

Abstract
Retroviruses express Gag and Pol proteins by translation of unspliced genome-length viral RNA. For some retroviruses, transport of unspliced viral RNA to the cytoplasm is mediated by small regulatory proteins such as human immunodeficiency virus Rev, while other retroviruses contain constitutive transport elements in their RNAs that allow transport without splicing. In this study, we found that the betaretrovirus Jaagsiekte sheep retrovirus (JSRV) encodes within the env gene a trans-acting factor (Rej) necessary for the synthesis of Gag protein from unspliced viral RNA. Deletion of env sequences from a JSRV proviral expression plasmid (pTN3) abolished its ability to produce Gag polyprotein in transfected 293T cells, and Gag synthesis could be restored by cotransfection of an env expression plasmid (ΔGP). Deletion analysis localized the complementing activity (Rej) to the putative Env signal peptide, and a signal peptide expression construct showed Rej activity. Two other betaretroviruses, mouse mammary tumor virus (MMTV) and human endogenous retrovirus type K, encode analogous factors (Rem and Rec, respectively) that are encoded from doubly spliced env mRNAs. Reverse transcriptase-PCR cloning and sequencing identified alternate internal splicing events in the 5′ end of JSRV env that could signify analogous doubly spliced Rej mRNAs, and cDNA clones expressing two of them also showed Rej activity. The predicted Rej proteins contain motifs similar to those found in MMTV Rem and other analogous retroviral regulatory proteins. Interestingly, in most cell lines, JSRV expression plasmids with Rej deleted showed normal transport of unspliced JSRV RNA to the cytoplasm; however, in 293T cells Rej modestly enhanced export of unspliced viral RNA (2.8-fold). Metabolic labeling experiments with [35S]methionine indicated that JSRV Rej is required for the synthesis of viral Gag polyprotein. Thus, in most cell lines, the predominant function of Rej is to facilitate translation of unspliced viral mRNA.
Jaagsiekte sheep retrovirus (JSRV) is the causative agent of a transmissible lung cancer (ovine pulmonary adenocarcinoma) in sheep. For all retroviruses, full-length viral RNAs are transcribed from integrated viral DNA in the nucleus; unspliced viral RNA then is exported to the cytoplasm where it is translated into viral Gag and Gag-Pol polyproteins or packaged as genomes into new virions. At the same time, full-length viral RNA is also spliced in the nucleus to give mRNA(s) for the synthesis of envelope and (for some retroviruses) other proteins. From this perspective, nuclear full-length viral RNA is an unspliced env mRNA precursor. For most cellular mRNAs, splicing of nuclear mRNA precursors is required for export to the cytoplasm. This results from binding of nuclear RNP splicing complexes to the intron-exon junctions during the splicing process, leading to deposition of cellular factors onto the mRNA that facilitate export (32). Unspliced or incompletely spliced RNAs are typically retained in the nucleus.
In order to export unspliced viral RNAs from the nucleus, retroviruses use one of two strategies to overcome the cellular barrier to export of unspliced RNAs (10). For complex retroviruses such as human immunodeficiency virus types 1 and 2 (HIV-1 and -2, respectively) and human T-cell leukemia virus types 1 and 2 (HTLV-1 and -2, respectively), export of full-length RNAs is facilitated by virally encoded trans-acting factors (Rev and Rex, respectively). Rev is encoded by a doubly spliced HIV-1 mRNA that is transported to the cytoplasm by the same mechanism used for spliced cellular mRNAs (10). Once Rev protein enters the nucleus (by way of a nuclear localization signal [NLS] [8, 22]), it mediates the export of full-length HIV-1 RNAs by binding to the RNA at a cis-acting Rev-responsive element (RRE) located in the env gene (19, 41, 54, 61). Rev bound to the RRE interacts with Crm1, a nucleocytoplasmic-transport factor (46), resulting in export of the unspliced viral RNA. Other retrovirus trans-acting factors that mediate unspliced viral RNA export include HTLV Rex (28), human endogenous retrovirus type K (HERV-K) Rec (38), and mouse mammary tumor virus (MMTV) Rem (27, 45).
Other retroviruses, notably simple retroviruses that encode only Gag, Pol, and Env, typically export full-length genomic RNA by recruitment of host cellular factors to viral RNA to mediate export, since they do not encode Rev-like proteins. The best-known example is the cis-acting constitutive transport element (CTE) found at the 3′ end of Mason-Pfizer monkey virus (MPMV) RNA. The MPMV CTE is a highly ordered stem-loop RNA structure (4) that binds to the cellular Tap (NXF1) protein (17), the major nuclear export receptor for cellular mRNAs (10). This binding of Tap, in conjunction with the cofactor p15 (NXT1) (18), facilitates export of unspliced MPMV RNAs from the nucleus. Another cis-acting element with export activity is the direct repeat regions (DRs) of Rous sarcoma virus and Rous-associated virus type 2 (35, 47).
JSRV is a simple retrovirus of the Betaretrovirus genus, whose strategy for unspliced viral RNA export and expression is unknown. Some betaretroviruses (MMTV and HERV-K) encode regulatory proteins necessary for export of full-length RNA (Rem and Rec, respectively). On the other hand, MPMV is also a betaretrovirus, and it contains a CTE. JSRV shows homology to both MMTV and MPMV, and indeed antisera directed against both MMTV and MPMV can recognize JSRV proteins (55, 60). At the nucleotide sequence level, there is higher homology between JSRV and MPMV in the gag and pol region, while there is more homology to MMTV in the env region (9). Thus, JSRV might use an unspliced RNA export and expression strategy resembling that of either MPMV or MMTV, or possibly a combination of both.
In this report, we demonstrate that JSRV encodes a trans-acting factor necessary for Gag protein synthesis, named Rej. Deletion analysis indicated that the necessary sequences for Rej activity map to the Env signal peptide. Further, we identified a pair of doubly spliced env mRNAs that could also encode Rej activity. Interestingly, we show that in most cell lines Rej does not facilitate export of unspliced retroviral RNA, but it is necessary for efficient synthesis of Gag protein. RNA sequences in the 3′ end of the env gene contain a JSRV expression/export element (JREE); the JREE contains both a CTE and sequences that respond to Rej (RejRE).
MATERIALS AND METHODS
Expression constructs.
The different JSRV-derived plasmids used in this study are shown in Fig. 1 and 5. Plasmids pCMV2JS21, which contains a full-length exogenous JSRV provirus, and pCMV2JS21ΔGP (ΔGP), which expresses only the env gene, and SU-env deletion constructs of ΔGP (i.e., ΔGP SUΔx) have previously been described (23, 40, 50). These plasmids are driven by the human cytomegalovirus (CMV) immediate-early promoter. Plasmid pCMV2JS21 SUΔ13-52 is a derivative of pCMV2JS21, encoding the full-length JSRV genome with deletion of Env residues corresponding to amino acids 13 to 52. It was generated by cloning the SUΔ13-52-env deletion from pCMV2JS21ΔGP SUΔ13-52 into the env reading frame of pCMV2JS21 by standard cloning techniques. Plasmid pTN3 is a derivative of pCMV2JS21 in which the env gene has been deleted (nucleotides [nt] 5347 to 7074) and carries only the gag, pro, and pol genes. ΔGP SUΔ85-612 (ΔGPSP) is a deletion of ΔGP designed to encode only the first 84 amino acids of Env, corresponding to the predicted signal peptide (9, 13). Deletion of Env coding sequences from ΔGP to make ΔGPSP was accomplished by overlapping PCR cloning. The BsmI site overlaps the first ATG of the JSRV env gene in plasmid ΔGP, while the SalI site is located in the multiple cloning site, downstream of JSRV 3′ long terminal repeat (LTR) sequences retained in this plasmid. In ΔGPSP, the Env signal peptide residues of JSRV were obtained from plasmid ΔGP by using oligonucleotide primers JSE(BsmI)F (5′-CACAGAATGCCGAAGCGCCGCGCTGGATTCCGG) and JSE(SP-CT)R (5′-GAAAATAGGCGGCCAAAAAGCCGCAGCTGCCCCATT), while the carboxy-terminal portion of the JSRV cytoplasmic tail (CT) (env sequences encoding the last three amino acids and Env stop codon) was amplified with JSE(CT-stop)F (5′-GCTTTTTGGGACGACGCGTGAAGGGTTAAG) and JSE(SalI)R (5′-CGACTCACTATAGGGCGAATTGGGTACCGGG). These two overlapping PCR products were then amplified with JSE(BsmI)F and JSE(SalI)R and subsequently subcloned into the BsmI and SalI sites of ΔGP.
FIG. 1.

JSRV expression constructs. (A) All constructs are driven by the CMV immediate-early promoter (bold arrows). The organization of the JSRV genome, along with its open reading frames, is detailed for the full-length provirus molecular clone of JSRV, pCMV2JS21. pTN3 has the majority of the env gene deleted, except for a portion that maps to the C terminus of Env. For the ΔGP deletions, deletions are shown as gaps. The nomenclature reflects amino acids deleted within the Env protein. The splice donor (sd) and splice acceptor (sa) nucleotide positions for the JSRV env transcript are indicated. The locations of the putative Env signal peptide (SP), the Env surface (SU) and transmembrane (TM) domains, and the Env membrane-spanning sequence (MS) and CT are shown. (B) The influenza virus HA epitope tag was fused immediately downstream of the Env CT domain for the JSRV Env and Env signal peptide expression constructs (ΔGPHA and ΔGPSPHA, respectively).
FIG. 5.

The alternatively spliced env transcripts encode Rej activity. (A) Schematic representation of the doubly spliced env cDNA expression vectors: ΔGPds-envcDNA2 and ΔGPds-envcDNA4 are shown relative to the other expression plasmids shown in Fig. 1. (B) 293T cells were transfected or cotransfected with the plasmids indicated, and at 40 h posttransfection, whole-cell lysates were analyzed on 7.5% SDS-polyacrylamide gels, followed by Western blot analysis with a monoclonal antibody to JSRV CA protein. The arrow indicates the cellular JSRV Gag polyprotein (74 kDa, top panel). Both doubly spliced expression constructs complemented pTN3 for Gag production equivalently to ΔGP and ΔGPSP. In pTN3-transfected cells, a slightly slower-migrating Gag-specific band was also detected, but it was distinct from the Gag polyprotein precursor. β-Tubulin (55 kDa) was used to control for equivalent loading of the cell lysates (bottom panel). (C) Northern blot hybridization analysis using a JSRV env probe indicated the authentic expression of env transcripts from ΔGP (2.4 kb, shown by an arrow) and the truncated env signal peptide from ΔGPSP or the ΔGP-spliced env transcripts expressed from ΔGPds-envcDNA2 and ΔGPds-envcDNA4 (0.7 kb, shown by an arrow).
The hemagglutinin (HA)-tagged version of the Env signal peptide construct, ΔGPSPHA, was constructed similarly to ΔGPSP but using primers JSE(BsmI)F and JSE(SP-CT-HA)R (5′-GTACCGGTCGTCCCAAAAAGCCGCAGCTGCCCCATT) to amplify the Env signal peptide portion and primers JSE(SP-CT-HA)F (5′-GCTTTTTGGGACGACCGGTACCCATACGAC) and JSE(SalI)R to amplify the HA-tag-containing C-terminal portion of the JSRV CT, from plasmid ΔGPHA.
Plasmids expressing the alternatively spliced env cDNAs (ΔGPenv-dscDNA2 and ΔGPenv-dscDNA4 [see Fig. 5A]) driven by the CMV promoter were derived by PCR amplification of JSRV env cDNA sequences, previously inserted into the TOPO-TA cloning vector (described below). JSRV env cDNA sequences were PCR amplified from pCR4-TOPO vectors using JSE(BsmI)F and rejcDNA (r) (5′-CTTAACCCTTCACTCGAGGATATGATATGTC) to obtain all env nucleotides, beginning with the 5′ start translation codon of env, encompassing the internal env splice region, and continuing through the second exon stop codon (see Fig. 4B). The 3′ JSRV LTR and downstream sequences were amplified from plasmid ΔGP using oligonucleotide primers rejcDNA (f) (5′-ATCCTCGAGTGAAGGGTTAAGTCCTGGGAGCTC) and JSE(SalI)R. Both of these PCR amplification products were then subjected to overlapping PCR using JSE(BsmI)F and JSE(SalI)R primers and subsequently subcloned into the BsmI and SalI sites of ΔGP (see Fig. 5A).
FIG. 4.

