mutation rates are low. However, if the mutation rate is increased (μ = 0.65), mutant strands dominate the population (Fig. 2c). However, two important differences between replication modes exist. While SMR still produces nonmutated genomes of both polarities and the population reaches a nontrivial equilibrium, GR fails to sustain replication and the population becomes extinct in the long term.
FIG. 2.

Solutions of the mathematical model. Shown are time series for master (black) and mutant (red) strands at the following mutation rates (μ): (a) 0.1, (b) 0.3, and (c) 0.65. Genomic and antigenomic strands are indicated with solid and dashed lines, respectively. In all the plots we show the time evolution for SMR (top; with Γ+ = 0.1) and GR (bottom; with Γ+ = 1). (d) Initial amplification phase for the genomic strands (in linear-log scale) for GR (thick line) and SMR (thin line) using a μ of 0.3. For the initial condition x0+(0) was 0.1 and all other parameters are as in Table 1.
Figure 2d illustrates the initial growth kinetics for each replication mode for the genomic strands (in linear-log scale). For GR the initial growth phase is exponential. For the SMR strategy we obtain subexponential growth kinetics. The model also shows a difference in the temporal dynamics of the ratio of positive to negative strands between both replication modes. For SMR, with the parameter values used in this study, such a ratio reaches an equilibrium at which the concentration of positive strands is approximately 3.5 times larger than that of negative strands (data not shown). However, as one may expect from the GR, this ratio flattens off at a ratio of 1, indicating equal production of both strands (data not shown).
In conclusion, SMR is the more efficient replication mechanism: for low mutation rates, it produces more genomic strands than GR without the necessity of generating equivalent amounts of antigenomic strands, while at high mutation rates, GR drives the population to extinction while SMR still generates small amounts of mutation-free genomes. In the next section, we explore under which mutation rates the master nonmutated strand disappears from the population.
(ii) SMR is compatible with a higher critical mutation rate.
One of the more important predictions of the quasispecies theory is the existence of a critical mutation rate, μc, beyond which the wild-type sequences does not exist anymore in the population. As already mentioned in the introduction, Eigen's model implicitly assumes GR. The question we address in this section is how the mode of replication may impact μc. As expected, for SMR the equilibrium concentrations for both master genomic (x0+) and antigenomic (x0−) strands as well as for both mutant strands (x1+ and x1−) are asymmetric (Fig. 3a). This actually means that we have a higher production of positive strands from the negative ones. On the other hand, for GR the equilibrium concentrations for the master and the mutant strands are the same for both genomic and antigenomic strands. The critical mutation rate, involving the extinction of the master sequences, is shown to be 10.07% larger for SMR (μc = 0.765) (Fig. 3a) than for GR (μc = 0.695) (Fig. 3b), suggesting that the extinction of master genomes under SMR takes place at mutation rates larger than those if genomes replicate according to GR.
FIG. 3.

Equilibrium concentrations for master (x0±) and mutant (x1±) strands versus mutation rate for SMR (a) and GR (b) modes. All other parameters are as in Table 1. To facilitate the comparison, the arrow indicates the critical mutation rate obtained for the SMR model.
From these results we can conclude that a virus replicating according to SMR would benefit from having a higher μc. In other words, it will be able to maintain a well-defined population structure at mutation rates for which a virus replicating according to GR would have undergone a collapse of the nonmutated strands.
(iii) SMR is more robust to the accumulation of slightly deleterious mutations.
