

Abstract
Fusogenic reoviruses utilize the FAST proteins, a novel family of nonstructural viral membrane fusion proteins, to induce cell-cell fusion and syncytium formation. Unlike the paradigmatic enveloped virus fusion proteins, the FAST proteins position the majority of their mass within and internal to the membrane in which they reside, resulting in extended C-terminal cytoplasmic tails (CTs). Using tail truncations, we demonstrate that the last 8 residues of the 36-residue CT of the avian reovirus p10 FAST protein and the last 20 residues of the 68-residue CT of the reptilian reovirus p14 FAST protein enhance, but are not required for, pore expansion and syncytium formation. Further truncations indicate that the membrane-distal 12 residues of the p10 and 47 residues of the p14 CTs are essential for pore formation and that a residual tail of 21 to 24 residues that includes a conserved, membrane-proximal polybasic region present in all FAST proteins is insufficient to maintain FAST protein fusion activity. Unexpectedly, a reextension of the tail-truncated, nonfusogenic p10 and p14 constructs with scrambled versions of the deleted sequences restored pore formation and syncytiogenesis, while reextensions with heterologous sequences partially restored pore formation but failed to rescue syncytiogenesis. The membrane-distal regions of the FAST protein CTs therefore exert multiple effects on the membrane fusion reaction, serving in both sequence-dependent and sequence-independent manners as positive effectors of pore formation, pore expansion, and syncytiogenesis.
The only examples of nonenveloped viruses that induce cell-cell fusion and syncytium formation occur within the family Orthoreoviridae, an extremely diverse group of viruses containing segmented double-stranded RNA genomes (9). In recent years, the viral proteins responsible for the syncytiogenic phenotype of the fusogenic orthoreoviruses and aquareoviruses have been identified and characterized (14, 18, 41, 46). These fusion-associated small transmembrane (FAST) proteins define a new family of viral fusogens with several unique biological and biophysical properties. Unlike the well-characterized enveloped virus fusion proteins, reovirus FAST proteins are nonstructural viral proteins and are therefore not involved in mediating virus-cell fusion and virus entry (18, 21, 46). The FAST proteins are instead dedicated to inducing cell-cell fusion and syncytium formation following their expression and trafficking to the plasma membrane of virus-infected or transfected cells (14, 17, 46). Data from previously reported studies also suggest that the FAST proteins serve as virulence factors for the fusogenic reoviruses, promoting virus dissemination and increased tissue destruction (6, 43). How this atypical family of viral fusogens functions to mediate cell-cell membrane fusion remains unclear.
The unusual biological role of the FAST proteins as nonstructural, virus-encoded, “cellular” fusogens is embodied in structural features that clearly distinguish the FAST proteins from the membrane fusion proteins of enveloped viruses. There are currently four distinct members of the FAST protein family, named according to their molecular masses: the homologous p10 proteins of avian reovirus (ARV) and Nelson Bay reovirus and the unrelated p14, p15, and p22 proteins of reptilian reovirus (RRV), baboon reovirus, and Atlantic salmon aquareovirus, respectively (14, 18, 41, 46). These proteins are the smallest known fusogens, ranging from 95 to 198 amino acids in size, and assume an asymmetric topology in the plasma membrane, with a single transmembrane domain that separates small N-terminal ectodomains of ∼20 to 41 residues from equal-sized or considerably larger C-terminal endodomains of ∼36 to 141 residues (Fig. 1A). A number of structural motifs in both the ecto- and endodomains of the FAST proteins have been identified, including sites of acylation, hydrophobic patches, a membrane-proximal polybasic region, and regions rich in proline, cysteine, or arginine, proline, and histidine. Each of the FAST proteins has its own signature repertoire and arrangement of these motifs. Determining how these various motifs contribute to the fusogenic activity of the FAST proteins remains an area of active investigation.
FIG. 1.

ARV p10 and RRV p14 FAST protein topologies and tail truncations. (A) Diagrammatic representation of the p10 and p14 FAST proteins showing their topology in the plasma membrane. Both are single-pass transmembrane proteins with N-terminal ectodomains on the surface of cells and C-terminal endodomains in the cytoplasm. Structural motifs include hydrophobic patches (HP), polybasic motifs (PB), fatty acid modifications (indicated by squiggly lines) that are either the N-terminal myristoylation or palmitoylation of a dicysteine motif (CC), and a polyproline motif (PP). The total number of residues in each protein is indicated by the numbers. (B) The amino acid sequences of the p10 and p14 endodomains are shown, along with the motifs described above. Progressive truncations of the CTs were constructed (arrows), with the numbers indicating the last amino acid present in the full-length proteins or each truncation.
Numerous studies of diverse fusion processes define five general steps of the pathway for membrane fusion and syncytium formation: membrane binding, close membrane apposition, hemifusion (i.e., the mixing of the outer leaflets of the two bilayers), stable pore formation, and pore expansion (12, 13, 44). The well-characterized enveloped virus fusion proteins utilize extensive structural rearrangement of their complex ectodomains to provide mechanical energy to draw membranes into close proximity and promote membrane merger (21, 53). The limited size of the FAST protein ectodomains precludes such a mechanical model for membrane fusion, necessitating the development of alternate models to explain how the diminutive FAST proteins breach the thermodynamic barriers that prevent the spontaneous merger of biological membranes. The FAST proteins are both necessary and sufficient to mediate membrane fusion (51). However, data from recent studies indicate that for maximal cell fusion activity, the FAST proteins rely on surrogate adhesins to mediate close membrane apposition (42). Data from recent studies also indicate that a small percentage of the p14 FAST protein expressed in virus-infected or transfected cells is proteolytically processed to generate a bioactive, soluble endodomain that recruits cellular pathways to drive the expansion of stable fusion pores into the extended fusion apertures needed for syncytium formation (50). The FAST proteins therefore utilize accessory proteins to mediate the prefusion (membrane binding and apposition) and postfusion (pore expansion) stages of syncytiogenesis, retaining within their rudimentary structures all that is required to mediate the actual process of membrane merger. This subdivision of the multistep process of syncytium formation is reflected in, and is perfectly suited to, the evolution of the FAST proteins as virus-encoded cellular fusogens.
The small size of the FAST protein ectodomains and their donor membrane-focused topology contrast markedly with enveloped virus fusion proteins that position the majority of their mass external to the membrane. While the complex ectodomains of the enveloped virus fusion proteins clearly play an essential role in the fusion reaction, the involvement of their cytoplasmic tails (CTs) is far less certain, and no consistent picture of the role of these C-terminal tails has emerged. The CTs of many enveloped viral fusion proteins, including baculovirus (31), severe acute respiratory syndrome coronavirus (5), vesicular stomatitis virus (36), parainfluenza virus type 2 (56), and influenza A virus subtype H3 (10), play no role in the membrane fusion reaction. Of the fusion protein tails that do modulate the fusion reaction, the majority serve inhibitory roles, including the F proteins of measles virus and parainfluenza virus type 5 SER (7, 45, 52), glycoprotein B from several herpesviruses (22, 24, 28), and the fusion proteins of numerous retroviruses (1, 8, 30, 32, 34, 47, 48). These inhibitory cytoplasmic domains alter the conformation of the fusion protein ectodomains, thereby coupling virion maturation to fusion competence (1, 2, 35, 52, 54). In the few cases where extensive tail truncations adversely affect fusion, these truncations generally decrease but do not eliminate syncytiogenesis, and it is the membrane-proximal portion of the tail that promotes pore formation or pore expansion (20, 25, 26, 32).