Identification of alternatively spliced transcripts within the JSRV env gene. (A) RT-PCR analysis for env sequences of cytoplasmic RNA from 293T cells transfected with ΔGP (lane 1). The expected 900-bp band was observed as well as three smaller amplification products (lane 1, bands of <850 bp). 293T cells transfected with pCDNA3.1 were used as a negative control (lane 2); to verify that amplification was from polyadenylated RNAs and reverse transcriptase dependent, an oligo(dT) primer was employed during cDNA synthesis prior to PCR amplification with SU primers (lane 3), and amplification without added reverse transcriptase was performed (lane 4), respectively. ΔGP plasmid DNA control was used as a template in PCR (lane 5); these primers did not amplify fragments of <900 bp. (B) Structure of doubly spliced env mRNAs corresponding to the 650-bp RT-PCR fragments (shown by an arrow in lane 1 of panel A) with the env splice donor (sd) and splice acceptor (sa) nucleotides indicated, as well as the location of the primer pairs (opposing arrows) used in the RT-PCRs. The locations of stop codons are indicated by asterisks. (C) Detection of the alternatively spliced JSRV env mRNAs in JSRV-infected cells and various cell lines by RT-PCR analysis. RNAs from sheep CP cells infected with JSRV particles were used to detect increased levels of spliced env transcripts (650-bp band, shown by an arrow) over endogenous levels (lanes 4 and 3 of left panel, respectively). RNAs from 293T cells transfected with the molecular clone of JSRV, pCMV2JS21, or RNAs from NIH 3T3, 208F, or HeLa cells transfected with ΔGP were used to detect the env (900-bp band) and the spliced env (650-bp band, shown by an arrow) transcripts (lane 2, left panel; lanes 2 and 4, middle panel; and lane 3, right panel). JSRV env-specific RT-PCRs used RNAs from cells transfected with the negative-control plasmid, pCDNA3.1 (lanes 3, left and middle panels, and lane 2, right panel).
The plasmid pDM128 containing the chloramphenicol acetyltransferase (CAT) gene and RRE under the control of the simian virus 40 promoter was provided by Thomas Hope (24). To make pDM/JREE, a portion of JSRV env and 3′ LTR was amplified from pCMV2JS21 by PCR with the primers 5′-AAAAAAAAAGGATCCGATTTTCTAAAGATGAGAGT and 5′-AAAAAAAAAGGATCCTTATTACAATGCTATATT. The PCR product was cloned into pCR4-TOPO (Invitrogen), and then the insert was extracted with BamHI digestion followed by ligation with BamHI-digested pDM128. The BamHI site at the junction of the JSRV 3′ LTR and downstream cellular sequences was then removed by site-directed mutagenesis methods (see Fig. 8A).
FIG. 8.

Nucleotides in the 3′ region of env (the JREE) are Rej responsive to cytoplasmic export of intron-containing RNA only in human 293T cells. (A) Schematic diagram of pDM128 and pDM/JREE reporter plasmids. The locations of splice donor and acceptor sites (sd and sa, respectively) are shown, as well as coding sequences for CAT, the HIV-1 RRE (RRE), and the JREE. The plasmids are driven by a simian virus 40 early promoter (SV40-P) and utilize the cleavage/poly(A) site of the HIV-1 LTR. (B) pDM128 and pDM/JREE were transfected into human 293T cells, and 48 h posttransfection, CAT activities were measured as described in Materials and Methods. As a positive control, pDM128 was cotransfected with the HIV-1 Rev expression vector, pRev1. To test if the JREE was responsive to Rej, pDM/JREE was cotransfected with the JSRV Rej expression vector, ΔGP, at a ratio of 3 μg to 1 μg DNA, respectively (left panel). This experiment was also performed with variable amounts of ΔGP DNA, ranging from 0.5 μg to 3 μg, while keeping pDM/JREE DNA amounts constant (3 μg) (right panel). To control for transfection efficiency, the pRL-null vector encoding Renilla luciferase was also cotransfected with each of the pDM CAT reporters and the CAT activities were normalized for luciferase activities measured on portions of each transfected cell extract. The results from triplicate experiments are shown (error bars represent standard deviations), and the P value (Student's t test) for the difference between the activities of pDM/JREE and pDM/JREE (cotransfected with Rej; ΔGP) is indicated. (C) Cytoplasmic RNA was isolated from 293T cells 16 h after transfection with the combinations of pDM CAT reporters indicated and subjected to agarose gel electrophoresis and Northern blot hybridization with a 32P-labeled HIV-1 LTR probe followed by phosphorimager analysis. Regions of the blot corresponding to the spliced and unspliced RNA in cytoplasmic RNA are shown (top panel). The blot was stripped and rehybridized with a GAPDH probe (1.2-kb band, arrow, middle panel) and a JSRV env probe (2.4-kb band, arrow, bottom panel) to control for equal loading and expression of rej-containing transcripts, respectively. Phosphorimaging analysis (bar graph) represents the ratio of cytoplasmic unspliced pDM/JREE RNA to pDM128 in the presence or absence of Rej in 293T cells. (D) CAT activity assay in human 293, rat 208F, mouse 3T3, and sheep CP cells using the same constructs and methods as those in the assay in 293T cells (B), described above. Note that a dose-response (for pDM/JREE plus variable amounts of ΔGP DNA) CAT assay was also performed in human 293 cells, identical to what was described above (top right panel).
Cell lines, DNA transfections, and infections.
Sheep choroid plexus (CP); rat 208F; and human 293T, 293, and HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Mouse NIH 3T3 cells were grown in DMEM supplemented with 10% calf serum. All cells were cultured at 37°C, 95% humidity, and 5% CO2. DNA transfections of 293T or 293 cells were performed using approximately 2.5 × 106 cells (24 h postseeding, 10-cm dish, ∼65% confluent) with variable amounts of DNA, ranging from 28 or 14 μg of DNA, as indicated, using the CalPhos mammalian transfection kit (BD Biosciences), as recommended by the manufacturer. For cotransfections, typically 14 μg of each plasmid DNA was used, unless indicated. Other cell lines were also seeded at densities to obtain 65% confluent growth 24 h prior to DNA transfection.
Released JSRV particles were obtained by transfection of pCMV2JS21 or derivative plasmids into 293T cells as previously described (50, 51). Briefly, cells were transfected with pCMV2JS21 DNA (28 μg/10-cm dish), as described above. Medium was changed at 12 h, and cell supernatants were harvested 48 h after the first medium change. The medium was filtered through a 0.45-μm-pore-size filter, and virus was concentrated by ultracentrifugation at 25,000 rpm in a Beckman SW41 Ti ultracentrifuge rotor for 1 h at 4°C. The resulting viral pellet was resuspended in 200 μl TNE buffer (100 mM NaCl, 10 mM Tris, 1 mM EDTA). For infection of sheep CP cells, viral stocks were adjusted to a 5-ml total volume in DMEM-10% FBS and 8 μg/ml Polybrene. CP cells (6.8 × 105 cells/10-cm dish) were seeded 24 h prior to infection, and then medium was replaced with a 5-ml viral preparation. The cells were then incubated for 2 h at 37°C and supplemented with 5 ml DMEM-10% FBS and 8 μg/ml Polybrene for 24 h at 37°C before being harvested for RNA isolation.
RNA fractionations and protein isolation.
Human 293T or 293 cells were transfected with plasmid DNA, and approximately 16 h or 40 h posttransfection, cells were harvested. Briefly, the growth medium was aspirated, cells were gathered with 5 ml 1× phosphate-buffered saline (PBS) (pH 7.3) and collected into a 15-ml conical tube, plates were washed with an additional 5 ml 1× PBS buffer, and the wash was collected as well. Cells were pelleted by centrifugation at 1,000 rpm (600 × g) at 4°C for 5 min in a Beckman Coulter model 6R centrifuge, washed with 5 ml 1× PBS buffer, resuspended in 10 ml 1× PBS buffer, and divided into two equal parts for isolation of total or cytoplasmic/nuclear RNA. For the isolation of total RNA, the supernatant was removed and 200 μl of RLT buffer (RNeasy kit lysis buffer [Qiagen, Valencia, CA]) was added. RNA fractions were obtained by completely removing the supernatant and resuspending cell pellets in 200 μl lysis-extraction buffer (10 mM HEPES, pH 7.6, 3 mM MgCl2, 40 mM KCl, 2 mM dithiothreitol, 5% glycerol, 0.5% NP-40, 0.5 mM phenylmethylsulfonyl fluoride, and 200 U/μl RNase inhibitor) (52) followed by incubation for 2 to 3 min on ice. Nuclei were pelleted at 2,400 rpm for 5 min at 4°C. Then, 30 μl of supernatant was removed for cytoplasmic protein-fractionation control analysis (see below); RNA was isolated from the remaining 170 μl of supernatant (cytoplasmic fraction) with the RNeasy kit (Qiagen, Valencia, CA) as recommended by the manufacturer, and cytoplasmic RNA was eluted twice in 30 μl RNase-free water. The nuclei (pellet) were aspirated of any residual supernatant, washed with 400 μl of lysis-extraction buffer, and pelleted as before. The supernatant was carefully aspirated, 200 μl of nuclear extraction buffer C (20 mM HEPES, pH 7.9, 450 mM NaCl, 1 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, and 200 U/μl RNase inhibitor) (58) was added, and the nuclear pellet was lysed by repetitive pipetting up and down. Nuclear samples were then transferred to a 1.5-ml tube and centrifuged at 14,000 rpm for 10 min at 4°C to pellet cell debris. Again, 30 μl of supernatant was removed for nuclear protein-fractionation control analysis (see below); RNA was isolated from the remaining 170 μl of supernatant (nuclear fraction) with the RNeasy kit (Qiagen, Valencia, CA) as recommended by the manufacturer, and nuclear RNA was also eluted twice in 30 μl RNase-free water.
For preparation of protein lysates, cells were harvested at 40 h posttransfection and cell pellets were lysed in 200 μl triple-detergent buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.02% sodium azide, 0.1% sodium dodecyl sulfate [SDS], 1.0% NP-40, 0.5% sodium deoxycholate, 20 mM sodium fluoride, 1 mM sodium orthovanadate, and 1 mM phenylmethylsulfonyl fluoride) for 20 min on ice, followed by centrifugation at 14,000 rpm in a microcentrifuge at 4°C for 30 min. Lysate supernatant (or supernatant from fractionation buffers [see above]) was collected and mixed with SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 5% sucrose, 0.012% bromophenol blue, and 5% β-mercaptoethanol). Concentrated cell supernatants (i.e., pelleted virus particles) were resuspended in a volume of SDS sample buffer equivalent to that for resuspended cell lysates. Both preparations were boiled for 5 min, and the supernatant was collected after centrifugation for 1 min in a microcentrifuge and subsequently used for SDS-polyacrylamide gel electrophoresis (SDS-PAGE), as previously described (39, 48).
RT-PCR and cDNA cloning.
Total or cytoplasmic RNA was obtained as described above and treated with amplification-grade DNase I (Invitrogen) prior to reverse transcriptase PCR (RT-PCR). JSRV env cDNAs were generated with the Titan One Tube RT-PCR system (Roche Applied Science, Indianapolis, IN) using the JSRV specific primer set env5348(f) and env6250(r) (5′-ATGCCGAAGCGCCGCGCTGGAT and 5′-CATGAATGGATACGGCACGC, respectively [900-bp product]) as recommended by the manufacturer. The RT-PCR was performed in a Stratagene Robocycler as follows: cDNA synthesis at 50°C for 30 min followed by 35 cycles of 94°C for 30 s, 53°C for 1 min, and 68°C for 1 min with a final extension of 68°C for 10 min. Identical RT-PCRs were also performed except that the primer poly(dT)17(r) was added in the cDNA synthesis step before the addition of env5348(f) and env6250(r) to the reaction mixture in the PCR phase, to ensure amplification of only polyadenylated cDNA transcripts. This JSRV primer set encompasses the majority of SU sequences in Env (see Fig. 4B). Primer pairs that detect β-actin unspliced pre-RNA, ActE5(f) and ActI5(r) (5′-TGGCACCCAGCACAATGAAG and 5′-TGGATGTGACAGCTCCCCAC, respectively [105-bp product]), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) spliced RNA, gapdh(f) and gapdh(r) (5′-GGTGAAGGTCGGAGTCAACG and 5′-GTTGAGGTCAATGAAGGGGTC, respectively [110-bp product]), were also used (as a method secondary to immunoblot analysis [see below]) to control for nuclear/cytoplasmic separation and/or RNA loading (33). A second primer set for GAPDH RNA, hGAPDH371(f) and hGAPDH810(r) (5′-CTGGCGTCTTCACCACCATGGA and 5′-CAGGTCCACCACTGACACGTT, respectively [440-bp product]), was also used in these studies.