So far, we have shown that a virus replicating according to SMR produces less mutant genomes and can sustain the population at mutation rates which are prohibitive for a virus replicating according to GR. Next, we sought to explore the effect of the severity of mutational effects on the accumulation of master and mutant strands of both polarities. The severity of mutations was computed as the ratio between the average replication rates of mutant (Λ+) and master (Γ+) genomic strands. This ratio will be 1 for neutral mutations (Λ+ = Γ+) and 0 for lethal ones. Γ+ was fixed at 0.1 for SMR and at 1 for GR, while Λ+ ∈ [0, Γ+]. Figure 4 shows the equilibrium population densities for each genomic class and their dependence on mutational severity. At relatively low mutation rates (μ = 0.15) the positive master sequence remains dominant regardless the replication mode. As expected, strong-effect mutations accumulate less than those causing mild effects, irrespective of the mode of replication. However, GR is more sensitive to the accumulation of mild mutations than SMR (Fig. 4a), as indicated by the steeper slope for the positive master strands. A similar situation occurs at intermediate mutation rates (μ = 0.25) (Fig. 4b): both replication modes accumulate more mild- than strong-effect mutations but the GR accumulates proportionally more mild mutations. At higher mutation rates (μ = 0.6) (Fig. 4c) results change for SMR in an important way, that is, positive master genomes are not dominant anymore for the entire range of mutation severities and, instead, the mutant ones become the most abundant class, although it is still possible to recover the master genome along all the range of mutational severities at ∼10% population frequency. Mild mutations are still the most commonly fixed ones. However, GR collapses at intermediate mutational severities (Λ+ ≈ 0.45), and all genotypes get extinguished due to the accumulation of small-effect mutations.
FIG. 4.

Severity of mutations and accumulation of the different strands. Shown is the relative effect of mutations on the equilibrium concentrations of the strands measured as the ratio Λ+/Γ+ for different values of the mutation rate (μ): (a) 0.15, (b) 0.25, and (c) 0.6. In all the plots, for SMR Γ+ is 0.1 and for GR Γ+ is 1. For the initial condition x0+(0) is 0.1 and all other parameters are as in Table 1.
Another difference between SMR and GR schemes is that at low mutation rates the second-most-abundant genotype for SMR is the antigenomic master strand, irrespective of the severity of mutations, whereas antigenomic mutants are the second-most-abundant class for GR. At intermediate mutation rates, the genomic mutants become the second-most-abundant class when replication occurs via SMR, and their frequency rises as mutation rate increases. These results are in agreement with those presented in the previous section and support the notion that the SMR model of virus replication is not only compatible with higher mutation rates but also more robust to the severity of mutations.
In general, strong-effect mutations will have very low impact on the fitness of populations, with the extreme case being lethal mutations, which do not contribute to the next generation. By contrast, mutations of mild effect will accumulate in the populations since selection is poorly efficient at removing them. We have shown here that viral populations replicating according to SMR accumulate less mild-effect mutations than those whose replication was according to GR and that, therefore, selection will be more efficient in keeping the population free of deleterious alleles (19).
Digital genomes.
To incorporate the stochastic characteristics inherent to viral infection and replication as well as to be able to analyze different fitness landscapes, in the following three sections we move from deterministic models based on differential equations to stochastic models of simulation with digital genomes.
(i) The equilibrium genotypic distributions differ between SMR and GR and show different dynamics for the loss of the master sequence.
We begin our study of the stochastic model by analyzing the effect of mutations across four different fitness landscapes for both replication modes. The variable we are measuring in this section is the equilibrium concentration for the master genomic sequences and its mutant spectrum using the per-bit mutation probability, μb, and an initial population size [N(0)] of 50 genomes. For the epistatic landscapes (Fig. 1d), the epistasis coefficients (ξ) were set to 1.4 and 0.6 for the synergistic and antagonistic cases, respectively. The critical mutation probability per bit, μbc, is defined as the lowest mutation value for which the master genomic strand (Si+) concentration is <10−4 (the assumed concentration at which extinction takes place). The results of these simulations are shown in Fig. 5. In the case of the Swetina-Schuster single-sharp-peak landscape (Fig. 1d), the extinction of the master sequence occurs when μbc is ≈0.156 for the SMR model and ≈0.070 for the GR model. Moreover, the composition of the mutant spectrum is shown to differ according to the replication strategy. It is well known that for the combination of a Swetina-Schuster landscape and GR the mutant spectrum suffers a sharp phase transition at μbc and each mutant genome reaches a steady-state concentration that depends only on its mutational coupling (13). However, we show that this is not the case for SMR, since such a sharp phase transition is never observed and different genomes rise and decrease in frequency depending on mutation rate (Fig. 5).
FIG. 5.