Since the FAST proteins are nonstructural viral proteins, their CTs (also referred to as endodomains) are not required to suppress fusion activity until after virus particle assembly. At the same time, the disproportionate size of their endodomains strongly suggests that these CTs play an important role in membrane fusion activity. Although one such role of the p14 CT is the generation of a soluble endodomain that recruits cellular factors involved in pore expansion, the majority of p14 is not proteolytically processed, suggesting that FAST protein CTs may serve additional roles as components of the intact protein (50). We now show that C-terminal truncations of the p10 and p14 FAST proteins reduced and eventually eliminated cell-cell fusion. Fluorescence-based pore formation assays coupled with tail reextension studies further revealed that FAST protein CTs drive fusion pore formation and expansion in both sequence-dependent and sequence-independent manners. The membrane-distal regions of FAST protein CTs therefore exert multiple effects on the mechanism of membrane fusion.
MATERIALS AND METHODS
Cells and antisera.
QM5 and Vero cells were maintained as previously described (14). Rabbit antisera specific for the full-length p14 FAST protein, the endodomain of the p10 FAST protein, or the ectodomain of the p14 FAST protein were previously described (14, 40, 51). Polyclonal rabbit antiserum specific for the full-length p10 protein was prepared using a purified maltose binding protein-p10 chimeric protein expressed in Escherichia coli cells, as previously described (46). Commercial antisera specific for actin (Sigma) and Alexa 647-conjugated (Invitrogen) or horseradish peroxidase-conjugated (KPL, Gaithersburg, MD) goat anti-rabbit immunoglobulin G (H+L) secondary antibodies were obtained from the indicated commercial sources.
Plasmids and cloning.
PCR cloning using Vent polymerase (Invitrogen) was used to subclone the full-length p14 and p10 FAST proteins and their respective C-terminal truncation constructs (p14c105, p14c78, p10c90, and p10c86) into pcDNA3 (Invitrogen). The 47 C-terminal amino acids of the p14 CT that were deleted in the p14c78 construct (RNSYRLSEIQRPISQHEYEDPYEPPSRRKPPPPPYSTYVNIDNVSAI) were replaced with a scrambled version (PRTSYIPNYQDPENAPIRSPESRNYQPVYSPRDPEYSIPIVKESRHL) of the same sequence (p14c78S) or with a random heterologous sequence of the same length (GENPYDTFLMHEDYCAFPHKCGSEIMLTEMAGWFYGPDQHVWLPNHL) (p14c78R). A second scrambled version of p14 (p14c78S2) was also created (PRTSYINYQDPPPENPAIRSESRNYQPVYPSRDPPYEHSIIVKESRPL). The C-terminal 12 amino acids deleted in the p10c86 construct (GKHNAMAPPYDV) were similarly replaced with a scrambled version of the same sequence (PHADMYGPVKAN) (p10c86S) or with a random heterologous sequence of the same length (QCYLEKNWLRAT) (p10c86R). All constructs were confirmed by sequencing (McLab, CA). The pEGFP plasmid (Clontech) served as a marker of transfection for pore formation assays.
Transfections and syncytial index.
QM5 monolayers were transfected with expression plasmids using Lipofectamine transfection reagent (Invitrogen) according to the manufacturer's instructions. At the designated times posttransfection, cells were methanol fixed and Wright-Giemsa stained (Dade Behring, NJ). The extent of syncytium formation was quantified by using a syncytial index based on determining the average number of syncytial nuclei per microscopic field (20× objective) from five random microscopic fields, as previously described (15). Results are reported as the means ± standard deviations (SD) from triplicate samples of a representative experiment.
Western blotting.
QM5 subconfluent monolayers in 10-cm dishes were transfected with the various expression plasmids, and transfected cells were resuspended using 50 mM EDTA at 8 or 24 h posttransfection for p14 or p10 constructs, respectively. Cell pellets were lysed in radioimmunoprecipitation assay buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, and 1 μM each of the protease inhibitors aprotinin, leupeptin, and pepstatin) for 45 min on ice, nuclei were removed by centrifugation, and samples were boiled for 5 min in protein sample buffer. Cell lysates were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (12.5% acrylamide), transferred onto polyvinylidene difluoride membranes, and probed with either p10 endodomain or p14 full-length antisera (1:30,000 and 1:20,000, respectively). Proteins were detected by using horseradish peroxidase-conjugated secondary antibody (1:10,000), visualized by staining with ECL plus reagent (GE healthcare), and analyzed by use of a Typhoon 9410 variable-mode imager (Amersham Biosciences).
FACS cell surface fluorescence.
QM5 cells in 24-well cluster plates were transfected with the various expression plasmids. Following a 3- to 6-h transfection in serum-free medium, the medium was replaced with Earle's 199 medium supplemented with 10% fetal bovine serum (FBS; Sigma) and a 1:25 dilution of complement-fixed antisera that recognize the p10 or p14 ectodomains to inhibit syncytium formation, and transfected cells were cultured overnight at 37°C. To normalize the cell surface expression of the various FAST protein constructs, the amount of expression plasmid DNA was decreased incrementally while being replaced with empty vector plasmid to maintain equal levels of total DNA transfected per well. Following culturing of cells grown overnight, live cells were blocked for 30 min at 4°C with fluorescence-activated cell sorter (FACS) surface staining buffer (Hanks' buffered saline solution [HBSS; Gibco] supplemented with 5% normal goat serum [Sigma], 1% bovine serum albumin [Sigma], and 0.02% NaN3 [Sigma]). Cells were washed twice with staining buffer, incubated with the same primary antibody (1:200) used to inhibit syncytium formation for 1 h at 4°C, extensively washed with staining buffer, and then incubated for 1 h at 4°C with Alexa 647-conjugated goat anti-rabbit secondary antibody (1:2,000). Stained cells were then washed extensively with staining buffer and phosphate-buffered saline (PBS; Gibco), suspended by using 10 mM EDTA in PBS, and fixed in suspension on ice by using 3.7% formaldehyde (Sigma). Fixed cells (10,000 to 20,000 cells) were analyzed for surface fluorescence by flow cytometry (FACSCalibur; Becton Dickinson) and analyzed by using FCS Express 2.0 software (DeNovo Software).
Fluorescent cell-cell pore formation.
Subconfluent QM5 monolayers in six-well plates were cotransfected with the indicated amounts of plasmid DNA expressing the various FAST protein constructs plus 0.2 μg of pEGFP, which served as a marker for transfected donor cells. Concurrently, target Vero cells were washed twice with HBSS and labeled with 10 μM calcein red-orange (Molecular Probes) in HBSS at 37°C for 35 min. The labeled Vero cells were washed twice with HBSS, incubated in Earle's 199 medium supplemented with 10% FBS for 1 to 2 h, washed twice with HBSS and twice with PBS, digested with trypsin, and resuspended in Earle's 199 medium with 10% FBS. An excess of Vero target cells (2 × 106 cells) was added to the QM5 donor cells prior to the onset of syncytium formation (3 and 13 h posttransfection for p14 and p10 constructs, respectively), and cells were cocultured at 37°C for 4 to 13 h to allow cell-cell fusion to proceed. Cells were then digested with trypsin, resuspended in PBS, and fixed in suspension on ice with 3.7% formaldehyde. Fixed cells were analyzed by flow cytometry, as described above, by examining the extent of calcein red fluorescence in 5,000 to 10,000 gated enhanced green fluorescent protein (GFP)-positive cells.
RESULTS
FAST protein CTs are essential for syncytium formation.