All PCR-amplified products were separated on a 2% agarose gel and visualized by ethidium bromide staining using a FluorChem SP imaging station (Alpha Innotech, San Leandro, CA). Bands appearing smaller than the expected size of 900 bp [defined by oligonucleotide primer pair env5348(f) and env6250(r)] were excised, and DNA was recovered and cloned using the TOPO-TA cloning system (Invitrogen). Inserts were sequenced in both directions with oligonucleotide M13 (r) (5′-CAGGAAACAGCTATGAC) and M13 (f) (5′-CTGGCCGTCGTTTTAC) primers using the Big Dye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA) as recommended by the manufacturer and elucidated by the University of California, Irvine, DNA sequencing core.
Western blotting and antibodies.
Protein lysates and cell supernatants (containing released virus particles) from transfected 293T or 293 cells were harvested as described above and analyzed by Western blotting essentially as previously described (39, 48). The primary antibody (anti-JSRV CA hybridoma C6 supernatant [monoclonal antibody]) was incubated with the membrane overnight at 4°C followed by three washes in Tris-buffered saline-Tween 20 (20 mM Tris-HCl, pH 7.6, 137 mM NaCl, and 0.1% Tween 20) (TBST) for 5 min each. The anti-CA hybridoma was generated by a commercial source (Genemed Synthesis, Inc.) using bacterially expressed JSRV CA protein. The secondary antibody (horseradish peroxidase-conjugated anti-mouse immunoglobulin G [IgG; GE Healthcare, Piscataway, NJ]) was diluted 1,000-fold in TBST containing 5% bovine serum albumin (BSA) and incubated with the membrane for 1 h at room temperature prior to three additional 5-min washes. All steps were performed with shaking. SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL) was used to detect antibody binding following the recommendations of the manufacturer, and detection used a FluorChem SP imaging station (Alpha Innotech). Western blot analysis was also used to validate the complete and authentic separation of cellular fractions by using primary antibodies specific to protein isolated from cytoplasmic and nuclear extracts (β-tubulin and lamin A/C, respectively). Briefly, equal protein fraction lysates (prepared as described above) were loaded onto 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes by standard methods. Membranes were blocked overnight in TBST containing 5% (wt/vol) powdered milk, incubated with primary rabbit anti-lamin A/C (Cell Signaling Technology, Danvers, MA) or primary rabbit anti-β-tubulin antibody (1:1,000 dilution in TBST) for 1 h at room temperature, washed, and then reacted with secondary antibody and detected via chemiluminescence as described above.
Northern blot analysis.
RNA fractions isolated from transfected 293T or 293 cells were analyzed by Northern blot analysis. Briefly, RNA (7 μg) was denatured in glyoxal-dimethyl sulfoxide loading dye (Ambion, Austin, TX) at 50°C for 30 min, subjected to agarose gel (0.9%) electrophoresis, and transferred to nylon membranes (Hybond-N Plus; GE Healthcare) using transfer buffer (Ambion) as recommended by the manufacturer. Membranes were incubated with rotation in hybridization buffer (GE Healthcare) at 42°C for at least 30 min followed by the addition of 32P-labeled JSRV gag- or mammalian GAPDH-specific probes (5 × 105 cpm/ml; carried in 4 mg salmon sperm DNA) and incubated at 58°C for 12 to 24 h. Membranes were subsequently washed two times in high-stringency wash buffer (300 mM NaCl, 30 mM sodium citrate, 0.1% SDS) at room temperature for 10 min and then washed two times in low-stringency wash buffer (15 mM NaCl, 1.5 mM sodium citrate, 0.1% SDS) at 55°C for 20 min. Membranes were exposed to phosphorimager plates, followed by phosphorimager analysis on an ABI Typhoon phosphorimager with ImageQuant version 4.2 software (Molecular Dynamics, Sunnyvale, CA). PCR-generated gag (nt 929 to 1110 [49]) or plasmid GAPDH DNA templates were labeled with [α-32P]dATP (Perkin-Elmer, Waltham, MA) by random hexamer-Klenow exonuclease using the DECA Prime II kit (Ambion), as recommended by the manufacturer. For Northern blot assays with pDM128 and pDM/JREE, a 32P-labeled probe corresponding to the HIV-1 LTR gene was made by digesting pDM128 with SacI and KpnI, followed by random hexamer-Klenow exonuclease using the DECA Prime II kit (Ambion), as recommended by the manufacturer. The probe for GAPDH was used to confirm the amount and quality of transferred RNA on the membranes.
Immunofluorescence.
The localization of HA epitope-tagged Env and the Env signal peptide in transfected cells was analyzed by indirect immunofluorescence assays. Briefly, 208F cells were seeded at 1.0 × 105 cells/well in four-well chamber slides and incubated for 24 h, followed by transfection with 1 μg of plasmid DNA, as previously described. At 48 h posttransfection, cells were washed with 1× PBS, dried, and fixed with 4% paraformaldehyde for 20 min. Cells were then permeabilized with 0.5% Triton X-100 for 5 min, washed, and blocked with 1% BSA in PBS for 30 min, prior to incubation with rabbit polyclonal antibody to HA tag (diluted 1:125 in 1× PBS containing 1% BSA [Sigma, St. Louis, MO]) and mouse monoclonal anti-nucleophosmin (B23; diluted 1:500; Sigma, St. Louis, MO), for 1 h at 37°C. After being washed with 1× PBS, cells were incubated with goat anti-rabbit IgG conjugated to fluorescein isothiocyanate (FITC) (diluted 1:200 in PBS containing 1% BSA [Sigma]) and donkey anti-mouse IgG conjugated to phycocyanin (diluted 1:200 in PBS containing 1% BSA [Sigma]) for 30 min at 37°C. After being washed with PBS, cells were covered with VectaShield (Vector Laboratories, Burlingame, CA) plus DAPI (4′,6-diamidino-2-phenylindole) and observed using a fluorescent microscope.
Metabolic labeling.
The JSRV expression constructs pCMV2JS21 (wild type), pCMV2JS21 SUΔ13-52, and pCDNA3 (control plasmid) were transfected (or cotransfected; pCMV2JS21 SUΔ13-52 with ΔGP or ΔGPSP) into 293T cells, and at 16 h or 40 h posttransfection, the cells were starved for 1 h with l-methionine and l-cysteine-deficient DMEM (Cellgro, Herndon, VA). The cells were then pulse-labeled with [35S]methionine and [35S]cysteine (200 μCi/ml) (Perkin-Elmer) for 20 min at 37°C. After the pulse-label, the cells were harvested, washed with PBS twice, and then lysed in 200 μl of ML lysis buffer (50 mM Tris [pH 7.5], 1% Triton X-100, 1% sodium deoxycholate, 150 mM NaCl, and 0.1% SDS). The cell lysates were incubated for 20 min on ice, and cell debris was removed by centrifugation at 20,000 × g for 20 min at 4°C. The viral proteins were immunoprecipitated from 150 μl of the supernatants using a polyclonal rabbit anti-CA serum. To normalize the amount of proteins used for immunoprecipitation, 5 μl of each sample was separated by SDS-PAGE and β-tubulin was detected by Western blotting using rabbit anti-β-tubulin (Cell Signaling Technology, Danvers, MA), as described above.
CAT and luciferase assays.
Transient transfections were performed on six-well plates (293T or 293 cells) or 6-cm tissue culture dishes (NIH 3T3, 208F, and CP-1 cells). For each well, 6.5 μg of plasmid DNA (4.5 μg of CAT reporter plasmids, pDM128 or pDM/JREE; 1.5 μg of pRev1 or ΔGP; and 0.5 μg of pRL-null) was used. The control plasmid, pCDNA3.1, was used to equalize the total amount of DNA transfected for all transfections. For each 6-cm-diameter tissue culture dish, 13 μg of plasmid DNA with the same plasmids described above was used. The transfections were performed using the CalPhos mammalian transfection kit for 293, 293T, NIH 3T3, and 208F cells and Lipofectamine 2000 transfection reagent (Invitrogen) for CP-1 cells. After 48 h, transfected cells were washed with 1× PBS and the same numbers of cells were separated into two tubes for CAT assay and luciferase assay.
The CAT assay was performed with the Fast CAT assay kit (Molecular Probes) following instructions from the manufacturer. In brief, the cells were lysed with 100 μl of 0.25 M Tris-HCl (pH 7.4) and 55 μl of the cleared lysates was mixed with 15 μl of Fast CAT substrate and 10 μl of 9 mM acetyl coenzyme A. After incubation for a fixed period of time depending on the cells, all reactions were stopped by adding ice-cold ethyl acetate. The samples were spotted on the silica gel thin-layer chromatography plates (PE SIL G/UV; Whatman), and the acetylated products were separated by chromatography. The separated products on the plate were visualized by photography under UV illumination and quantified with AlphaEase FC. Renilla luciferase activity was also measured to adjust for transfection efficiency. The procedures for the luciferase assay using the dual-luciferase reporter assay system (Promega) were described previously (44).
RESULTS
A trans-acting factor encoded in the env region is necessary for expression of Gag polyprotein.
Since JSRV has homology with MMTV in the env region, we hypothesized that JSRV may also encode a Rem-like factor required for synthesis of JSRV Gag protein. MMTV Rem is encoded from a doubly spliced mRNA from the env region; MMTV deletion mutants of Rem are incapable of Gag polyprotein synthesis (45). We deleted env coding sequences (with the exception of a short stretch from the 3′ end) from a human CMV promoter-driven full-length JSRV expression plasmid (pCMV2JS21 [50]) to give the plasmid pTN3 (Fig. 1A). We then tested the ability of pTN3 to encode synthesis of Gag protein by transient transfection in human 293T cells followed by SDS-PAGE and Western blotting of cell lysates and cell supernatants with a monoclonal antibody specific for JSRV capsid (CA) protein (Fig. 2A). pCMV2JS21 (but not the empty vector pCDNA3.1) transfection resulted in expression of Gag polyprotein (74 kDa) in cell lysates and release of virions with cleaved CA (27-kDa) protein into the supernatant as expected. In contrast, transfection with pTN3 alone did not result in detectable Gag polyprotein or cleaved CA, either in cell extracts or in released virus, even though the Gag-Pol reading frame is intact in this plasmid. These results indicated that JSRV sequences within the env region are required for efficient Gag protein synthesis and virion production.
FIG. 2.

JSRV encodes a trans-acting factor necessary for Gag synthesis. 293T cells were transiently transfected (or cotransfected) with the plasmids indicated, and at 40 h posttransfection, intracellular JSRV Gag polyprotein and/or JSRV particle production (in whole-cell lysates or cell supernatants, respectively) was resolved on 7.5% (lysate) or 10% (supernatant) SDS-polyacrylamide gels followed by Western blotting with a monoclonal antibody to JSRV CA. (A) Transfection with pCMV2JS21, pTN3, and pCDNA3.1. Intracellular JSRV Gag polyprotein (lysate, 74 kDa) and released virion CA (supernatant, 27 kDa) are indicated. A slower-migrating intracellular Gag-specific band (80 kDa) was also detected in cell lysates from both pCMV2JS21- and pTN3-transfected 293T cells and may be a minor nonfunctional form of Gag polyprotein (lanes 2 and 3). (B) Cotransfection of pTN3 with the JSRV env expression construct ΔGP. Intracellular Gag and released virion CA are shown. (C) Cotransfection of pTN3 with different internal deletions of ΔGP. Gag polyprotein in the cellular lysates only is shown. In this initial survey, equivalent amounts of cell lysates were loaded into each lane. (D) Cotransfection of pTN3 with the signal peptide expression construct ΔGPSP rescues Gag synthesis as efficiently as does full-length ΔGP (top panel). To control for equal loading, cell lysates were immunoblotted for β-tubulin (arrow, middle panel). Expression of the processed JSRV Env HA-tagged protein (from ΔGPHA; TM HA, 40 kDa) or from the JSRV Env signal peptide (HA-tagged) region of Env (from ΔGPSPHA; SP HA, 15 kDa) from cotransfection with pTN3 is shown in the cell lysates (bottom panel).
If the JSRV env region encodes a trans-acting factor analogous to MMTV Rem, we hypothesized that coexpression of the JSRV env gene would rescue the ability of pTN3 to express Gag protein. Therefore, we cotransfected pTN3 and an env-only expression construct (pCMV3JS21ΔGP [ΔGP] [Fig. 1A]) into 293T cells and found that ΔGP was able to complement pTN3 for intracellular Gag protein synthesis and virus production (Fig. 2B). ΔGP also could complement pTN3 for Gag protein synthesis and JSRV particle production in murine NIH 3T3 fibroblasts (not shown). These results indicated that the JSRV env region encodes a trans-acting factor necessary for synthesis of Gag protein. We have termed this activity/factor Rej, for regulator of JSRV expression.