Average equilibrium concentration of genomic strands in the bit string model for the four fitness landscapes studied as a function of the per-bit mutation rate. ξ values for antagonistic, additive, and synergistic epistasis were 0.6, 1, and 1.4, respectively. For the Swetina-Schuster landscape, ɛ was 0.01, and ɛ was 0.001 for all other landscapes. In all the diagrams we show the normalized population numbers (averaging over 200 independent replicates when τ = 20,000) for genomic master strands (thick line) and their mutant spectrum (thin lines). N(0) was 50 in all simulations.
For more-realistic landscapes, μbc is also larger for SMR than for GR. The difference between both replication strategies is maximized for the antagonistic landscape (7.43-fold difference) and minimized for the synergistic landscape (4.72-fold difference), while the additive landscape produces a 6.41-fold difference in μbc. No sharp phase transition is observed for any of these three landscapes.
Therefore, a very important conclusion can be drawn from this section: the sharp phase transition usually known as the error threshold is characteristic of populations of GR replicators and its occurrence depends on whether the fitness landscapes conform to the Swetina-Schuster model. Failure to meet either of these requirements results in a lack of a sharp phase transition at the critical mutation rate.
(ii) Statistical analysis of the distribution of the number of mutations per genome.
Next we sought to explore the effect of the two extreme models of RNA virus replication and of landscape topology on the properties of the cloud of mutants generated around the master sequence. Figure 6a shows the population frequency of master genomes for both replication modes and the four different fitness landscapes. A clear effect of the replication mode is observed. While a master genome replicating according to SMR will remain in the population at frequencies over the extinction threshold (10−4) for a μb of ≤0.148, irrespective of the landscape employed, a master genome using a GR strategy always crosses the extinction threshold at much lower mutation rates (e.g., μb ≈ 0.031 for the synergistic landscape) (Fig. 6a). Indeed, irrespective of the fitness landscape and the mutation rate employed in the simulations, the distributions of the number of mutations per genome were significantly different both in location (Fig. 6b) (Mann-Whitney test; P < 0.001 in all comparisons) and shape (Fig. 6c) (Kolmogorov-Smirnov test; P < 0.001 in all comparisons), with GR always generating a much larger average mutational load and a larger variance in the number of mutations per genome. Figure 6b reflects once again the sharp phase transition characteristic of the GR in a Swetina-Schuster landscape, with genomes increasing their average number of mutations up to 16 (Fig. 6b). For the other landscapes, the average number of mutations per genome increases smoothly. For SMR, the landscape topology does not affect to a large extent the average number of mutations per genome. However, for GR, the antagonistic landscape generates the largest mutational load for all mutation rates tested. By contrast, the synergistic landscape generates the lowest mutational loads. Therefore, the magnitude of the benefit of SMR over GR (i.e., the difference between the surfaces under both curves) varies across landscapes: it is largest for the Swetina-Schuster landscape, followed by, in order of size, the antagonistic landscape, the additive landscape, and finally the synergistic landscape.
FIG. 6.

Interplay between mutation rate, mode of replication, and the topology of the fitness landscape. (a) Frequency of the mutation-free genomic strands. The dotted horizontal line represents the extinction threshold. (b) Average number of mutations per genome as a function of mutation rate. (c) Standard deviation of the number of mutations per genome.
Similarly, the standard deviation of the number of mutations per genome is also affected by the interplay between the mode of replication and the landscape topology (Fig. 6c). Again, the sharp phase transition characteristic of the combination of Swetina-Schuster landscape and GR is reflected in the observed sudden increase in variability in the number of mutations per genome as the mutation rate approaches the critical value. However, this fast increase was transient only until the average mutational load reached its maximum value. Afterwards, the variance in the number of mutations sharply decreased and asymptotically approached the standard deviation value observed for the SMR (Fig. 6c). For the other fitness landscapes, the standard deviation of the number of mutations per genome does not sharply change, because its increase is more monotonous. At low mutation rates GR produces larger variances than SMR regardless of the landscape topology, with the following rank order: antagonistic, additive, and synergistic. As mutation rate increases, however, the differences between GR and SMR are reduced and the curves intersect at some value of μb (Fig. 6c) beyond which GR produces less variable populations than SMR. The precise value at which SMR loses the advantage over GR depends on the landscape topology: synergistic first, followed by antagonistic and finally additive.