We previously reported that C-terminal truncations of the p14 FAST protein resulted in a decrease in or loss of syncytium formation in Vero cells, although the basis for this phenotype was not determined (14). To examine cell-specific effects of the p14 CT, the p14c105 and p14c78 constructs that were previously analyzed using Vero cells were similarly analyzed with QM5 quail fibroblasts. These constructs lack 20 and 47 residues, respectively, of the 68-residue p14 CT (Fig. 1B). As previously reported for Vero cells, p14c105 displayed diminished syncytiogenic activity in QM5 cells when assayed using a quantitative syncytial index (Fig. 2A). This construct, however, still fused monolayers to completion with only a ∼2-h time lag relative to full-length p14 (Fig. 2B). The C-terminal 20 residues of p14 therefore affect the kinetics of, but are not required for, p14-induced syncytium formation. In contrast, the p14c78 construct displayed no fusion activity in either QM5 cells (Fig. 2) or Vero cells (14), indicating that the C-terminal 47 residues of p14 are essential for syncytiogenesis regardless of the cell type.
FIG. 2.

C-terminal truncations progressively reduce, and eventually eliminate, fusion activity. (A) QM5 cells were transfected with the indicated constructs (p14, p14c105, p14c78, p10, p10c90, or p10c86) and Giemsa stained at either 8 h or 26 h posttransfection for the p14 and p10 constructs, respectively. Syncytial nuclei present in five random microscopic fields were quantified, and results are presented as the mean numbers of syncytial nuclei per field ± SD from one of two experiments conducted in triplicate. (B) The same experiment described above (A) except that cells were Giemsa stained, and images were captured by bright-field microscopy at a ×200 magnification at different time points (p14, 8 h; p14c105, 10 h; p14c78, 24 h; p10, 20 h; p10c90, 38 h; p10c86, 48 h) to show the progression of syncytium formation.
To determine whether the concept of an essential CT is generally applicable to the FAST proteins, the 36-residue C-terminal tail of the 98-residue p10 FAST protein was truncated by 8 or 12 residues to generate the p10c90 and p10c86 constructs (Fig. 1B), and the syncytiogenic properties of the truncated proteins were assessed by using QM5 cells. Despite the difference in their lengths, the absence of any sequence similarity, and the dramatically different syncytiogenic kinetics of the p10 and p14 FAST proteins (43), the same trend observed with the p14 truncations was also manifested by the p10 truncations. The deletion of the C-terminal 8 residues of p10 resulted in delayed syncytium formation, while the removal of an additional 4 residues eliminated the cell-cell fusion capacity (Fig. 2). A residual CT of 21 to 24 residues that contains the conserved FAST protein polybasic motif is therefore insufficient for p10 or p14 to mediate syncytium formation. Similar results were obtained with the p15 FAST protein (data not shown), indicating that the CTs of the FAST proteins are essential for syncytium formation.
Tail truncations have varied effects on protein expression.
Decreasing the plasma membrane concentration of activated enveloped virus fusion proteins can result in a progressive transition from the full-fusion phenotype to pore formation, hemifusion, and loss of fusion (11). Protein expression levels of the FAST protein constructs were therefore analyzed to determine whether the truncations exerted an indirect effect on the fusion phenotype by altering the steady-state expression levels of the FAST proteins. To examine total protein expression, QM5-transfected cells were lysed when syncytium formation induced by the full-length proteins was approaching maximum levels (8 and 24 h posttransfection for p14 and p10 constructs, respectively). Lysates were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and steady-state protein levels were determined by Western blotting using anti-FAST protein antibodies. The majority of the tail truncations had little, if any, effect on p10 or p14 expression levels, with the exception of p14c78, whose steady-state levels were noticeably reduced (Fig. 3A).
FIG. 3.

C-terminal truncations have variable effects on expression of FAST proteins. (A) QM5 cells were transfected with the indicated FAST protein constructs or with empty vector, and cell lysates were harvested at either 8 h or 26 h posttransfection for the p14 and p10 constructs, respectively. Lysates were analyzed by Western blotting using antisera specific for p10 or p14 or using anti-actin antibody as a loading control. (B) Expression of the various FAST protein constructs in the plasma membrane was quantified by labeling intact transfected cells with anti-p14 ectodomain or anti-p10 full-length antibodies at 24 h posttransfection, as described in Materials and Methods, followed by goat anti-rabbit Alexa 647-conjugated secondary antibody. Labeled cells were analyzed by flow cytometry, and results are presented as relative fluorescence (FL4-H) versus cell counts. Shaded gray histograms indicate background fluorescence from empty vector-transfected cells, while the gray and black line tracings indicate duplicate samples of cells transfected with the indicated FAST protein constructs from a single representative experiment.
To determine whether the reduced fusion activity of the tail truncations reflected decreased cell surface protein expression levels, transfected cells expressing the various FAST proteins were analyzed by flow cytometry. Antisera that recognize the p10 or p14 ectodomains were added to transfected cells just prior to the onset of syncytium formation (∼3 to 12 h posttransfection). These antisera inhibited syncytium formation, allowing transfected cells to be incubated overnight at 37°C to improve the sensitivity of the cell surface fluorescence assay, and simultaneously permitted the determination of both FAST protein cell surface expression levels and p14 topology in the plasma membrane. As evident from the distribution of cell surface fluorescence (Fig. 3B), the p10 truncations exhibited similar levels of cell surface expression that differed by only ∼20 to 40% relative to full-length p10. The same situation applied to the p14c105 construct, which showed no difference in cell surface expression relative to full-length p14 (Fig. 3B). The cell surface expression of p14c78 was reduced by ∼75% relative to full-length p14 (Fig. 3B), a reduction consistent with the decreased steady-state expression levels of p14c78 observed in Western blots (Fig. 3A). Tail truncations of the FAST proteins therefore have modest, though somewhat variable, effects on protein expression or trafficking to the plasma membrane in the correct topology.
CTs affect membrane fusion independent of effects on cell surface protein levels.
To exclude the possibility that variable cell surface expression levels of the truncated FAST proteins, particularly p14c78, were responsible for the fusion phenotypes, plasmid DNA transfection conditions were modified to normalize FAST protein expression in the plasma membrane. Decreasing the dose of p14 plasmid from 1 μg to 0.2 μg yielded cell surface expression levels of p14 that were equivalent to those observed for cells transfected with 1 μg of the p14c78 plasmid, as determined by flow cytometry (Fig. 4A). Similar cell surface expression levels of p14c105 were achieved using 0.1 μg of plasmid DNA. As shown in Fig. 4B, the previously characterized fusion phenotypes of the p14 constructs were maintained under these normalized cell surface expression conditions; syncytium formation induced by p14c105 at the midpoint of the syncytial time course was ∼20% of that induced by full-length p14, while p14c78 was devoid of syncytiogenic activity. A similar situation applied to the p10 tail truncations under normalized cell surface expression levels. Although normalized cell surface expression levels of p10c90 did increase the fusion efficiency, the syncytiogenic activity of p10c90 was still reduced by ∼20% relative to that induced by full-length p10 (Fig. 4), while p10c86 was still nonfusogenic. The results for both p10 and p14 indicate that the membrane-distal portions of the CTs of the FAST proteins exert a direct positive effect on fusion activity independent of any effects that they may have on protein expression or trafficking to the plasma membrane.
FIG. 4.

C-terminal truncations reduce fusion activity independent of cell surface expression levels. (A) QM5 cells were transfected with the indicated amounts of plasmid DNA expressing the p10 or p14 constructs, and nonpermeabilized cells were analyzed for cell surface fluorescence by flow cytometry using antisera specific for p10 or p14, as described in Materials and Methods. Gray histograms are the fluorescence profiles of cells transfected with empty vector and stained with the same antisera. (B) Cells were transfected with quantities of plasmid DNA that yielded equivalent levels of surface expression of the p10 or p14 constructs, and syncytial nuclei were quantified at 8 h or 24 h posttransfection for the p14 and p10 mutants, respectively, as described in the legend of Fig. 2. Results are presented as the mean numbers of syncytial nuclei per microscopic field ± SD from one of two experiments conducted in triplicate.
FAST protein CTs are required for fusion pore formation.