N-terminal Env coding sequences are sufficient for Rej activity.
In order to identify the region of the env region necessary for Rej activity, we employed a series of in-frame deletions of ΔGP that we have described previously (23) (Fig. 1A). These deletions were tested for Rej activity by cotransfection with pTN3 into 293T cells, followed by analysis for Gag protein production (Fig. 2C). Deletion of the most-amino-terminal amino acids of Env abolished the ability of ΔGP to complement pTN3 for Gag protein synthesis (Fig. 2C, lanes 5 and 6, corresponding to deletion of residues 13 to 102). In contrast, deletion of amino acids downstream of residue 103 did not abolish Rej activity (Fig. 2C, lanes 7 and 8, corresponding to deletion of residues 103 to 402), although the amount of detectable Gag was reduced somewhat in comparison to the wild type. The residues in this region (between 103 and 402) of Env might stabilize Rej protein or contribute to optimal function. Nonetheless, Rej activity mapped to the amino-terminal 100 amino acids of the Env coding sequences—largely corresponding to the putative signal peptide for Env (residues 1 to 77 [9, 13]). To test if these sequences were sufficient for Rej activity, we generated a derivative of ΔGP that expressed only the JSRV Env signal peptide (residues 1 to 84, ΔGP Δ85-612, also called ΔGPSP [Fig. 1A]), as well as an influenza virus HA epitope-tagged version of the Env signal peptide (ΔGPSPHA, Fig. 1B). Cotransfection of pTN3 and ΔGPSPHA resulted in expression of Gag protein (Fig. 2D), which indicated that the Env coding sequences corresponding to the amino-terminal signal peptide are necessary and sufficient for Rej activity.
The Env signal peptide localizes primarily to the nucleus.
Since the isolated JSRV Env signal peptide showed Rej activity, we hypothesized that it would have subcellular localization patterns similar to those of other retroviral accessory proteins that facilitate unspliced RNA nuclear export and/or Gag protein synthesis (11, 27, 38, 45, 56). In particular, the MMTV Rem protein was shown to localize primarily to the nucleoplasm of cells when expressed as a fusion with green fluorescent protein (27, 45). Moreover, Mertz et al. (45) showed that a Rem truncation consisting of the N-terminal 100 amino acids fused to green fluorescent protein localized entirely to the nucleus. To study the cellular localization of JSRV proteins with Rej activity, we employed influenza virus HA epitope-tagged versions of full-length Env protein or the signal peptide (ΔGPHA and ΔGPSPHA, Fig. 1B). Use of the epitope-tagged proteins was necessary since an antibody for the N terminus of JSRV Env that reliably detects proteins in Western blots or immunofluorescence is not currently available. Rat 208F cells were transiently transfected with the epitope-tagged expression constructs or with an empty vector control (pCDNA3.1), and fixed cells were subjected to immunofluorescence microscopy after being stained with an FITC-conjugated anti-HA antibody (Fig. 3). The cells were also stained with DAPI to show locations of the cell nuclei. As expected, cells transfected with pCDNA3.1 did not show any HA-specific fluorescence (Fig. 3, bottom panels). The full-length epitope-tagged Env protein (ΔGPHA, Fig. 3, top panels) showed predominantly cytoplasmic staining, with concentration at the periphery, characteristic of the plasma membrane localization of JSRV Env (23, 26). In contrast, the epitope-tagged signal peptide-only construct (ΔGPSPHA, Fig. 3, middle panels) showed localization exclusively within the nucleus with some subnuclear concentration that corresponds to nucleoli, as demonstrated by staining with the nucleolar marker nucleophosmin (B23) (7). This result was consistent with the localization patterns of other Rev-like proteins that are predominantly localized in the nucleus (11, 27, 38, 45, 56). Analysis of the JSRV Env signal peptide sequences revealed the presence of several motifs present in other RNA export proteins (see below and Fig. 6A).
FIG. 3.

The JSRV Env signal peptide localizes to the cell nucleus. 208F cells were transfected with the expression constructs for HA-tagged JSRV Env, HA-tagged JSRV Env signal peptide, or empty vector control (ΔGPHA [top panels], ΔGPSPHA [middle panels], or pCDNA3.1 [bottom panels], respectively) and grown on chamber slides for 40 h prior to fixation and incubation with an FITC-conjugated polyclonal antibody to HA and a phycocyanin-conjugated monoclonal antibody to nucleophosmin (B23). To visualize the nuclei of all cells, the cells were also stained with DAPI. Immunofluorescence images were recorded at excitation wavelengths of 350 nm (DAPI), 490 nm for FITC (HA), and 620 nm for phycocyanin (B23); individual and merged images are shown. Bar, 5 μm. Magnification, ×630.
FIG. 6.

Domain structures of the signal peptide and the putative translation products of the doubly spliced JSRV env transcripts. (A) The JSRV Env signal peptide sequence is diagramed, and the locations of predicted sequence motifs are indicated by arrows under the diagram. (B) Amino acid composition of the predicted translations from the doubly spliced env transcripts. The locations of the alternative splice donor and splice acceptor sites are indicated, as well as the location of the stop codon (asterisks) within the second exons (shaded).
Identification of internally spliced env mRNAs with Rej activity.
Since MMTV Rem and HERV-K Rec proteins are encoded by doubly spliced mRNAs from the env region, we tested if JSRV Rej might also be encoded by a doubly spliced mRNA. In particular, we sought identification of a second splice event within the env gene. Therefore, we transfected 293T cells with the ΔGP Env/Rej expression plasmid and isolated cellular (cytoplasmic) RNA for RT-PCR analysis. RT-PCR was carried out with a reverse oligonucleotide primer corresponding to the middle of env (nt 6231 to 6250) and a forward primer from the 5′ end (nt 5348 to 5369, Fig. 4B). A major RT-PCR product of 900 bp was detected (Fig. 4A, lane 1) that corresponded to an unspliced JSRV env transcript; this fragment comigrated with a PCR product from ΔGP DNA using the same primers (Fig. 4A, lane 5). The RT-PCR product was specific for JSRV since cells transfected with the backbone vector lacking JSRV sequences (pCDNA3.1) did not show any RT-PCR products (Fig. 4A, lane 2) and was reverse transcriptase dependent (Fig. 4A, lane 4). Interestingly, in addition to the major 900-bp RT-PCR product from the ΔGP-transfected cells, several smaller products of ca. 800, 650, and 525 bp were detected (Fig. 4A, lane 1); these appeared to be specific RT-PCR products since PCR amplification of ΔGP DNA with the same primers did not yield them (Fig. 4A, lane 5). These smaller RT-PCR products were also obtained when the first reverse transcriptase step employed an oligo(dT) primer, followed by PCR amplification with the JSRV env primers (Fig. 4A, lane 3), which further indicated that they were derived from polyadenylated RNAs.
The smaller RT-PCR products were cloned into TA cloning vectors and sequenced. The sequence analysis revealed that the 650-bp RT-PCR fragment was actually two fragments corresponding to internal splicing events within the env sequences utilizing the same splice donor site at nt 5548 and two closely spaced splice acceptor sites at nt 5785 and 5803 (Fig. 4B). It was noteworthy that these two transcripts had the canonical GT-AG nucleotides at the splice donor and acceptor junctions, which supported the conclusion that they represented an authentic splicing event. The cDNAs corresponding to these internally spliced RNAs were designated ds-env cDNA2 and ds-env cDNA4. Moreover, RT-PCRs using a reverse primer that anneals to the nucleotides immediately adjacent to the internal env splice site on both the 5′ and 3′ ends and using a forward primer upstream from the native env splice donor revealed that the 650-bp RT-PCR fragments are truly doubly spliced env transcripts (data not shown). Translation of doubly spliced env mRNAs containing these second splices would yield proteins that share the N-terminal signal peptide sequences of the Env precursor protein up to residue 66 (the first coding exon) fused to 17 (ds-env cDNA2) or 12 (ds-env cDNA4) amino acids from the second coding exon (out of the env reading frame) before a termination codon is encountered (Fig. 4B; see also Fig. 6B). Doubly spliced mRNAs with these internal env splices would be candidate Rej mRNAs since they encode a substantial part of the Env signal peptide, where Rej activity is localized.
Sequencing of the 525-bp fragment revealed that it was also two fragments that resulted from internal env splicing. However, these fragments resulted from the utilization of a splice donor site in env at nt 5448 but used the same splice acceptor sites at nt 5785 and 5803 described above. The deduced translations from these constructs would encode proteins of 42 and 37 amino acids, respectively, again sharing the same sequences of JSRV Env in exon 1, but would have amino acids from exon 2 different than those of the env reading frame before a termination codon (data not shown). These mRNAs encode less than one-half of the Env signal peptide and therefore were not tested for Rej activity. The 800-bp fragment had the lowest intensity (as detected by ethidium bromide staining) of all the subfragments in RT-PCR, and our attempts to clone this fragment were not successful.
We also tested if cell lines other than 293T also showed evidence for internal env RNA splicing or doubly spliced transcripts. Murine NIH 3T3 and rat 208F fibroblasts, human HeLa cervical carcinoma cells, and sheep CP cells were transiently transfected with ΔGP or pCDNA3.1, and cellular RNAs were subjected to RT-PCR analysis for internal env RNA splicing as shown in Fig. 4C. All cells showed evidence for the 650-bp RT-PCR products at various levels. Thus, the internal env RNA splicing occurred in all cell lines examined. The 650-bp RT-PCR product was also detected in RNA from CP cells infected with JSRV (Fig. 4C, left panel, lane 4), which indicated that the internal env splicing was observed not only under conditions of transient transfection. Minor amounts of the 650-bp RT-PCR fragment were observed in CP cells transfected with pCDNA3.1 (Fig. 4C, left panel, lane 3), which could have reflected the fact that sheep harbor and express endogenous betaretroviruses highly homologous to JSRV (60). In any event, the amounts of 650-bp products from the JSRV21-infected CP cells were substantially higher, indicating that the majority of the product represented internal splicing of exogenous JSRV RNA. Figure 4C also shows RT-PCR of RNA from 293T cells transfected with the full-length JSRV proviral expression clone pCMV2JS21 (left panel, lane 2); the 650-bp product was also observed.
To test if doubly spliced mRNAs corresponding to the 650-bp RT-PCR product could encode active Rej protein, we inserted the ds-env cDNA2 and cDNA4 sequences in place of the full-length env gene in ΔGP to give the constructs ΔGPds-envcDNA2 and ΔGPds-envcDNA4 (Fig. 5A). These constructs would retain the splice donor and acceptor sites between the first (noncoding) and second exons, which would allow nuclear export of the transcripts; however, they would encode only the proteins corresponding to the putative doubly spliced mRNAs. These two constructs expressed their respective RNAs at levels equivalent to those expressed by ΔGP and ΔGPSP in Northern blot experiments (Fig. 5C). They were then tested for Rej activity by cotransfection with pTN3 into 293T cells and testing for production of Gag protein as shown in Fig. 5B. ΔGPds-envcDNA2 and ΔGPds-envcDNA4 complemented pTN3 for synthesis of Gag protein to the same extent as did wild-type ΔGP or ΔGPSP. The Gag polyprotein band in Fig. 5B migrated slightly faster than a background band evident in all lanes, including transfection with pCDNA3.1 or pTN3 alone.
Sequence comparisons of JSRV Env signal peptide and translation products from doubly spliced mRNAs.
As shown above, both the JSRV Env signal peptide and the doubly spliced mRNAs could express Rej activity. Also, the Env signal peptide localized to the nucleus (Fig. 3), a characteristic of other retroviral transactivators of Gag synthesis (11, 27, 38, 45, 56). The signal peptide region of JSRV Env protein was found to contain several putative domains present in other retroviral export proteins, most notably MMTV Rem (Fig. 6A). These included overlapping putative nuclear and nucleolar localization signals (NLS, three basic amino acids and a proline, residues 2 to 5, and NoLS, region of 30 amino acids with at least nine basic residues, residues 2 to 29, respectively). In addition the JSRV Env signal peptide contains a putative arginine-rich motif (ARM), an RNA-binding domain characteristic of Rev-like proteins (42) (residues 4 to 26). The signal peptide also contains a putative nuclear export signal (NES, a leucine-rich domain; residues 66 to 73). Finally, the signal peptide contains a signal recognition particle (SRP) sequence (stretch of hydrophobic amino acids, residues 66 to 84) that would normally conduct the nascent polypeptide chain into the rough endoplasmic reticulum (ER) for export. The presence of these motifs further supported the idea of the 5′ end of JSRV env encoding Rej activity.