As described in the introduction, under a purely SMR mechanism, the number of mutations per genome should conform to a Poisson distribution, whereas for a GR mechanism the distribution departs from the Poisson model and fits the more complex Luria-Delbrück distribution. To confirm this expectation, we ran Kolmogorov-Smirnov tests for deviations of the Poisson null hypothesis. As expected, under the SMR, the numbers of mutations per genome were Poisson distributed for all combinations of μb and landscapes (P ≥ 0.050 in all tests). Furthermore, the distributions of mutations per genome generated under the GR scheme departed from the null Poisson model for values of μb that were ≥0.035 for the Swetina-Schuster, additive, and antagonistic landscapes (P ≤ 0.019 in all tests) and 0.015 for the synergistic one (P = 0.011).
The take home message from this section is that, if one looks for differences between SMR and GR in terms of where in sequence space the cloud of mutants is placed and how big it is, the results strongly depend on the fitness landscape. Whereas SMR is quite insensitive to the topology, GR is greatly affected.
(iii) Interplay between epistasis and critical mutation rate.
In the previous section, we observed that for a given replication mode the most divergent results were always observed for the two extremes of the epistatic landscape. To further characterize the effect of the epistasis on the replication dynamics, next we sought to explore the effect of the fitness landscape curvature (ξ) on μbc. To do so, we ran simulation experiments similar to those described in the above sections but varying ξ in 0.1 intervals (Fig. 7). Irrespective of the mode of replication, the shapes of the relationship were similar: antagonistic epistasis (ξ < 1) barely affects μbc, whereas increasing the strength of synergistic epistasis (ξ > 1) allows for almost linear or exponential increases in μbc of up to 14% for an ξ of 2. In good agreement with the results shown in previous sections, what most strongly determines the magnitude of μbc is the mechanism of replication, with μbc being around 1 order of magnitude larger for SMR than for GR across the entire range of ξ.
FIG. 7.

Mean critical mutation rate, μ̄bc, as a function of the sign and strength of epistasis, ξ. Each data point is the result of averaging 20 independent runs at equilibrium. Error bars represent standard deviations. The population equilibrium values were obtained by averaging the entire population at generation 20000 over 200 independent runs. The vertical dotted line at a ξ of 1 corresponds to the additive fitness landscape. (a) SMR; (b) GR.
From these results we conclude that the critical mutation rate is insensitive to antagonistic epistasis but that it dramatically increases as epistasis becomes more and more synergistic. The biological meaning of this difference is clear. Antagonistic epistasis means that genomes with multiple mutations are fitter than expected under a multiplicative model and, therefore, may persist in the population for long periods of time before being removed by selection. Therefore, the extinction of the master genotype is only weakly affected by changes in μbc. By contrast, increasing synergistic epistasis means that the pernicious effect of multiple mutations goes beyond the multiplicative expectation and thus that mutant genomes will be removed from the population in a very efficient way by natural selection; henceforth, only the master sequence survives and produces progeny. This “population robustness” effect allows μbc to increase up to the point at which it is not possible anymore to produce progeny of master sequences from a master template.
DISCUSSION
It is known that the mode of virus replication can change the rates at which deleterious mutations occur. Depending on whether genomes replicate according to a stamping machine model or geometrically one may observe different fractions of mutation-free genomes in the offspring. Luria's SMR (25) implies that the copies produced during the first round of replication will be the only templates for the generation of the entire viral population. Experimental data suggest that φX174 replicates in such a way (7). However, in GR, as for example in the case of phage T2 (25), all the synthesized copies during infection serve as templates in the following generations. Intermediate situations have also been described for other viruses like the phage φ6 (4). As far as we know, previous attempts to model virus replication have not integrated the many features that we have explored here: mode of replication involving genomic and antigenomic strands, topology of fitness landscape, and mutation rates.