While the CTs of p10 and p14 are clearly required for syncytiogenesis, it was unclear whether the tails exerted a similar effect on the formation of stable fusion pores. This question was particularly germane in view of recent results indicating that the coexpression of soluble FAST protein endodomains augments syncytium formation induced by various fusogens, influencing an event that occurs downstream of stable pore formation (50). The C-terminally truncated FAST proteins were therefore analyzed for their ability to induce stable pore formation using a quantitative FACS-based fluorescent cell-cell pore formation assay.
Donor QM5 cells were cotransfected with pEGFP (as a marker for transfection) and plasmid doses of the various p14 protein constructs that normalized surface protein expression levels. At 3 h posttransfection the donor cells were overseeded with calcein red-labeled Vero target cells, and the cells were cocultured for 4 h at 37°C to allow cell-cell fusion to proceed. Cell fluorescence was analyzed by flow cytometry, gating the donor cells based on enhanced GFP fluorescence and determining the percentage of these cells that acquired the small aqueous calcein red fluor via fusion with target cells. As shown in the two-dimensional (2D) scatter plots (Fig. 5A) and the histograms of fluorescence distribution (Fig. 5B), low background transfer of the aqueous fluor was observed in mock-transfected cells, while p14-induced cell-cell fusion resulted in extensive dye transfer, which is indicative of stable pore formation. Paralleling their syncytiogenic phenotypes, p14c78 failed to induce dye transfer above background levels, while p14c105-induced pore formation was reduced relative to that of full-length p14. By setting the extent of dye transfer induced by p14 at 100%, dye transfer induced by the truncation mutants was quantified by either threshold analysis of the 2D scatter plots or Overton subtraction of the histograms versus mock-transfected histograms (Fig. 5C). Both approaches revealed that the fusion pore formation induced by p14c105 was ∼35 to 40% of that induced by full-length p14, while p14c78 was incapable of mediating the formation of either stable fusion pores or syncytia. The C-terminal 47 residues of p14 are therefore essential for membrane fusion, with the extreme C terminus (i.e., residues 106 to 125) serving an enhancing, though nonessential, role in pore formation and syncytiogenesis.
FIG. 5.

The membrane-distal region of the p14 CT drives fusion pore formation. (A) QM5 cells cotransfected with pEGFP and the indicated p14 constructs were cocultured with Vero target cells labeled with calcein red-orange. Cells were resuspended, fixed 9 h posttransfection, and analyzed by flow cytometry. The GFP-positive transfected cells were gated, and the percentages of GFP-positive cells that acquired the aqueous calcein red fluor due to pore formation (indicated above the horizontal threshold line) were quantified and are shown relative to the forward scatter (FSC-H). (B) A similar experiment was performed as described above (A), and results are presented as the calcein red fluorescence distribution of the gated GFP-positive cells (black line tracing). The percentage of cells with calcein red fluorescence above the background fluorescence profile of empty-vector-transfected cells (filled gray histogram) was determined by Overton subtraction of the fluorescence profiles of the two cell populations and is indicated. (C) The dot plots in A and Overton subtraction analysis of the fluorescence profiles in B were quantified and are presented as the means ± SD from one of three experiments conducted in triplicate.
Similar studies were conducted using the p10 truncations. As shown by quantifying the 2D scatter plots (Fig. 6), prolonged coculture of the p10 donor cells with target cells (for 13 h), which was required due to the slower kinetics of p10-induced cell-cell fusion, resulted in a higher level of spontaneous dye transfer than that observed under p14 fusion conditions (Fig. 5C). This background, however, did not interfere with the clear detection of dye transfer mediated by p10-induced pore formation. The inability of p10c86 to induce syncytium formation (Fig. 4) was mirrored by the inability of this truncated construct to induce pore formation and dye transfer above background levels (Fig. 6), indicating that the C-terminal 12 residues of p10 are essential for pore formation. In contrast, the p10c90 construct induced pore formation and dye transfer at levels equivalent to those induced by full-length p10 (Fig. 6), even though this construct displayed a modest, though reproducible, decrease in levels of syncytium formation (Fig. 4). The C-terminal 12 residues of p10 are therefore required for pore formation and syncytiogenesis, with the extreme C terminus (i.e., residues 91 to 98) influencing events that occur after the formation of small, stable fusion pores to increase the efficiency of syncytium formation.
FIG. 6.
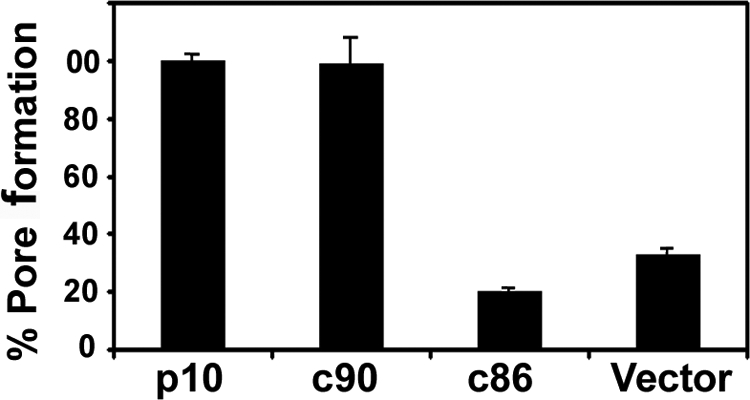
The membrane-distal region of the p10 CT drives fusion pore formation. QM5 cells cotransfected with pEGFP and the indicated p14 constructs were cocultured with Vero target cells labeled with calcein red-orange. Cells were resuspended and fixed 26 h posttransfection, and the percentages of GFP-positive cells that acquired the aqueous calcein red fluor due to pore formation were quantified from the dot plots as described in the legend of Fig. 5. Results are presented as the means ± SD from one of three experiments conducted in triplicate.
A sequence-independent, amino acid content-dependent role for the C termini of FAST protein CTs in membrane fusion.
While truncation of FAST protein CTs resulted in a loss of membrane fusion activity, these results provided no information on how the deleted sequences contribute to the fusion reaction. More specifically, do the C termini of the CTs contain an essential amino acid motif, or is there merely a minimal-length requirement for the CTs? To determine whether FAST protein CTs function in a size- or sequence-dependent manner, the truncated constructs that displayed no fusion activity were reextended to their full length by using various sequences, and the fusion activities of these reextended constructs were assessed by using normalized cell surface protein expression levels, as described above.
The p14c78S construct replaced the C-terminal 47 residues deleted from p14c78 with a scrambled version of the same sequence. The scrambled sequence was screened using the Eukaryotic Linear Motifs database (http://elm.eu.org) to confirm the disruption of predicted linear sequence motifs. A time course analysis of syncytiogenesis revealed that replacing the C-terminal 47 residues of p14 with a scrambled version of the same sequence restored the syncytiogenic ability of p14c78 albeit with reduced kinetics that directly paralleled those displayed by p14c105 (Fig. 7A). A second scrambled version of p14 (see Materials and Methods) displayed a similar restoration of fusion activity (data not shown). The scrambled sequence in p14c78 also restored cell-cell pore formation to a level equivalent to, or slightly higher than, that of p14c105 (Fig. 7B). Most interestingly, replacing the 47 residues deleted from p14c78 with a heterologous, random series of amino acids (p14c78R construct) had no effect on restoring the syncytiogenic activity of p14c78 (Fig. 7A). The p14c78R construct did, however, consistently induce levels of dye transfer that exceeded background levels (Fig. 7B), indicating that this random sequence of amino acids partially restored the pore formation capability of p14c78 to ∼25% of the level induced by authentic p14.
FIG. 7.