It was interesting that the proteins encoded by the doubly spliced mRNAs show Rej activity and that they would contain some but not all of the motifs in the signal peptide. In particular, they would contain the NLS, NoLS, and ARM motifs, but since they are effectively truncated at amino acid 67, they would lack the NES and SRP motifs (Fig. 6B). The lack of the SRP motif would result in proteins that are not conducted into rough ER; such proteins would remain cytosolic and possibly have a higher likelihood of transport to the nucleus and/or nucleolus than would the full signal peptide. On the other hand, the fact that these doubly spliced mRNAs encode functional Rej activity indicates that the putative NES in the signal peptide is not required for Rej function.
The effect of JSRV Rej on nuclear export of unspliced viral RNA is cell type specific.
By analogy to MMTV Rem and other retroviral regulatory proteins that facilitate expression of unspliced mRNAs, we hypothesized that JSRV Rej acts at the level of unspliced mRNA export. To test this, we constructed a derivative of the pCMV2JS21 full proviral expression construct, in which env coding sequences corresponding to amino acids 13 to 52 were deleted (pCMV2JS21 SUΔ13-52, Fig. 1A). As shown in Fig. 2C and Fig. 7C, deletion of these amino acids from ΔGP or pCMV2JS21, respectively, abolished Rej activity. Wild-type or mutant pCMV2JS21 was transfected in 293T cells, and total, cytoplasmic, and nuclear RNAs were prepared both 16 and 40 h posttransfection. The RNAs were then analyzed by agarose gel electrophoresis and Northern blot hybridization with a radioactive JSRV gag probe. As shown in Fig. 7A, when assayed in 293T cells, the deletion of Rej diminished the amount of unspliced cytoplasmic JSRV RNA; simultaneously, unspliced JSRV RNA accumulated in the nuclear fraction. Because export of unspliced JSRV RNA might be affected by the high levels of expression in 293T cells, we also carried out these measurements at different levels of input transfected DNA, at 28 μg per 10-cm dish (our standard conditions) and also 14 μg/dish. The same results were obtained under both conditions. Quantification of the Northern blot data from six experiments by phosphorimage analysis indicated an approximately 2.8-fold difference in unspliced cytoplasmic RNA export between the pCMV2JS21 SUΔ13-52 construct with Rej deleted and the wild-type, pCMV2JS21 construct. Western blotting for cytoplasmic and nuclear proteins (β-tubulin and lamin A/C, respectively) confirmed the fractionations (Fig. 7B). This was also confirmed by RT-PCR analysis (not shown) (33). Although the deletion of Rej reduced JSRV unspliced cytoplasmic RNA export by 2.8-fold, pCMV2JS21 SUΔ13-52 showed less than 1% of the Gag synthesized compared to pCMV2JS21, as determined by phosphorimaging (Fig. 7C). Taken together, these results indicate that even though Rej facilitates unspliced JSRV RNA export to the cytoplasm, residual export occurs in these cells in the absence of Rej. However, Rej is absolutely required for Gag production.
FIG. 7.
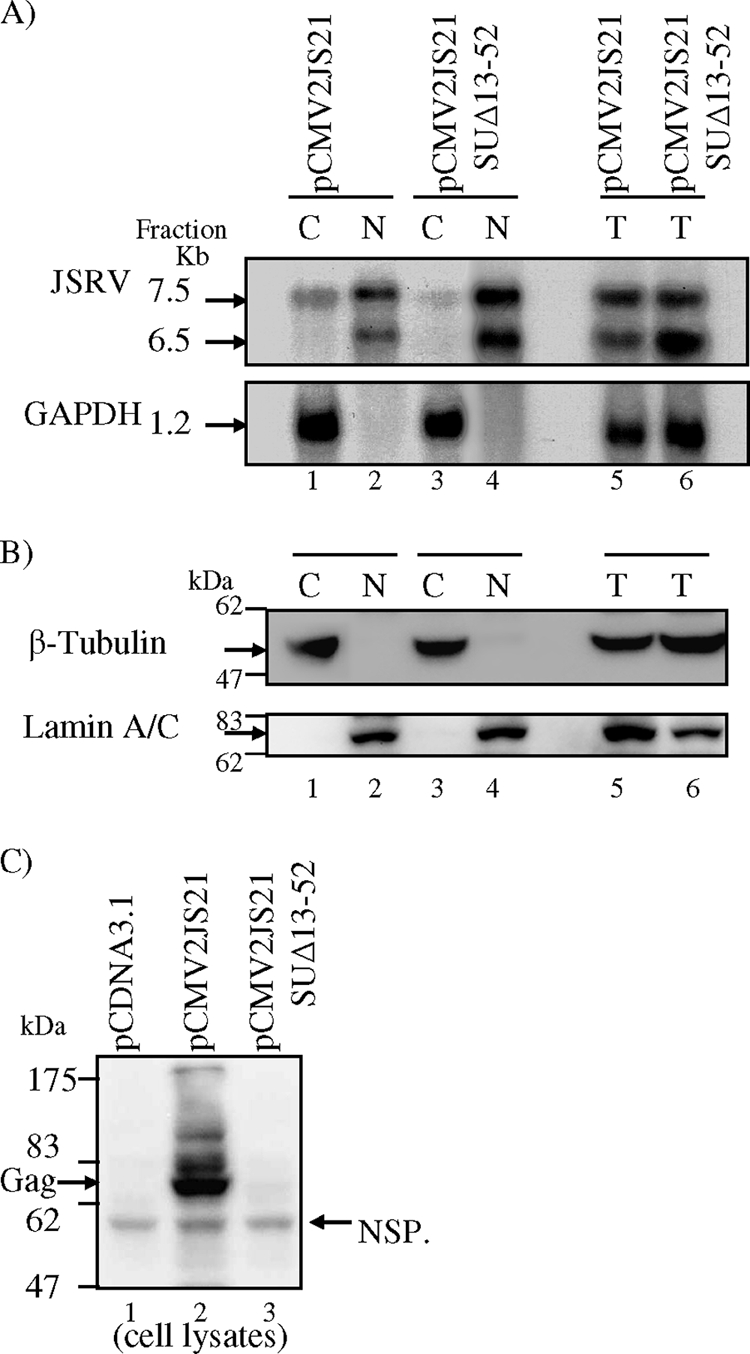
Rej facilitates export of unspliced cytoplasmic RNA in human 293T cells. 293T cells (106) were transfected with 14 μg of pCMV2JS21, pCMV2JS21 SUΔ13-52, or pCDNA3.1; at 40 h posttransfection, the cells were harvested and total RNA or RNA from nuclei and cytoplasm was extracted. (A) Total, nuclear, and cytoplasmic RNAs from equivalent numbers of cells analyzed by agarose gel electrophoresis and Northern blot hybridization with a JSRV gag probe. Bands corresponding to the unspliced 7.5-kb and the 6.5-kb prematurely polyadenylated JSRV genome RNAs are shown (arrows, top panel). The blots were stripped and rehybridized with a GAPDH probe (1.2-kb band, arrow, bottom panel). (B) Western blot analysis of cell lysate fractions 1 to 6 of panel A with antibodies that detect β-tubulin (55 kDa, as a cytoplasmic marker) and lamin A/C (74 kDa, as a nuclear marker) was used to control for nuclear/cytoplasmic separation. Cell lysates were run on 10% SDS-polyacrylamide gels and visualized by immunoblotting; a 55-kDa specific band corresponding to β-tubulin was detected only in cytoplasmic fractions (lanes 1 and 3, top panel), whereas a 74-kDa specific band corresponding to lamin A/C was detected in only nuclear fractions (lanes 2 and 4, bottom panel); both proteins were detected in the total fractions (lanes 5 and 6, top and bottom panels). (C) Cell lysates at 40 h posttransfection from 293T cells transiently transfected with 14 μg of the plasmids indicated were analyzed by Western blotting with anti-JSRV CA monoclonal antibody. Gag polyprotein (74 kDa) was expressed from pCMV2JS21 (Gag, arrow, lane 2) but not from pCMV2JS21 SUΔ13-52 (lane 3) or pCDNA3.1 (lane 1). A nonspecific protein (NSP) which migrated faster than the JSRV poly-Gag protein was detected in all lanes and demonstrates equivalent loading of the cell lysates (60 kDa, arrow).
Since Rej facilitates unspliced viral RNA export in 293T cells, we hypothesized that Rej will bind to JSRV sequences, likely in the 3′ region of full-length retroviral RNAs, to facilitate the export, analogously to other retroviral proteins that play a role in unspliced RNA export (15, 27, 28, 41, 45). To test this, we employed the pDM128 system (24). The pDM128 plasmid contains the bacterial CAT gene within an intron that also contains an HIV-1 RRE. In the absence of HIV Rev, mRNA splicing will remove the CAT coding sequences so that cytoplasmic RNA will not encode CAT. However, in the presence of Rev, unspliced pDM128 RNA will be exported to the cytoplasm, resulting in expression of CAT enzyme activity. Moreover, insertion of CTEs from MPMV or Rous sarcoma virus within the intron of pDM128 results in Rev-independent expression of CAT (3, 47). We inserted JSRV env sequences containing nt 7075 to 7441 (termed the JREE) into the pDM128 intron downstream of the CAT gene to give the plasmid pDM/JREE (Fig. 8A). pDM128 and pDM/JREE were transiently transfected into 293T cells, and the amounts of CAT enzyme activity were measured. To control for transfection efficiency, the pRL-null vector encoding Renilla luciferase was cotransfected along with these plasmids, and the amount of Renilla luciferase activity was also measured; CAT activities were normalized for Renilla luciferase activities from each transfected cell extract.
Transfection with pDM/JREE in 293T cells showed threefold-more CAT activity than did that with pDM128 (Fig. 8B, top left panel), indicating that the JSRV JREE can confer cytoplasmic export on unspliced pDM128 mRNA. Thus, the JREE had constitutive export (CTE) activity. In addition, in agreement with our hypothesis, when Rej was provided in trans (cotransfection with ΔGP), Rej further increased the CAT activity of pDM128/JREE approximately twofold (Fig. 8B). The enhancement of CAT activity by Rej was significant (P < 0.05 by Student's t test). We also tested the ability of Rej to increase the cytoplasmic RNAs from the transfected cells by Northern blotting (Fig. 8C). The levels of unspliced mRNAs paralleled the CAT enzyme activities, and phosphorimaging analysis indicated a twofold increase of unspliced cytoplasmic RNA in the presence of Rej (Fig. 8C). These results indicated that the JREE has both CTE activity for unspliced RNA export and also a response to Rej for enhanced export in human 293T cells.
We also used the pDM128 assay to test if the JREE is functional in other cell lines including human 293 cells, rat 208F fibroblasts, mouse NIH 3T3 cells, and sheep CP-1 choroid plexus-derived cells (Fig. 8D). The CP-1 cells were of particular interest since the normal host for JSRV is sheep. As shown, the JREE was able to facilitate export of unspliced pDM128 RNA without addition of Rej (CTE activity) in all of these cell lines, as measured by enhanced CAT enzyme activity. Moreover, in these other cells, the CTE activity of the JREE was equivalent to the cytoplasmic export activity of HIV-1 Rev working on the RRE. Most interestingly, when we supplied Rej in trans by cotransfection of pDM/JREE with ΔGP, there was no additional increase of unspliced RNA export to the cytoplasm in these cell lines. Thus, the JREE had functional CTE activity in all cell lines tested, but 293T cells were the only ones to show enhancement of unspliced RNA export by Rej.
To confirm the pDM128 assay indicating that Rej does not facilitate unspliced viral RNA export in many cell lines, we tested for cytoplasmic unspliced RNA in 293 cells by Northern blot assay. Wild-type or mutant pCMV2JS21 was transfected in 293 cells, and total, cytoplasmic, and nuclear RNAs were prepared both 16 and 40 h posttransfection as described above for 293T cells. As shown in Fig. 9A, the deletion of Rej did not diminish the amount of unspliced cytoplasmic JSRV RNA in 293 cells (compare lanes 1 and 3, top panel), as also confirmed by phosphorimaging (not shown). Western blot analysis confirmed effective cytoplasmic and nuclear fractionation (Fig. 9B). Although deletion of Rej did not affect the amount of unspliced cytoplasmic RNA in 293 cells, pCMV2JS21 ΔSU13-52 failed to produce detectable Gag polyprotein, even though the unspliced RNA was present in the cytoplasm (Fig. 9A and C). As determined by phosphorimaging analysis, the amount of Gag present in pCMV2JS21 SUΔ13-52-transfected cells was less than 1% of that in pCMV2JS21-transfected cells (Fig. 9C).