In this work we analyze a model describing the within-cell replication of positive-sense RNA viruses that make no subgenomic mRNAs and encode a single polyprotein that is self-processed into structural and nonstructural components. We develop a mathematical model using as key variables the genomic and antigenomic RNA strands (considering mutation-free and mutant genomes), the viral polyprotein, the translational complexes formed by viral RNA and cellular ribosomes, and the mature virions. However, the model is defined in quite general terms, so it can be easily modified to account for any other genomic organization. Genetic heterogeneity is introduced with a simplified quasispecies structure of the strands considering master genomes (which have the higher fitness) and the pool of deleterious mutants for each strand's polarity, which are grouped into a single variable denoting an average mutant sequence different from the master one. We analyze a Swetina-Schuster single-peak-fitness landscape (41), which assumes that mutations have a large deleterious effect. We explore the critical mutation rate at which master sequences disappear and the sensitivity to mutations of viral populations replicating according to SMR or GR. We have shown that the extinction of the master genome takes place at a higher mutation rate for SMR, indicating that this strategy is less sensitive to the effect of deleterious mutations. On the other hand, GR displays a lower critical mutation rate, being more sensitive to mutation. Consistently, we have also shown that mild mutational effects tend to accumulate more in GR and that under such a growth kinetics the population collapses at intermediate mutation rates. However, the SMR strategy is less sensitive to the accumulation of mild mutations, and viral strands continue to exist at mutation rates that produce a collapse of viral populations replicating geometrically.
A critical assumption of our model is that mutations have two immediate effects. First, they preclude the right interaction of mutant RNAs with ribosomes, thus fully preventing their translation, and, second, they reduce their efficiency of interaction with the wild-type replicase, thus negatively affecting their transcription. Certainly, with the exception of mutations generating stop codons, severely modifying ribosomal entry sites, or altering signals for replicase binding, it is assumed that mutations will not necessarily have such a severe effect. By removing these assumptions and moving to the proteins the effect of mutations, we clearly increase the realism of our models. However, this increase in realism will come with a cost in terms of model complexity due to the necessity of incorporating interactions between mutant proteins and the four RNA species, each interaction governed by a different set of parameters. On the one hand, complementation of defective genomes by wild-type proteins will increase the chances of mutant genomes persisting in the viral population, effectively reducing the strength of purifying selection (44). However, this possibility does not represent a problem in our formulation because we assume that mutant genotypes are replicated and encapsidated only by wild-type proteins. On the other hand, the presence of proteins with impaired functions will reduce the replication/encapsidation efficiency of both wild-type and mutant genomic RNAs. This second option has not been taken into consideration in our current formulation and is the subject of ongoing research. Finally, readers should keep in mind that our model is an extension of the basic quasispecies model (12, 13) that has impacted the virology community so much. As in Eigen's original formulation, the fitness of mutant genotypes does not depend on mutations affecting translated proteins but is an inherent property of the RNA molecules.
To gain further insights into the replication process and the genesis and fate of variability, we complement the analysis of the mathematical model with a model of digital genomes which considers the intrinsic noise due to small population sizes and incorporates different fitness landscapes, including the above-mentioned Swetina-Schuster single-peaked landscape plus the more realistic and rugged additive and epistatic ones. This model consistently shows that, for the SMR case, the critical per-bit mutation rate is higher and the mutational load is lower regardless of the fitness landscape used in the simulations. However, the topology of the fitness landscapes does indeed affect the results. The error threshold and the sharp transition to error catastrophe predicted by the quasispecies theory (13) only hold for the Swetina-Schuster landscape and for GR. However, by assuming an SMR model, which should be the norm for many viruses, even with this unrealistic fitness landscape the sharp transition disappears. Furthermore, neither the additive nor the epistatic (on its two configurations) landscape shows such a sharp transition. Therefore, all the results here shown suggest that the existence of a sudden error threshold is very sensitive to the selection of the topology of the fitness landscape and of the mode of replication and that, thus, caution needs to be used in its application to real RNA virus populations for which nothing is known about their fitness landscapes or their mode of replication.