Reextending the C terminus of p14 with scrambled sequences, but not random sequences, restores pore formation and syncytiogenesis. (A) The nonfusogenic p14c78 truncation was reextended to full length with either a scrambled version of the deleted amino acids (Sc) or the same number of random amino acids (Ra). These constructs, along with p14, p14c105, and p14c78, were transfected into QM5 cells using amounts of DNA to give equalized surface expression levels. Transfected cells were fixed and Giemsa stained at the indicated times posttransfection, and a syncytial index based on the numbers of syncytial nuclei/microscopic field was determined. Results are the means ± SD from one of three experiments conducted in triplicate. (B) QM5 cells were cotransfected with pEGFP and the indicated p14 constructs at plasmid doses that yielded equivalent cell surface expression levels, and transfected cells were cocultured with Vero target cells labeled with calcein red-orange. Cells were resuspended and fixed 7 h posttransfection, and the percent pore formation was determined by flow cytometry and Overton subtraction as described in the legend of Fig. 5. Results are the means ± SD from one of two experiments conducted in triplicate.
Similar reextension studies were performed on the p10 FAST protein by using the nonsyncytiogenic p10c86 truncation as a backbone (Fig. 8). The p10c86S construct was reextended with a scrambled version of the C-terminal 12 residues of p10, while p10c86R was reextended with random heterologous amino acids. The results obtained with these p10 constructs showed trends that are remarkably similar to those observed for the p14 constructs: scrambled sequences partially restored both syncytiogenesis (Fig. 8A) and pore formation (Fig. 8B), while random amino acids had no such enhancing effect on syncytium formation. As with p14, the replacement of the C-terminal sequences of p10 with random amino acids partially restored the pore formation capacity, with levels of dye transfer that were consistently above background levels and ∼20% of the levels induced by authentic p10 (Fig. 8B). The membrane-distal regions of FAST protein CTs are therefore required for fusion activity and exert multiple sequence-dependent and sequence-independent effects on the mechanism by which the FAST proteins mediate membrane fusion.
FIG. 8.

Scrambled, but not random, amino acids at the C terminus of p10 support pore formation and syncytiogenesis. (A) The nonfusogenic p10c86 truncation was reextended to full length with either a scrambled version of the deleted amino acids (Sc) or the same number of random amino acids (Ra). These constructs, along with p10, p10c90, and p10c86, were transfected into QM5 cells using amounts of DNA to give equalized surface expression levels. Transfected cells were fixed and Giemsa stained at the indicated times posttransfection, and a syncytial index based on the numbers of syncytial nuclei/microscopic field was determined. Results are the means ± SD from one of three experiments conducted in triplicate. (B) QM5 cells were cotransfected with pEGFP and the indicated p10 constructs at plasmid doses that yielded equivalent cell surface expression levels, and transfected cells were cocultured with Vero target cells labeled with calcein red-orange. Cells were resuspended and fixed 27 h posttransfection, and the percent pore formation was determined by flow cytometry and threshold analysis of the dot plots as described in the legend of Fig. 5. Results are the means ± SD from one of two experiments conducted in triplicate.
DISCUSSION
The unusual, asymmetric membrane topology of the reovirus FAST proteins results in extended C-terminal endodomains and comparatively smaller N-terminal ectodomains. The FAST proteins are also nonstructural viral fusion proteins not involved in virus assembly or maturation, and their CTs have evolved to function from the cytosol and not the interior of a virus particle. The extended size of their endodomains and unusual biological properties of the FAST proteins suggested that their CTs likely play a role in the fusion activity of the FAST proteins and that these tails may function in a different manner than the tails of enveloped virus fusion proteins. Based on analyses of both the p10 and p14 FAST proteins, we now show that (i) the membrane-distal portion of the FAST protein CTs play an essential role in the membrane fusion reaction, (ii) the tails are required for pore formation and enhance pore expansion, (iii) FAST protein CTs function both in cis and in trans to promote various stages of the fusion reaction, (iv) the fusion activity of the FAST proteins is contingent upon the amino acid content of the tails and not merely their length or primary sequence, and (v) pore formation is less restricted than syncytiogenesis in regard to the length or amino acid content of the extreme C termini of these tails. These attributes are unique to FAST protein CTs and further highlight the distinct evolutionary origins of the FAST proteins and the enveloped virus fusion proteins, which may in turn reflect divergent mechanisms of action of these distinct classes of viral fusion proteins.
Truncation analysis indicates that the membrane-distal portions of FAST protein CTs play an essential role in fusion pore formation. The progressive truncation of the C-terminal tails from both ARV p10 and RRV p14 resulted in the reduction, and eventual abrogation, of both syncytiogenesis and pore formation. The loss of fusion activity due to the tail truncations occurred independently of the modest effects of these truncations on the cell surface expression of the FAST proteins (Fig. 4). Furthermore, a residual tail of 21 to 24 residues that included the conserved, membrane-proximal polybasic motif present in the endodomains of all the FAST proteins was insufficient for fusion activity. The ability of the FAST proteins to mediate the formation of stable fusion pores therefore requires an extended CT, and the membrane-distal portion of these tails is essential for membrane fusion activity. Various hemifusion assays developed for enveloped virus fusion proteins, which detect hemifusion in the absence of pore formation, have been unsuccessful when applied to analyses of the FAST protein-mediated fusion reaction (our unpublished results). We therefore cannot formally exclude the possibility that FAST protein endodomains may also play some role in the hemifusion stage of the fusion reaction. However, the location of the CTs on the distal side of the membrane, coupled with the fact that the CTs of enveloped viruses generally influence the pore formation or pore expansion stages of the fusion reaction (4, 20, 23, 29, 37), makes it more likely that the membrane-distal portion of FAST protein CTs play a direct, essential role in the formation of stable fusion pores.
In addition to the essential role of the CTs of the FAST proteins in pore formation, the tails also exert an additional enhancing effect on pore expansion. There was a modest though consistent and statistically significant decrease in the extent of syncytium formation induced by p10c90 versus full-length p10. This decrease occurred throughout the time course of syncytium formation, resulting in levels of syncytiogenesis that ranged from 76 to 87% of the levels induced by authentic p10 (Fig. 4 and 8). In contrast, pore formation induced by p10c90 was indistinguishable from that induced by p10 (Fig. 6 and 8), suggesting that the removal of the C-terminal 8 residues from p10 leads to a defect in the expansion of fusion apertures needed for syncytiogenesis. The involvement of FAST protein CTs in pore expansion is further supported by the observation that reextending the truncated, nonfusogenic p10 and p14 constructs with random amino acids restored pore formation to 23 to 26% of the levels induced by the authentic p10 or p14 FAST proteins but had no effect on syncytiogenesis (Fig. 7 and 8). In addition, syncytium formation induced by p14c105 and p14c78S was ∼20% of that induced by p14, while pore formation ranged from ∼42 to 60% (Fig. 7). While we would caution against overinterpreting differences in the efficiencies of these two very different fusion assays, these results are certainly consistent with the concept that tail alterations can exert a more pronounced effect on syncytiogenesis (and, by inference, pore expansion) than pore formation. Taken together, the differential effects of tail alterations on pore formation versus pore expansion (i.e., syncytium formation) imply that FAST protein CTs exert overlapping or independent effects on both of these stages of the fusion reaction.