FIG. 9.
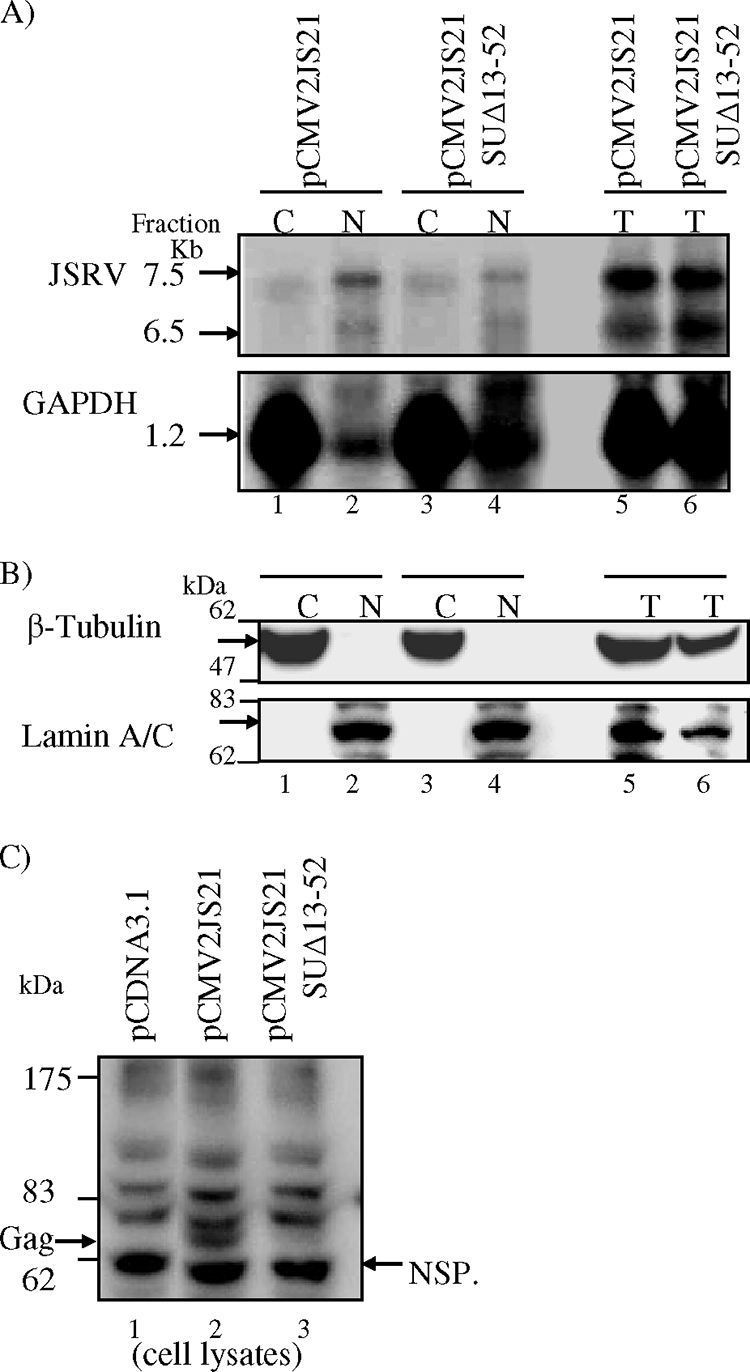
Rej does not facilitate the transport of unspliced cytoplasmic RNA in human 293 cells but is absolutely required for JSRV poly-Gag protein synthesis. 293 cells (106) were transfected with 14 μg of pCMV2JS21, pCMV2JS21 SUΔ13-52, or pCDNA3.1; at 40 h posttransfection, the cells were harvested and total RNA or RNA from nuclei and cytoplasm was extracted. (A) Total, nuclear, and cytoplasmic RNAs from equivalent numbers of cells analyzed by agarose gel electrophoresis and Northern blot hybridization with a JSRV gag probe. Bands corresponding to the unspliced 7.5-kb and the 6.5-kb prematurely polyadenylated JSRV genome RNAs are shown (arrows, top panel). The blots were stripped and rehybridized with a GAPDH probe (1.2-kb band, arrow, bottom panel). (B) Western blot analysis of cell lysate fractions 1 to 6 of panel A with antibodies that detect β-tubulin (55 kDa, as a cytoplasmic marker) and lamin A/C (74 kDa, as a nuclear marker) was used to control for nuclear/cytoplasmic separation. Cell lysates were run on 10% SDS-polyacrylamide gels and visualized by immunoblotting; a 55-kDa specific band corresponding to β-tubulin was detected only in cytoplasmic fractions (lanes 1 and 3, top panel), whereas a 74-kDa specific band corresponding to lamin A/C was detected only in nuclear fractions (lanes 2 and 4, bottom panel); both proteins were detected in the total fractions (lanes 5 and 6, top and bottom panels). (C) Cell lysates at 40 h posttransfection from 293 cells transiently transfected with 14 μg of the plasmids indicated were analyzed by Western blotting with anti-JSRV CA monoclonal antibody. Gag polyprotein (74 kDa) was expressed from pCMV2JS21 (Gag, arrow, lane 2) but not from pCMV2JS21 SUΔ13-52 (lane 3) or pCDNA3.1 (lane 1). A nonspecific protein (NSP) that migrated faster than the JSRV poly-Gag protein was detected in all lanes and was used to show equivalent loading of the cell lysates (60 kDa, NSP, arrow).
JSRV Rej is required for the translation of gag-pol RNA.
The results shown in Fig. 7C and 9C suggested that Rej is necessary for the synthesis of Gag polyprotein from unspliced JSRV RNA, since deletion of the Rej-encoding region from pCMV2JS21 SUΔ13-52 resulted in no Gag polyprotein, as assayed by Western blotting. However, it was also possible that Rej stabilizes newly synthesized Gag polyprotein, rather than mediating its translation. Indeed the HIV-1 Vpu protein stabilizes the Pr55 Gag polyprotein and prevents its degradation (20) but does not affect Gag translation. To test if Rej facilitates translation of Gag polyprotein, we used 35S pulse-labeling to measure the rates of Gag synthesis in the presence and absence of Rej. 293T cells transfected with pCMV2JS21, pCMV2JS21 SUΔ13-52, or pCDNA3.1 were pulse-labeled with [35S]cysteine-methionine for 20 min followed by immunoprecipitation of cell extracts with anti-JSRV CA antibody, SDS-PAGE, and phosphorimaging (Fig. 10A, top panel). We detected a 74-kDa JSRV Gag-specific protein in pCMV2JS21-transfected cells that was not detected in cells transfected with the empty vector control, pCDNA3.1, corresponding to the Gag precursor polyprotein (Fig. 10A, top panel). Of note, this band was not detected in pCMV2JS21 SUΔ13-52-transfected cell lysates, indicating that Rej facilitates translation of Gag-Pol proteins (Fig. 10A, top panel). To control for equivalent amounts of proteins in the cell lysates, we used Western blot analysis for endogenous β-tubulin protein. All samples showed similar levels of cell lysate proteins (Fig. 10A, bottom panel).
FIG. 10.
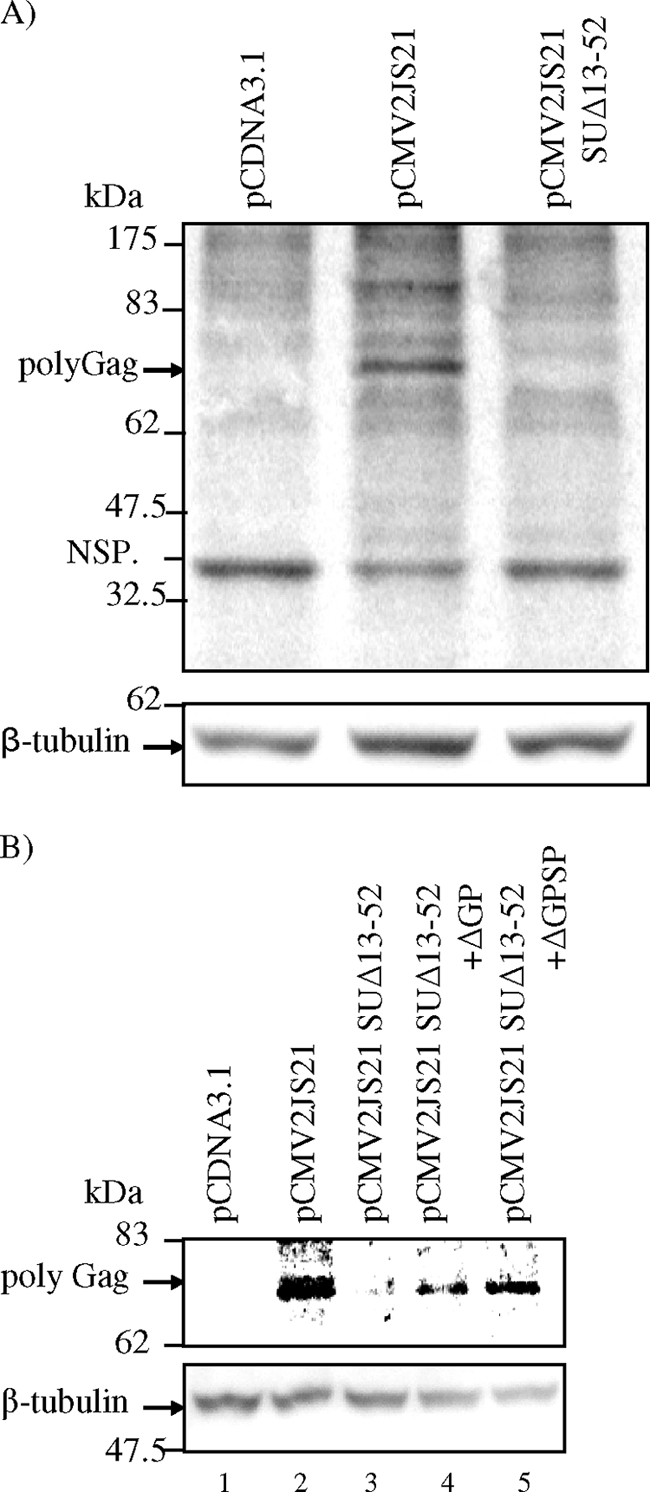
(A) Rej is necessary for translation of gag-pol RNA. 293T cells were transfected with pCDNA3.1, pCMV2JS21, or pCMV2JS21 SUΔ13-52; at 16 h posttransfection, the cells were starved for 1 h and then pulse-labeled with [35S]cysteine-methionine for 20 min. Cell lysates were prepared and subjected to immunoprecipitation assay using a rabbit anti-JSRV CA antibody and then resolved by SDS-PAGE followed by phosphorimaging. JSRV-specific 35S-labeled Gag was detected in immunoprecipitates of cells transfected with pCMV2JS21 (middle lane, top panel) but not in cells transfected with pCDNA3.1 or pCMV2JS21 SUΔ13-52 (left and right lanes, respectively, top panel). NSP, nonspecific protein band, immunoprecipitated from all extracts. To control for equivalent amounts of cellular proteins subject to immunoprecipitation, Western blot analysis with anti-β-tubulin antibody was performed (bottom panel). (B) Rej can complement the translation of Gag polyprotein from mutant constructs having Rej deleted. 293T cells were transfected with pCDNA3.1, pCMV2JS21, or pCMV2JS21 SUΔ13-52, or pCMV2JS21 SUΔ13-52 was cotransfected with ΔGP or ΔGPSP (to supply Rej in trans); the cells were treated, labeled, and immunoprecipitated as described above. Gag polyprotein (74 kDa) was expressed from pCMV2JS21 (Gag, arrow, lane 2) but not from pCMV2JS21 SUΔ13-52 (lane 3) or pCDNA3.1 (lane 1); Gag polyprotein was detected from pCMV2JS21 SUΔ13-52 when cotransfected with ΔGP (Gag, arrow, lane 4) or ΔGPSP (Gag, arrow, lane 5).
We then asked if we could rescue Gag synthesis by pCMV2JS21 SUΔ13-52 if we supplied Rej in trans. As shown in Fig. 10B, Rej complements for Gag synthesis from pCMV2JS21 SUΔ13-52 when expressed in trans from either ΔGP or ΔGPSP.