Recent results suggest that antagonistic epistasis should dominate in viral genomes (2, 30, 34, 42). This being the case, our results suggest that natural selection would have favored viruses using an SMR mechanism, as a way of reducing mutational load. With the combination of antagonistic epistasis and SMR, the average number of mutations per genome will be minimized and the critical mutation rate increased and also the range of mutation rates at which SMR is still a better choice than GR will be larger than for a synergistic landscape. It is generally assumed that RNA viruses have mutation rates on the order of magnitude of the inverse of genome length (11). For the digital genomes used here, this would be equivalent to a mutation rate of 1/32 (0.031). Even at this value, the combination of an antagonistic landscape and an SMR model provides the best solution. Antagonistic epistasis usually appears in small genomes as a consequence of the lack of genetic redundancy that also makes genomes extremely sensitive to the effect of mutations (20, 32). Due to their parasitic lifestyle, RNA viruses have been selected for fast replication at the cost of an increased sensitivity to mutations. However, a pure SMR strategy may minimize the impact of mutation accumulation and preserve in the population mutation-free genomes at frequencies that would not be possible by adopting a pure GR strategy. In this sense, SMR may represent a mechanism of genetic robustness.
Whether RNA viruses may have evolved some mechanisms to buffer the deleterious effects of mutations has recently attracted the attention of researchers (5, 27, 28, 31, 43). Robustness is defined as a reduced sensitivity to perturbations affecting phenotypic expression (8). RNA virus populations may owe their robustness to several of the following mechanisms (reviewed in reference 16). First, the above-mentioned hypersensitivity to mutational effects of individual genomes translates into robustness at the population level as a consequence of a more efficient purifying selection that maintains average fitness high (20). Second, high mutation rates characteristic of RNA viruses may impose a strong selective pressure that pushes virus populations toward regions of the sequence space where the density of neutral mutations is higher (5, 31). Third, the variable and random ploidy of viruses and frequent coinfection events enhance the possibility of genetic complementation. Fourth, segregation of segments during mixed infections and homologous recombination are forms of sex that may re-create mutation-free genomes (28). Fifth, cellular buffering mechanisms (e.g., heat shock proteins) can be utilized by the viruses to their own benefit as an extrinsic source of robustness. The results reported in this study suggest that, in addition to these five potential mechanisms, compacted genomes characterized by an excess of antagonistic epistasis may benefit from an SMR model because they will accumulate less deleterious mutations, have a higher critical mutation rate, and suffer to a lesser extent from the effect of deleterious mutations, that is, they will have increased their robustness.
Acknowledgments
We appreciate the constructive comments and suggestions from the three reviewers of a previous version of this work.
This work was supported by Human Frontier Science Program Organization grant RGP12/2008. Work in València was also funded by the Spanish Ministerio de Ciencia e Innovación, grant BFU2006-14819-CO2-01/BMC. We also acknowledge support from the Santa Fe Institute.
Footnotes
Published ahead of print on 23 September 2009.
REFERENCES
- 1.Ball, L. A. 2001. Replication strategies of RNA viruses, p. 105-118. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 2.Bonhoeffer, S., C. Chappey, N. T. Parkin, J. M. Whitcomb, and C. J. Petropoulos. 2004. Evidence for positive epistasis in HIV-1. Science 306:1547-1550. [DOI] [PubMed] [Google Scholar]