The truncation and reextension analyses also indicate that the FAST protein membrane fusion reaction is dependent on both cis- and trans-acting activities of the CTs. We recently demonstrated that the p14 protein, expressed in virus-infected or transfected cells, is proteolytically processed in or near the transmembrane domain to generate a soluble CT (50). In cell-cell fusion assays, this soluble endodomain increases the syncytiogenic activity of different fusogens by ∼60 to 80%, functioning in trans to promote events that occur downstream of stable pore formation. Several lines of evidence indicate that the FAST protein endodomains also play a cis-acting role in membrane fusion. First and foremost, the present truncation results indicate that the CT is required for pore formation (Fig. 5 and 7), while the soluble endodomain promotes pore expansion but has no effect on pore formation (50). Second, the soluble endodomain loses all trans-acting activity when scrambled (50), while p14c78S and p10c86S, although delayed in kinetics, retain substantial cell-cell fusion activity (Fig. 7). Third, only a small proportion of p14 is processed to generate the bioactive endodomain fragment (50), suggesting that the majority of the CTs are retained in a cis configuration to serve in the fusion reaction. FAST protein CTs therefore function both in cis and in trans to promote various stages of the membrane fusion reaction. The endodomain of Jagged-1, a membrane-spanning ligand for Notch receptors, exhibits a similar dual cis/trans activity, functioning both in a membrane-tethered form in the context of the full-length protein and as a soluble nucleocytoplasmic signaling polypeptide following cleavage via regulated intramembrane proteolysis (38, 39).
The complex overlapping functions of FAST protein CTs complicate the interpretation of the fusion phenotypes of various FAST protein constructs. For example, the ∼70 to 80% decrease in syncytiogenesis manifested by p14c105 (Fig. 7) inversely correlates with the approximate level of enhanced syncytium formation mediated by the soluble endodomain (50). Since the 20 C-terminal residues deleted in p14c105 include 11 residues that influence the ability of the soluble p14 endodomain to enhance syncytiogenesis (50), the loss of the bioactive trans-acting cytoplasmic fragment could contribute to the p14c105 fusion phenotype. However, since the trans-acting p14 endodomain affects only pore expansion while p14c105 shows reduced pore formation and syncytiogenesis (Fig. 4 and 5), the fusion phenotype of p14c105 (and other constructs with altered CTs) likely reflects the combined loss of both cis- and trans-acting activities that are required for pore formation and that augment pore formation. Parsing out the distinct cis and trans functions of FAST protein CTs and further definitive insights into the complex, multifaceted role of FAST protein CTs in the fusion reaction will require further comprehensive structural and functional analyses.
Results further indicate that the mechanisms by which FAST protein CTs influence pore formation and pore expansion are differentially influenced by the amino acid content versus the length and/or primary sequence of these tails. Replacing the membrane-distal portions of the p10 or p14 CTs with scrambled versions of the deleted sequences substantially restored pore formation and syncytiogenesis, while similar replacements with heterologous random sequences had no such restorative capacity for syncytium formation (Fig. 7 and 8). The essential role of the CT in pore formation and syncytiogenesis is therefore dependent on the amino acid content of the tail and not on the length or primary sequence of the tail. The fact that scrambled sequences only partially restored fusion activity also indicates that the tail exerts a second, nonessential role in the fusion reaction, influencing the efficiency of pore formation and syncytiogenesis in a length- and/or primary sequence-dependent manner. At the same time, random reextensions partially restored pore formation in the absence of syncytium formation, indicating that pore expansion leading to syncytium formation is more sensitive than pore formation to the amino acid content of the membrane-distal region of FAST protein CTs. The corollary is that while there is no linear motif in the membrane-distal portion of the tail that is required for membrane fusion activity, the linear motif(s) in this region may exert a secondary enhancing effect on pore formation and syncytiogenesis.
It is presently unclear exactly how FAST protein CTs function to drive pore formation and enhance pore expansion. The precise role of enveloped virus fusion protein CTs also remains elusive, although several models have been proposed to explain how CTs might influence membrane fusion activity. One possibility is that the CT influences the conformation of the ectodomains. This inside-out signaling model has been speculated or demonstrated to occur in the Env proteins of retroviruses such as Moloney murine leukemia virus, human immunodeficiency virus, and simian immunodeficiency virus (1, 2, 32, 49); in the gB protein of herpes simplex virus (25); and in the F proteins of paramyxoviruses such as simian virus 5 and Nipah virus (3, 52). Sequences in the membrane-distal portions of these CTs or the overall length and hydrophobicity of the tail are presumed to influence interactions with other structural proteins or with other monomers of the fusion protein to inhibit fusion-inducing structural rearrangements of the ectodomain, thereby restricting fusion activity to mature virus particles (37, 52, 54, 55). However, not only are FAST protein CTs fusion inducing rather than inhibitory, but the FAST proteins are also nonstructural proteins with simple, primarily unstructured ectodomains (15). It therefore seems unlikely that the membrane-distal portions of FAST protein CTs could function in both an essential and an ancillary fashion to promote pore formation and expansion via inside-out signaling.
A second hypothesis to explain the involvement of CTs in enveloped virus membrane fusion revolves around the concept of elastic coupling between the ectodomain and the transmembrane domain (33). Dramatic conformational changes in the complex ectodomain structures translate into the tilting of the transmembrane domain, imparting stress on the membrane that leads to a rupture of the hemifusion diaphragm and pore formation and expansion. These transmembrane movements are speculated to require the partial insertion of the CT into the membrane, a process that would be facilitated by the hydrophobicity or presence of an amphipathic helix in the membrane-proximal region of the CT (23, 29, 32). This model, however, is inconsistent with several features of FAST proteins, including the small size of their ectodomains (making it unlikely that FAST proteins use mechanical stress to drive the fusion reaction) and our demonstration that the membrane-distal, but not the membrane-proximal, regions of their CTs promote fusion activity.
Perhaps the most intriguing result from the present study was that stable pore formation and expansion are contingent upon the amino acid content, but not the primary sequence, of the C-terminal portion of the CTs. Aside from conservation in the overall hydropathy of the authentic and scrambled sequences, there were no other obvious conserved features of these functional sequences that were absent from the nonfusogenic random sequences. There are, however, several significant differences in the amino acid compositions of these sequences. For p14 (Table 1), over half of the residues differ by more than twofold between the authentic and random sequences, with the authentic sequence containing a higher percentage of Arg, Ile, Pro, and Ser, while the random sequence contains a higher proportion of Cys, Gly, His, Leu, Met, Phe, and Trp. Authentic and scrambled p14 CTs are therefore more hydrophilic overall than the CT containing the random heterologous sequence (53% versus 42% hydrophilic residues, respectively). This is not the case for p10, where the authentic and scrambled sequences are more hydrophobic than the nonfunctional random sequence (67% versus 42% hydrophobic residues, respectively). How the overall hydropathy of the membrane-distal portions of FAST protein CTs could exert such a profound effect on the fusion activity of p14 is therefore not apparent.
TABLE 1.
Percent amino acid content of p14 and p14c78R FAST protein CTsa
| Amino acid | % Amino acid content |
|
|---|---|---|
| Random | FAST | |
| p14 | ||
| Ala | 4.2 | 2.1 |
| Arg | 0 | 10.6 |
| Asn | 4.2 | 6.4 |
| Asp | 6.4 | 4.2 |
| Cys | 4.2 | 0 |
| Glu | 8.5 | 8.5 |
| Gln | 2.1 | 4.2 |
| Gly | 8.5 | 0 |
| His | 8.5 | 2.1 |
| Ile | 2.1 | 8.5 |
| p14c78R | ||
| Leu | 8.5 | 2.1 |
| Lys | 2.1 | 2.1 |
| Met | 6.4 | 0 |
| Phe | 6.4 | 0 |
| Pro | 8.5 | 19.1 |
| Ser | 2.1 | 12.8 |
| Thr | 4.2 | 2.1 |
| Trp | 4.2 | 0 |
| Tyr | 6.4 | 10.6 |
| Val | 2.1 | 4.2 |
The percent amino acid contents of the C-terminal 47 residues of the p14 and p14c78R CTs are indicated. Underlined residues indicate amino acid residues that differ by more than twofold between the p14 and p14c78R CTs. In disordered structures, residues in boldface type are overrepresented, while those in italic type are depleted.