DISCUSSION
In the studies described here, we have identified a factor(s) encoded in the JSRV genome that is necessary for Gag protein synthesis from unspliced viral mRNA. We have named this trans-acting factor Rej, for regulator of JSRV expression. JSRV Rej is analogous in some ways to the Rem and Rec proteins of MMTV and HERV-K, two other betaretroviruses, in that these proteins are also required for Gag protein synthesis. Similarly to Rem and Rec, the coding sequences for Rej activity also map to the signal peptide of the envelope protein precursor (27, 38, 45) (Fig. 2C). However, the mechanism of Rej action may differ from that of Rem and Rec. The latter proteins function similarly to the well-characterized retroviral export proteins HIV-1 Rev (15, 41) and HTLV Rex (28) in that they facilitate nuclear export of unspliced viral RNA, thus allowing its translation into Gag and Gag-Pol proteins. However, in most cell lines Rej does not appear to facilitate export of unspliced viral RNA, as measured by the pDM128/JREE assays. Also in 293 cells transfection of a viral expression construct having Rej deleted showed normal levels of full-length JSRV RNA in the cytoplasm (Fig. 9). Nevertheless, the rej-negative expression construct did not produce Gag protein (Fig. 2A and 7C). Taken together, these results suggest that the main function of Rej is to facilitate translation of unspliced viral RNA rather than its nuclear export. This concept was further supported by metabolic labeling experiments in which Gag polyprotein was synthesized only in the presence of Rej (Fig. 10).
Human 293T cells were exceptional in that in these cells Rej also modestly enhanced transport of unspliced viral RNA or reporter RNAs containing the JREE (2.8-fold). Thus, within the JREE there are RNA sequences that respond to Rej (the RejRE). We have characterized the RejRE through study of several single or double nucleotide substitutions within the JREE that reduce unspliced viral RNA export in 293T cells (46a). These sequences appear to be involved in binding of Rej to viral RNA since Rej protein (normally nuclear) is relocalized to the cytoplasm by wild-type viral RNA but not by RejRE mutant RNA. The physical relationship of the CTE and the RejRE within the JREE remains to be determined. Presumably 293T cells express factors (missing from most other cell lines) that can interact with Rej bound to the RejRE and facilitate RNA export.
The betaretrovirus genus includes viruses that export unspliced viral RNA and synthesize Gag and Gag-Pol proteins by different mechanisms. As mentioned above, HERV-K and MMTV encode Rec and Rem. On the other hand, MPMV does not appear to encode an analogous protein. Instead, the export of full-length MPMV RNA is facilitated by the presence of a CTE in the RNA. JSRV has more homology to MMTV in the env gene than with MPMV (60); it is thus reasonable that both JSRV and MMTV encode analogous regulatory proteins (Rej and Rem). Both Rej and Rem can be encoded by doubly spliced mRNAs within the env gene as well. Furthermore, the JSRV Rej protein has localization patterns (Fig. 3) and motifs (NLS and NoLS, Fig. 6A) similar to those of MMTV Rem (27, 45) and HERV-K Rec (38). On the other hand, in most cells Rej protein is not required for nuclear export of unspliced viral RNA. As shown in Fig. 8, a CTE in the 3′ end of the JSRV env gene is responsible for unspliced RNA export, analogously to the CTE of MPMV. Thus, JSRV has a mechanism for expression of Gag and Pol proteins that has features in common with those of other betaretroviruses but is unique. JSRV resembles MPMV in that unspliced RNAs are constitutively transported to the cytoplasm by way of the JREE. On the other hand, the viral protein Rej is necessary for expression of Gag and Pol proteins, analogously to MMTV Rem and HERV-K Rec, although its mechanism of action predominantly appears to be facilitating translation of cytoplasmic unspliced viral RNA.
While the predominant mechanism of action of the retroviral Rev/Rex-like proteins has been considered to be nuclear export of unspliced viral RNA, several reports indicate a role for HIV-1 Rev protein in viral RNA translation (1, 12, 34, 53). Rev directs the association of RRE-containing RNAs with the polysomes in the cytoplasm (1, 12, 30). Moreover, Rev is required for the colocalization of HIV-1 gag mRNA and cytoskeletal proteins into perinuclear clusters, a site where viral protein synthesis occurs via association with cytoskeleton polyribosomes (12, 30, 31). Further support for a translational role for Rev comes from comparing the levels of Rev-induced full-length cytoplasmic RNAs with the amounts of Gag synthesis from these same RNA transcripts. One report indicated only a 4- to 16-fold increase by Rev in the concentration of unspliced gag mRNA from a reporter plasmid carrying an RRE-containing gag gene but a more-than-800-fold increase in the production of Gag protein (12). A similar effect has been reported for Rev-mediated translation of HIV-1 env mRNA (15, 53). Rev expression resulted in a 1.6-fold increase in env mRNA transport (15), whereas translation from this mRNA was increased more than 100-fold (53). Other studies have also demonstrated a role for the Rev protein in mRNA translation (34). Thus, there is precedence for JSRV Rej functioning at the level of translation. Future studies of Rej may elucidate general mechanisms by which all of these small retroviral proteins facilitate viral mRNA translation; studies of Rej in cells other than 293T cells may be more definitive, without complications from effects on nuclear export of the mRNAs.
While we have demonstrated the essential role of Rej for Gag protein expression, the exact form(s) of the protein in infected cells remains to be determined, since antibodies for Rej are not yet available. On one hand, the Env signal peptide by itself (residues 1 to 84) has Rej activity (Fig. 2D), and this protein would have all of the motifs present in MMTV Rem protein. The HA epitope-tagged version of this signal peptide will be valuable in future mechanistic experiments on Rej action. On the other hand, we detected two internal splicing events that reflect doubly spliced mRNAs, encoding Rej, and mRNAs containing the second splice encoded Rej activity (Fig. 5). As mentioned above, Rej proteins encoded by these doubly spliced mRNAs would lack the SRP motif, which would allow them to be translated from cytoplasmic polysomes and released into the cytoplasm. On the other hand, since they would also lack the NES, their export would require interaction with a cellular or viral protein or RNA. One mechanism for nuclear export of these Rej forms would be for them to bind to the RejRE on unspliced viral RNA, followed by export of the unspliced RNA-protein complex via the CTE.
Alternatively, the Rej protein could arise from cleavage of the signal peptide from JSRV Env polyprotein, and indeed artificial expression of the signal peptide sequence (ΔGPSP) is sufficient for Rej activity (Fig. 2D). Recently intracellular regulatory and/or signaling functions of cleaved cellular signal peptide fragments have been described. Typically, signal sequences target proteins to the ER membrane by interaction with the SRP, which conducts the protein into the lumen of the ER (57). In the lumen, signal peptidase cleaves the signal peptide from the preprotein; as a result, both the cleaved protein and the signal peptide are localized in the ER (57). However, some signal sequences are liberated from the ER membrane back into the cytosol if cleavage is by intramembrane-cleaving proteases (I-CliPs) (43, 59). Thus, these signal peptides could have activities in the cytoplasm. An example is the generation of signal sequence-derived human leukocyte antigen E (HLA-E) epitopes by an I-CliP (36). The cleaved epitope-containing signal sequence fragment is released into the cytosol, allowing it to be bound by the transporter associated with antigen presentation (TAP) complex and loaded onto a major histocompatibility complex class I molecule for presentation (36, 43). It is possible that Rej could be cleaved from the Env precursor polyprotein in the ER by an I-CliP, resulting in release of the signal peptide into the cytosol, while the Env protein is transferred into the lumen of the ER.
Another possibility is that the signal peptide could be liberated into the cytosol after cleavage by a cellular furin-like protease. This has been described for the cleavage of the leader peptide of the Env glycoprotein (Elp) of feline foamy virus; the resultant Elp protein is required for feline foamy virus particle budding (16). Alternatively, the JSRV Env signal peptide may be liberated into the cytosol by a mechanism analogous to the signal peptide of the MMTV Env and its alternative splice variant Rem protein (14). However, these authors demonstrated that the cleavage of the Rem signal peptide and its accumulation into the cytosol are independent of signal peptide peptidase, suggesting a third pathway by which a signal peptide can be released from the ER membrane to fulfill a posttargeting function in a different compartment (14). Inhibitors of signal peptide peptidase or the furin protease may be useful in future studies of how the JSRV Env signal peptide functions as Rej (14, 16). Additionally, it will be desirable to generate antisera to Env signal sequences or to attach an N-terminal epitope tag to the preenvelope protein in future experiments.
We found that the epitope-tagged JSRV Env signal peptide localizes to the cell nucleus when expressed by itself (Fig. 3). This suggests that Rej may affect unspliced viral RNA capability for translation by acting from the nucleus (ticketing RNA for translation), or alternatively, Rej and associated proteins bound to cytoplasmic unspliced viral RNA could facilitate translation. Future experiments will test if cytoplasmic Rej protein in infected cells is associated with polyribosomes. In addition, it will be interesting to determine if Rej directly binds to the RejRE or if binding is through a cellular protein.
The coupling of nuclear export and translation has been reported for simple retroviral unspliced RNAs. The Tap/p15 (NXF1/NXT1) complex functions as a general mRNA export receptor (32) as well as a mediator of export of simple retroviral unspliced RNAs containing a CTE (17). This complex also promotes the translation of unspliced viral mRNAs (29). Further, the Tap protein could be immunoprecipitated from the polyribosome fractions of sucrose sedimentation gradients, indicating that Tap associates with polyribosomes (29). Similarly, cellular RNA helicase A (RHA) has recently been shown to enhance translation of retroviral RNAs containing a 5′-terminal posttranscriptional control element, found in avian spleen necrosis virus (5) and MPMV (25). RHA binds the highly structured posttranscriptional control element to mediate the translational enhancement (21). RHA is a member of the DEAD-box family of RNA helicases that function at many levels of RNA processing and metabolism, including export and translation (37). Therefore, RHA is also likely to load onto the mRNA in the nucleus and enhance translation in the cytoplasm. It will be interesting to investigate the effect of deleting the Rej NLS on Rej activity.
One of the unique features of JSRV is that the env gene functions as an oncogene, as demonstrated by its ability to transform rodent fibroblasts in vitro (40). While there is an absolute requirement for sequences in the cytoplasmic tail of the transmembrane (TM) domain of JSRV Env for transformation, we also found that deletions or insertions in the N-terminal SU domain also abolished transformation (23). Of note, deletions or linker insertions in the signal peptide region also abolished transformation, even though they should not have affected the sequence of the mature SU protein. The experiments reported here therefore raised the possibility that Rej may be involved in the JSRV transformation, since signal peptide mutations would affect Rej activity but not SU protein. Indeed, expression of the HERV-K Rec protein in NIH 3T3 cells increased their tumorigenicity, although it did not cause morphological transformation (2). Therefore, we tested whether expression constructs for JSRV Rej (for the signal peptide [ΔGPSP] or for the doubly spliced env transcripts [Fig. 5]) could complement N-terminal env mutants for transformation in cotransfection assays (23). However, none of the JSRV Rej expression constructs showed complementation (A. Hofacre and H. Fan, unpublished data), which did not support a role for Rej in JSRV transformation.
After this work was submitted for publication, a similar study was reported by Caporale et al. (6). They also described a JSRV regulatory protein necessary for expression of Gag protein and which facilitates export of unspliced viral RNA in 293T cells, termed the Env signal peptide. This report is consistent with our results, and we additionally show that in most cell lines the predominant mechanism of action is through mRNA translation. In this study doubly spliced mRNAs that can encode functional Rej activity were also identified.
Acknowledgments
This work was supported by NIH grant R01CA94188. The support of the UCI Cancer Research Institute and the DNA sequencing core and the Optical Biology core of the Chao Family Comprehensive Cancer Center is acknowledged. T.N. was supported in part by fellowships from the Ishidu Shun Memorial Scholarship and the YASUDA Medical Research Foundation.
We thank James Goedert of the National Cancer Institute for facilitating production of the JSRV CA-specific hybridoma. Thomas J. Hope is also acknowledged for the kind gift of plasmid DNA pDM128. The advice of Stacey Hull and Mina Kalantari-Dehaghi is greatly appreciated, as well the assistance of Joshua Chung in producing some of the expression constructs used in these studies.
Footnotes
Published ahead of print on 23 September 2009.