- 3.Carrasco, P., F. de la Iglesia, and S. F. Elena. 2007. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in tobacco etch virus. J. Virol. 81:12979-12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chao, L., C. U. Rang, and L. E. Wong. 2002. Distribution of spontaneous mutants and inferences about the replication mode of the RNA bacteriophage φ6. J. Virol. 76:3276-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Codoñer, F. M., J. A. Daròs, R. V. Solé, and S. F. Elena. 2006. The fittest versus the flattest: experimental confirmation of the quasispecies effect with subviral pathogens. PLoS Pathog. 2:e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dahari, H., R. M. Ribeiro, C. M. Rice, and A. S. Perelson. 2007. Mathematical modeling of subgenomic hepatitis C virus replication in Huh-7 cells. J. Virol. 81:750-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denhardt, D., and R. B. Silver. 1966. An analysis of the clone size distribution of ϕX174 mutants and recombinants. Virology 30:10-19. [DOI] [PubMed] [Google Scholar]
- 8.de Visser, J. A. G. M., J. Hermisson, G. P. Wagner, L. Ancel Meyers, H. Bagheri-Chaichian, J. L. Blanchard, L. Chao, J. M. Cheverud, S. F. Elena, W. Fontana, G. Gibson, T. F. Hansen, D. C. Krakauer, R. C. Lewontin, C. Ofria, S. H. Rice, G. von Dassow, A. Wagner, and M. C. Whitlock. 2003. Evolution and detection of genetic robustness. Evolution 57:1959-1972. [DOI] [PubMed] [Google Scholar]
- 9.Dewanji, A., E. G. Luebeck, and S. H. Moolgavkar. 2005. A generalized Luria-Delbrück model. Math. Biosci. 197:140-152. [DOI] [PubMed] [Google Scholar]
- 10.Domingo, E., E. Baranowski, C. M. Ruiz-Jarabo, A. M. Martín-Hernández, J. C. Sáiz, and C. Escarmís. 1998. Quasispecies structure and persistence of RNA viruses. Emerg. Infect. Dis. 4:521-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drake, J. W., and J. J. Holland. 1999. Mutation rate among RNA viruses. Proc. Natl. Acad. Sci. USA 96:13910-13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eigen, M., C. K. Biebricher, M. Gebinoga, and W. C. Gardiner. 1991. The hypercycle. Coupling of RNA and protein synthesis in the infection cycle of an RNA bacteriophage. Biochemistry 30:11005-11018. [DOI] [PubMed] [Google Scholar]
- 13.Eigen, M., J. McCaskill, and P. Schuster. 1989. The molecular quasispecies. Adv. Chem. Phys. 75:149-263. [Google Scholar]
- 14.Elena, S. F. 2001. Evolutionary history conditions the timing of transmission in Vesicular stomatitis virus. Infect. Genet. Evol. 1:151-159. [DOI] [PubMed] [Google Scholar]
- 15.Elena, S. F., P. Agudelo-Romero, P. Carrasco, F. M. Codoñer, S. Martín, C. Torres-Barceló, and R. Sanjuán. 2008. Experimental evolution of plant RNA viruses. Heredity 100:478-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elena, S. F., P. Carrasco, J. A. Daròs, and R. Sanjuán. 2006. Mechanisms of genetic robustness in RNA viruses. EMBO Rep. 7:168-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Endy, D., D. Kong, and J. Yin. 1997. Intracellular kinetics of a growing virus: a genetically structured simulation for the bacteriophage T7. Biotechnol. Bioeng. 55:375-389. [DOI] [PubMed] [Google Scholar]
- 18.French, R., and D. C. Stenger. 2003. Evolution of Wheat streak mosaic virus: dynamics of population growth within plants may explain limited variation. Annu. Rev. Phytopathol. 41:199-214. [DOI] [PubMed] [Google Scholar]
- 19.Krakauer, D. C., and N. L. Komarova. 2003. Levels of selection in positive-strand virus dynamics. J. Evol. Biol. 16:64-73. [DOI] [PubMed] [Google Scholar]
- 20.Krakauer, D. C., and J. B. Plotkin. 2002. Redundancy, antiredundancy, and the robustness of genomes. Proc. Natl. Acad. Sci. USA 99:1405-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landau, D. P., and K. Binder. 2000. A guide to Monte Carlo simulations in statistical physics, p. 70-73. Cambridge University Press, Cambridge, United Kingdom.