An intriguing second possibility is that FAST protein CTs function as intrinsically disordered domains. Intrinsically disordered domains are common in proteins involved in regulation and signaling, with their flexible disordered regions facilitating binding to multiple targets, either proteins or membranes (16, 39). Furthermore, a recent survey of single-pass membrane receptors with type I topology found evidence that regions with predicted intrinsic disorder were concentrated in their cytoplasmic endodomains (19). Amino acid composition directly contributes to intrinsic disorder in protein structures, with disordered regions generally rich in Arg, Glu, Gln, Lys, Pro, and Ser and depleted of Cys, Ile, Leu, Phe, Trp, Tyr, and Val (27). For p14, functional CTs are substantially enriched in residues that favor disorder (57% disordered to 25% ordered), a compositional bias that is lost in CTs containing the random sequences (30% disordered to 34% ordered) (Table 1). The same situation applies to p10: 33% are disordered and 16% are ordered in authentic and scrambled sequences, versus 33% disordered to 42% ordered in the random sequences, respectively. Whether the amino acid content dependence of FAST protein CTs reflects the preference for an intrinsically disordered C terminus and how such a property might contribute to an essential role in pore formation and an ancillary role in pore expansion remain to be determined.
Our present results demonstrate that the CTs of the FAST proteins serve multiple independent or overlapping roles in the cell-cell fusion reaction that are influenced by distinct features of the tail. Functioning in cis, the tails exert essential, amino acid content-dependent and auxiliary primary sequence-dependent effects on pore formation (i.e., tail truncations are nonfusogenic, while scrambled reextensions restore pore formation and syncytiogenesis but not to an equivalent level as the authentic primary sequence). CTs also enhance pore expansion, functioning in cis in an amino acid content- and sequence-independent manner (i.e., random reextensions form pores but do not induce syncytium formation) while operating in trans in a sequence-specific, nonessential manner (i.e., scrambling of the soluble endodomain leads to a loss of trans-complementing pore expansion activity). Given the minimal size of the FAST proteins, the disproportionate size of their CTs, and the need for these tails to function from the cytosol and not from within a virus particle, it may not be surprising that the CTs of these virus-encoded cellular fusogens have evolved to serve more than one role in promoting cell-cell fusion and syncytium formation, nor is it surprising that these CTs might serve a different function than the CTs of enveloped virus fusion proteins in the membrane fusion reaction.
Acknowledgments
We thank Jingyun Shou for excellent technical assistance.
This research was supported by grants from the Canadian Institutes of Health Research (CIHR). C.B. was funded by scholarships from the Nova Scotia Health Research Foundation and the Cancer Research Training Program. R.D. was the recipient of a CIHR-RPP Investigators award.
Footnotes
Published ahead of print on 16 September 2009.
REFERENCES
- 1.Abrahamyan, L. G., S. R. Mkrtchyan, J. Binley, M. Lu, G. B. Melikyan, and F. S. Cohen. 2005. The cytoplasmic tail slows the folding of human immunodeficiency virus type 1 Env from a late prebundle configuration into the six-helix bundle. J. Virol. 79:106-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguilar, H. C., W. F. Anderson, and P. M. Cannon. 2003. Cytoplasmic tail of Moloney murine leukemia virus envelope protein influences the conformation of the extracellular domain: implications for mechanism of action of the R peptide. J. Virol. 77:1281-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguilar, H. C., K. A. Matreyek, D. Y. Choi, C. M. Filone, S. Young, and B. Lee. 2007. Polybasic KKR motif in the cytoplasmic tail of Nipah virus fusion protein modulates membrane fusion by inside-out signaling. J. Virol. 81:4520-4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagai, S., and R. A. Lamb. 1996. Truncation of the COOH-terminal region of the paramyxovirus SV5 fusion protein leads to hemifusion but not complete fusion. J. Cell Biol. 135:73-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broer, R., B. Boson, W. Spaan, F. L. Cosset, and J. Corver. 2006. Important role for the transmembrane domain of severe acute respiratory syndrome coronavirus spike protein during entry. J. Virol. 80:1302-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown, C. W., K. B. Stephenson, S. Hanson, M. Kucharczyk, R. Duncan, J. C. Bell, and B. D. Lichty. 2009. The p14 FAST protein of reptilian reovirus increases vesicular stomatitis virus neuropathogenesis. J. Virol. 83:552-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cathomen, T., H. Y. Naim, and R. Cattaneo. 1998. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J. Virol. 72:1224-1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Celma, C. C., M. G. Paladino, S. A. Gonzalez, and J. L. Affranchino. 2007. Importance of the short cytoplasmic domain of the feline immunodeficiency virus transmembrane glycoprotein for fusion activity and envelope glycoprotein incorporation into virions. Virology 366:405-414. [DOI] [PubMed] [Google Scholar]
- 9.Chappell, J. D., R. Duncan, P. P. C. Mertens, and T. S. Dermody. 2005. Orthoreovirus, Reoviridae. Elsevier/Academic Press, London, United Kingdom.
- 10.Chen, B. J., M. Takeda, and R. A. Lamb. 2005. Influenza virus hemagglutinin (H3 subtype) requires palmitoylation of its cytoplasmic tail for assembly: M1 proteins of two subtypes differ in their ability to support assembly. J. Virol. 79:13673-13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chernomordik, L. V., V. A. Frolov, E. Leikina, P. Bronk, and J. Zimmerberg. 1998. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 140:1369-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chernomordik, L. V., and M. M. Kozlov. 2008. Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 15:675-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chernomordik, L. V., J. Zimmerberg, and M. M. Kozlov. 2006. Membranes of the world unite! J. Cell Biol. 175:201-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corcoran, J. A., and R. Duncan. 2004. Reptilian reovirus utilizes a small type III protein with an external myristylated amino terminus to mediate cell-cell fusion. J. Virol. 78:4342-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corcoran, J. A., R. Syvitski, D. Top, R. M. Epand, R. F. Epand, D. Jakeman, and R. Duncan. 2004. Myristoylation, a protruding loop, and structural plasticity are essential features of a nonenveloped virus fusion peptide motif. J. Biol. Chem. 279:51386-51394. [DOI] [PubMed] [Google Scholar]
- 16.Cortese, M. S., V. N. Uversky, and A. K. Dunker. 2008. Intrinsic disorder in scaffold proteins: getting more from less. Prog. Biophys. Mol. Biol. 98:85-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawe, S., J. A. Corcoran, E. K. Clancy, J. Salsman, and R. Duncan. 2005. Unusual topological arrangement of structural motifs in the baboon reovirus fusion-associated small transmembrane protein. J. Virol. 79:6216-6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dawe, S., and R. Duncan. 2002. The S4 genome segment of baboon reovirus is bicistronic and encodes a novel fusion-associated small transmembrane protein. J. Virol. 76:2131-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Biasio, A., C. Guarnaccia, M. Popovic, V. N. Uversky, A. Pintar, and S. Pongor. 2008. Prevalence of intrinsic disorder in the intracellular region of human single-pass type I proteins: the case of the notch ligand Delta-4. J. Proteome Res. 7:2496-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutch, R. E., and R. A. Lamb. 2001. Deletion of the cytoplasmic tail of the fusion protein of the paramyxovirus simian virus 5 affects fusion pore enlargement. J. Virol. 75:5363-5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Earp, L. J., S. E. Delos, H. E. Park, and J. M. White. 2005. The many mechanisms of viral membrane fusion proteins. Curr. Top. Microbiol. Immunol. 285:25-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan, Z., M. L. Grantham, M. S. Smith, E. S. Anderson, J. A. Cardelli, and M. I. Muggeridge. 2002. Truncation of herpes simplex virus type 2 glycoprotein B increases its cell surface expression and activity in cell-cell fusion, but these properties are unrelated. J. Virol. 76:9271-9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer, C., B. Schroth-Diez, A. Herrmann, W. Garten, and H. D. Klenk. 1998. Acylation of the influenza hemagglutinin modulates fusion activity. Virology 248:284-294. [DOI] [PubMed] [Google Scholar]