- 22.Leuthäusser, I. 1986. An exact correspondence between Eigen's evolution model and a two-dimensional Ising system. J. Chem. Phys. 84:1884-1885. [Google Scholar]
- 23.Leuthäusser, I. 1987. Statistical mechanics of Eigen's evolution model. J. Stat. Phys. 48:343-360. [Google Scholar]
- 24.Lim, K., V. Lang, T. Lam, and J. Yin. 2006. Model-based design of growth-attenuated viruses. PLoS Comput. Biol. 2:e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luria, S. E. 1951. The frequency distribution of spontaneous bacteriophage mutants as evidence for the exponential rate of phage production. Cold Spring Harbor Symp. Quant. Biol. 16:463-470. [DOI] [PubMed] [Google Scholar]
- 26.Malpica, L. M., A. Fraile, I. Moreno, C. I. Obies, J. W. Drake, and F. García-Arenal. 2002. The rate and character of spontaneous mutation in an RNA virus. Genetics 162:1505-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martín, V., A. Grande-Pérez, and E. Domingo. 2008. No evidence of selection for mutational robustness during lethal mutagenesis of Lymphocytic choriomeningitis virus. Virology 378:185-192. [DOI] [PubMed] [Google Scholar]
- 28.Montville, R., R. Froissart, S. K. Remold, O. Tenaillon, and P. E. Turner. 2005. Evolution of mutational robustness in an RNA virus. PLoS Biol. 3:e381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reddy, B., and J. Yin. 1999. Quantitative intracellular kinetics of HIV type 1. AIDS Res. Hum. Retrovir. 15:273-283. [DOI] [PubMed] [Google Scholar]
- 30.Sanjuán, R. 2006. Quantifying antagonistic epistasis in a multifunctional RNA secondary structure of the Rous sarcoma virus. J. Gen. Virol. 87:1595-1602. [DOI] [PubMed] [Google Scholar]
- 31.Sanjuán, R., J. M. Cuevas, V. Furió, E. C. Holmes, and A. Moya. 2007. Selection for robustness in mutagenized RNA viruses. PLoS Genet. 3:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanjuán, R., and S. F. Elena. 2006. Epistasis correlates to genomic complexity. Proc. Natl. Acad. Sci. USA 103:14402-14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanjuán, R., A. Moya, and S. F. Elena. 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. USA 101:8396-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanjuán, R., A. Moya, and S. F. Elena. 2004. The contribution of epistasis to the architecture of fitness in an RNA virus. Proc. Natl. Acad. Sci. USA 101:15376-15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sardanyés, J., S. F. Elena, and R. V. Solé. 2008. Simple quasispecies models for the survival-of-the-flattest effect: the role of space. J. Theor. Biol. 250:560-568. [DOI] [PubMed] [Google Scholar]
- 36.Shukla, D. D., C. W. Ward, and A. A. Brunt. 1994. The Potyviridae. CAB International, Wallingford, United Kingdom.
- 37.Sidorenko, Y., and U. Reichl. 2004. Structured model of influenza virus replication in MDCK cells. Biotech. Bioeng. 88:1-14. [DOI] [PubMed] [Google Scholar]
- 38.Solé, R. V., J. Sardanyés, J. Díez, and A. Mas. 2006. Information catastrophe in RNA viruses through replication thresholds. J. Theor. Biol. 240:353-359. [DOI] [PubMed] [Google Scholar]
- 39.Srivastava, R., L. You, J. Summers, and J. Yin. 2002. Stochastic vs. deterministic modeling of intracellular viral kinetics. J. Theor. Biol. 218:309-321. [DOI] [PubMed] [Google Scholar]
- 40.Strogatz, S. H. 2000. Nonlinear dynamics and chaos with applications to physics, biology, chemistry, and engineering. Westview Press, Boulder, CO.
- 41.Swetina, J., and P. Schuster. 1982. Self-replication with errors. A model for polynucleotide replication. Biophys. Chem. 16:329. [DOI] [PubMed] [Google Scholar]
- 42.van Opijnen, T., M. C. Boerlijst, and B. Berkhout. 2006. Effects of random mutation in the human immunodeficiency virus type 1 transcriptional promoter on viral fitness in different host cell environments. J. Virol. 80:6678-6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagner, A., and P. F. Schuster. 1999. Viral RNA and evolved mutational robustness. J. Exp. Zool. 285:119-127. [PubMed] [Google Scholar]
- 44.Wilke, C. O., and I. S. Novella. 2003. Phenotypic mixing and hiding may contribute to memory of viral quasispecies. BMC Microbiol. 3:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.You, L., P. F. Suthers, and J. Yin. 2002. Effects of Escherichia coli physiology on growth of phage T7 in vivo and in silico. J. Bacteriol. 184:1888-1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhdanov, V. P. 2004. Bifurcation in a generic model of intracellular viral kinetics. J. Phys. A Math. Gen. 37:L63-L66. [Google Scholar]