- 24.Foster, T. P., J. M. Melancon, and K. G. Kousoulas. 2001. An alpha-helical domain within the carboxyl terminus of herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) is associated with cell fusion and resistance to heparin inhibition of cell fusion. Virology 287:18-29. [DOI] [PubMed] [Google Scholar]
- 25.Gage, P. J., M. Levine, and J. C. Glorioso. 1993. Syncytium-inducing mutations localize to two discrete regions within the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B. J. Virol. 67:2191-2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haan, K. M., S. K. Lee, and R. Longnecker. 2001. Different functional domains in the cytoplasmic tail of glycoprotein B are involved in Epstein-Barr virus-induced membrane fusion. Virology 290:106-114. [DOI] [PubMed] [Google Scholar]
- 27.Hansen, J. C., X. Lu, E. D. Ross, and R. W. Woody. 2006. Intrinsic protein disorder, amino acid composition, and histone terminal domains. J. Biol. Chem. 281:1853-1856. [DOI] [PubMed] [Google Scholar]
- 28.Klupp, B. G., R. Nixdorf, and T. C. Mettenleiter. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J. Virol. 74:6760-6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kozerski, C., E. Ponimaskin, B. Schroth-Diez, M. F. Schmidt, and A. Herrmann. 2000. Modification of the cytoplasmic domain of influenza virus hemagglutinin affects enlargement of the fusion pore. J. Virol. 74:7529-7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li, M., C. Yang, and R. W. Compans. 2001. Mutations in the cytoplasmic tail of murine leukemia virus envelope protein suppress fusion inhibition by R peptide. J. Virol. 75:2337-2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long, G., X. Pan, M. Westenberg, and J. M. Vlak. 2006. Functional role of the cytoplasmic tail domain of the major envelope fusion protein of group II baculoviruses. J. Virol. 80:11226-11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melikyan, G. B., R. M. Markosyan, S. A. Brener, Y. Rozenberg, and F. S. Cohen. 2000. Role of the cytoplasmic tail of ecotropic Moloney murine leukemia virus Env protein in fusion pore formation. J. Virol. 74:447-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melikyan, G. B., J. M. White, and F. S. Cohen. 1995. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J. Cell Biol. 131:679-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mulligan, M. J., G. V. Yamshchikov, G. D. Ritter, Jr., F. Gao, M. J. Jin, C. D. Nail, C. P. Spies, B. H. Hahn, and R. W. Compans. 1992. Cytoplasmic domain truncation enhances fusion activity by the exterior glycoprotein complex of human immunodeficiency virus type 2 in selected cell types. J. Virol. 66:3971-3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murakami, T., S. Ablan, E. O. Freed, and Y. Tanaka. 2004. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J. Virol. 78:1026-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Odell, D., E. Wanas, J. Yan, and H. P. Ghosh. 1997. Influence of membrane anchoring and cytoplasmic domains on the fusogenic activity of vesicular stomatitis virus glycoprotein G. J. Virol. 71:7996-8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohuchi, M., C. Fischer, R. Ohuchi, A. Herwig, and H. D. Klenk. 1998. Elongation of the cytoplasmic tail interferes with the fusion activity of influenza virus hemagglutinin. J. Virol. 72:3554-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pintar, A., A. De Biasio, M. Popovic, N. Ivanova, and S. Pongor. 2007. The intracellular region of Notch ligands: does the tail make the difference? Biol. Direct 2:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Popovic, M., A. De Biasio, A. Pintar, and S. Pongor. 2007. The intracellular region of the Notch ligand Jagged-1 gains partial structure upon binding to synthetic membranes. FEBS J. 274:5325-5336. [DOI] [PubMed] [Google Scholar]
- 40.Racine, T., C. Barry, K. Roy, S. J. Dawe, M. Shmulevitz, and R. Duncan. 2007. Leaky scanning and scanning-independent ribosome migration on the tricistronic S1 mRNA of avian reovirus. J. Biol. Chem. 282:25613-25622. [DOI] [PubMed] [Google Scholar]
- 41.Racine, T., T. Hurst, C. Barry, J. Shou, F. S. Kibenge, and R. Duncan. 2009. Aquareovirus effects syncytiogenesis by using a novel member of the FAST protein family translated from a noncanonical translation start site. J. Virol. 83:5951-5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salsman, J., D. Top, C. Barry, and R. Duncan. 2008. A virus-encoded cell-cell fusion machine dependent on surrogate adhesins. PLoS Pathog. 4:e1000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salsman, J., D. Top, J. Boutilier, and R. Duncan. 2005. Extensive syncytium formation mediated by the reovirus FAST proteins triggers apoptosis-induced membrane instability. J. Virol. 79:8090-8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sapir, A., O. Avinoam, B. Podbilewicz, and L. V. Chernomordik. 2008. Viral and developmental cell fusion mechanisms: conservation and divergence. Dev. Cell 14:11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seth, S., A. Vincent, and R. W. Compans. 2003. Mutations in the cytoplasmic domain of a paramyxovirus fusion glycoprotein rescue syncytium formation and eliminate the hemagglutinin-neuraminidase protein requirement for membrane fusion. J. Virol. 77:167-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shmulevitz, M., and R. Duncan. 2000. A new class of fusion-associated small transmembrane (FAST) proteins encoded by the non-enveloped fusogenic reoviruses. EMBO J. 19:902-912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song, C., K. Micoli, and E. Hunter. 2005. Activity of the Mason-Pfizer monkey virus fusion protein is modulated by single amino acids in the cytoplasmic tail. J. Virol. 79:11569-11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spies, C. P., and R. W. Compans. 1994. Effects of cytoplasmic domain length on cell surface expression and syncytium-forming capacity of the simian immunodeficiency virus envelope glycoprotein. Virology 203:8-19. [DOI] [PubMed] [Google Scholar]
- 49.Spies, C. P., G. D. Ritter, Jr., M. J. Mulligan, and R. W. Compans. 1994. Truncation of the cytoplasmic domain of the simian immunodeficiency virus envelope glycoprotein alters the conformation of the external domain. J. Virol. 68:585-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Top, D., C. Barry, T. Racine, C. L. Ellis, and R. Duncan. 2009. Enhanced fusion pore expansion mediated by the trans-acting endodomain of the reovirus FAST proteins. PLoS Pathog. 5:e1000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Top, D., R. de Antueno, J. Salsman, J. Corcoran, J. Mader, D. Hoskin, A. Touhami, M. H. Jericho, and R. Duncan. 2005. Liposome reconstitution of a minimal protein-mediated membrane fusion machine. EMBO J. 24:2980-2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waning, D. L., C. J. Russell, T. S. Jardetzky, and R. A. Lamb. 2004. Activation of a paramyxovirus fusion protein is modulated by inside-out signaling from the cytoplasmic tail. Proc. Natl. Acad. Sci. USA 101:9217-9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White, J. M., S. E. Delos, M. Brecher, and K. Schornberg. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43:189-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wyma, D. J., J. Jiang, J. Shi, J. Zhou, J. E. Lineberger, M. D. Miller, and C. Aiken. 2004. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J. Virol. 78:3429-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wyss, S., A. S. Dimitrov, F. Baribaud, T. G. Edwards, R. Blumenthal, and J. A. Hoxie. 2005. Regulation of human immunodeficiency virus type 1 envelope glycoprotein fusion by a membrane-interactive domain in the gp41 cytoplasmic tail. J. Virol. 79:12231-12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yao, Q., and R. W. Compans. 1995. Differences in the role of the cytoplasmic domain of human parainfluenza virus fusion proteins. J. Virol. 69:7045-7053. [DOI] [PMC free article] [PubMed] [Google Scholar]