REFERENCES
- 1.Arrigo, S. J., and I. S. Chen. 1991. Rev is necessary for translation but not cytoplasmic accumulation of HIV-1 vif, vpr, and env/vpu 2 RNAs. Genes Dev. 5:808-819. [DOI] [PubMed] [Google Scholar]
- 2.Boese, A., M. Sauter, U. Galli, B. Best, H. Herbst, J. Mayer, E. Kremmer, K. Roemer, and N. Mueller-Lantzsch. 2000. Human endogenous retrovirus protein cORF supports cell transformation and associates with the promyelocytic leukemia zinc finger protein. Oncogene 19:4328-4336. [DOI] [PubMed] [Google Scholar]
- 3.Bogerd, H. P., A. Echarri, T. M. Ross, and B. R. Cullen. 1998. Inhibition of human immunodeficiency virus Rev and human T-cell leukemia virus Rex function, but not Mason-Pfizer monkey virus constitutive transport element activity, by a mutant human nucleoporin targeted to Crm1. J. Virol. 72:8627-8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bray, M., S. Prasad, J. W. Dubay, E. Hunter, K. T. Jeang, D. Rekosh, and M. L. Hammarskjold. 1994. A small element from the Mason-Pfizer monkey virus genome makes human immunodeficiency virus type 1 expression and replication Rev-independent. Proc. Natl. Acad. Sci. USA 91:1256-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butsch, M., S. Hull, Y. Wang, T. M. Roberts, and K. Boris-Lawrie. 1999. The 5′ RNA terminus of spleen necrosis virus contains a novel posttranscriptional control element that facilitates human immunodeficiency virus Rev/RRE-independent Gag production. J. Virol. 73:4847-4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caporale, M., F. Arnaud, M. Mura, M. Golder, C. Murgia, and M. Palmarini. 2009. The signal peptide of a simple retrovirus envelope functions as a posttranscriptional regulator of viral gene expression. J. Virol. 83:4591-4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan, W. Y., Q. R. Liu, J. Borjigin, H. Busch, O. M. Rennert, L. A. Tease, and P. K. Chan. 1989. Characterization of the cDNA encoding human nucleophosmin and studies of its role in normal and abnormal growth. Biochemistry 28:1033-1039. [DOI] [PubMed] [Google Scholar]
- 8.Cochrane, A. W., A. Perkins, and C. A. Rosen. 1990. Identification of sequences important in the nucleolar localization of human immunodeficiency virus Rev: relevance of nucleolar localization to function. J. Virol. 64:881-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cousens, C., E. Minguijon, R. G. Dalziel, A. Ortin, M. Garcia, J. Park, L. Gonzalez, J. M. Sharp, and M. de las Heras. 1999. Complete sequence of enzootic nasal tumor virus, a retrovirus associated with transmissible intranasal tumors of sheep. J. Virol. 73:3986-3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cullen, B. R. 2003. Nuclear mRNA export: insights from virology. Trends Biochem. Sci. 28:419-424. [DOI] [PubMed] [Google Scholar]
- 11.Cullen, B. R., J. Hauber, K. Campbell, J. G. Sodroski, W. A. Haseltine, and C. A. Rosen. 1988. Subcellular localization of the human immunodeficiency virus trans-acting art gene product. J. Virol. 62:2498-2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D'Agostino, D. M., B. K. Felber, J. E. Harrison, and G. N. Pavlakis. 1992. The Rev protein of human immunodeficiency virus type 1 promotes polysomal association and translation of gag/pol and vpu/env mRNAs. Mol. Cell. Biol. 12:1375-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dirks, C., F. M. Duh, S. K. Rai, M. I. Lerman, and A. D. Miller. 2002. Mechanism of cell entry and transformation by enzootic nasal tumor virus. J. Virol. 76:2141-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dultz, E., M. Hildenbeutel, B. Martoglio, J. Hochman, B. Dobberstein, and K. Kapp. 2008. The signal peptide of the mouse mammary tumor virus Rem protein is released from the endoplasmic reticulum membrane and accumulates in nucleoli. J. Biol. Chem. 283:9966-9976. [DOI] [PubMed] [Google Scholar]
- 15.Felber, B. K., M. Hadzopoulou-Cladaras, C. Cladaras, T. Copeland, and G. N. Pavlakis. 1989. rev protein of human immunodeficiency virus type 1 affects the stability and transport of the viral mRNA. Proc. Natl. Acad. Sci. USA 86:1495-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiselhart, V., P. Bastone, T. Kempf, M. Schnolzer, and M. Lochelt. 2004. Furin-mediated cleavage of the feline foamy virus Env leader protein. J. Virol. 78:13573-13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gruter, P., C. Tabernero, C. von Kobbe, C. Schmitt, C. Saavedra, A. Bachi, M. Wilm, B. K. Felber, and E. Izaurralde. 1998. TAP, the human homolog of Mex67p, mediates CTE-dependent RNA export from the nucleus. Mol. Cell 1:649-659. [DOI] [PubMed] [Google Scholar]
- 18.Guzik, B. W., L. Levesque, S. Prasad, Y. C. Bor, B. E. Black, B. M. Paschal, D. Rekosh, and M. L. Hammarskjold. 2001. NXT1 (p15) is a crucial cellular cofactor in TAP-dependent export of intron-containing RNA in mammalian cells. Mol. Cell. Biol. 21:2545-2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hadzopoulou-Cladaras, M., B. K. Felber, C. Cladaras, A. Athanassopoulos, A. Tse, and G. N. Pavlakis. 1989. The rev (trs/art) protein of human immunodeficiency virus type 1 affects viral mRNA and protein expression via a cis-acting sequence in the env region. J. Virol. 63:1265-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harila, K., I. Prior, M. Sjöberg, A. Salminen, J. Hinkula, and M. Suomalainen. 2006. Vpu and Tsg101 regulate intracellular targeting of the human immunodeficiency virus type 1 core protein precursor Pr55gag. J. Virol. 80:3765-3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hartman, T. R., S. Qian, C. Bolinger, S. Fernandez, D. R. Schoenberg, and K. Boris-Lawrie. 2006. RNA helicase A is necessary for translation of selected messenger RNAs. Nat. Struct. Mol. Biol. 13:509-516. [DOI] [PubMed] [Google Scholar]
- 22.Henderson, B. R., and P. Percipalle. 1997. Interactions between HIV Rev and nuclear import and export factors: the Rev nuclear localisation signal mediates specific binding to human importin-beta. J. Mol. Biol. 274:693-707. [DOI] [PubMed] [Google Scholar]
- 23.Hofacre, A., and H. Fan. 2004. Multiple domains of the Jaagsiekte sheep retrovirus envelope protein are required for transformation of rodent fibroblasts. J. Virol. 78:10479-10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hope, T. J., X. J. Huang, D. McDonald, and T. G. Parslow. 1990. Steroid-receptor fusion of the human immunodeficiency virus type 1 Rev transactivator: mapping cryptic functions of the arginine-rich motif. Proc. Natl. Acad. Sci. USA 87:7787-7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hull, S., and K. Boris-Lawrie. 2002. RU5 of Mason-Pfizer monkey virus 5′ long terminal repeat enhances cytoplasmic expression of human immunodeficiency virus type 1 gag-pol and nonviral reporter RNA. J. Virol. 76:10211-10218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hull, S., and H. Fan. 2006. Mutational analysis of the cytoplasmic tail of Jaagsiekte sheep retrovirus envelope protein. J. Virol. 80:8069-8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Indik, S., W. H. Gunzburg, B. Salmons, and F. Rouault. 2005. A novel, mouse mammary tumor virus encoded protein with Rev-like properties. Virology 337:1-6. [DOI] [PubMed] [Google Scholar]
- 28.Inoue, J., M. Yoshida, and M. Seiki. 1987. Transcriptional (p40x) and post-transcriptional (p27x-III) regulators are required for the expression and replication of human T-cell leukemia virus type I genes. Proc. Natl. Acad. Sci. USA 84:3653-3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin, L., B. W. Guzik, Y. C. Bor, D. Rekosh, and M. L. Hammarskjold. 2003. Tap and NXT promote translation of unspliced mRNA. Genes Dev. 17:3075-3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimura, T., I. Hashimoto, M. Nishikawa, and J. I. Fujisawa. 1996. A role for Rev in the association of HIV-1 gag mRNA with cytoskeletal beta-actin and viral protein expression. Biochimie 78:1075-1080. [DOI] [PubMed] [Google Scholar]
- 31.Kimura, T., I. Hashimoto, A. Yamamoto, M. Nishikawa, and J. I. Fujisawa. 2000. Rev-dependent association of the intron-containing HIV-1 gag mRNA with the nuclear actin bundles and the inhibition of its nucleocytoplasmic transport by latrunculin-B. Genes Cells 5:289-307. [DOI] [PubMed] [Google Scholar]
- 32.Kohler, A., and E. Hurt. 2007. Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell Biol. 8:761-773. [DOI] [PubMed] [Google Scholar]
- 33.Kusuhara, K., M. Anderson, S. M. Pettiford, and P. L. Green. 1999. Human T-cell leukemia virus type 2 Rex protein increases stability and promotes nuclear to cytoplasmic transport of gag/pol and env RNAs. J. Virol. 73:8112-8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrence, J. B., A. W. Cochrane, C. V. Johnson, A. Perkins, and C. A. Rosen. 1991. The HIV-1 Rev protein: a model system for coupled RNA transport and translation. New Biol. 3:1220-1232. [PubMed] [Google Scholar]
- 35.LeBlanc, J. J., S. Uddowla, B. Abraham, S. Clatterbuck, and K. L. Beemon. 2007. Tap and Dbp5, but not Gag, are involved in DR-mediated nuclear export of unspliced Rous sarcoma virus RNA. Virology 363:376-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lemberg, M. K., F. A. Bland, A. Weihofen, V. M. Braud, and B. Martoglio. 2001. Intramembrane proteolysis of signal peptides: an essential step in the generation of HLA-E epitopes. J. Immunol. 167:6441-6446. [DOI] [PubMed] [Google Scholar]
- 37.Linder, P. 2004. Molecular biology. The life of RNA with proteins. Science 304:694-695. [DOI] [PubMed] [Google Scholar]
- 38.Löwer, R., R. R. Tönjes, C. Korbmacher, R. Kurth, and J. Löwer. 1995. Identification of a Rev-related protein by analysis of spliced transcripts of the human endogenous retroviruses HTDV/HERV-K. J. Virol. 69:141-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maeda, N., Y. Inoshima, D. A. Fruman, S. M. Brachmann, and H. Fan. 2003. Transformation of mouse fibroblasts by Jaagsiekte sheep retrovirus envelope does not require phosphatidylinositol 3-kinase. J. Virol. 77:9951-9959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeda, N., M. Palmarini, C. Murgia, and H. Fan. 2001. Direct transformation of rodent fibroblasts by Jaagsiekte sheep retrovirus DNA. Proc. Natl. Acad. Sci. USA 98:4449-4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malim, M. H., J. Hauber, S. Y. Le, J. V. Maizel, and B. R. Cullen. 1989. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature 338:254-257. [DOI] [PubMed] [Google Scholar]
- 42.Malim, M. H., L. S. Tiley, D. F. McCarn, J. R. Rusche, J. Hauber, and B. R. Cullen. 1990. HIV-1 structural gene expression requires binding of the Rev trans-activator to its RNA target sequence. Cell 60:675-683. [DOI] [PubMed] [Google Scholar]
- 43.Martoglio, B. 2003. Intramembrane proteolysis and post-targeting functions of signal peptides. Biochem. Soc. Trans. 31:1243-1247. [DOI] [PubMed] [Google Scholar]
- 44.McGee-Estrada, K., and H. Fan. 2007. Comparison of LTR enhancer elements in sheep beta retroviruses: insights into the basis for tissue-specific expression. Virus Genes 35:303-312. [DOI] [PubMed] [Google Scholar]
- 45.Mertz, J. A., M. S. Simper, M. M. Lozano, S. M. Payne, and J. P. Dudley. 2005. Mouse mammary tumor virus encodes a self-regulatory RNA export protein and is a complex retrovirus. J. Virol. 79:14737-14747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neville, M., F. Stutz, L. Lee, L. I. Davis, and M. Rosbash. 1997. The importin-beta family member Crm1p bridges the interaction between Rev and the nuclear pore complex during nuclear export. Curr. Biol. 7:767-775. [DOI] [PubMed] [Google Scholar]
- 46a.Nitta, T., A. Hofacre, S. Hull, and H. Fan. 2009. Identification and mutational analysis of a Rej response element in Jaagsiekte sheep retrovirus RNA. J. Virol. 83:12499-12511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paca, R. E., R. A. Ogert, C. S. Hibbert, E. Izaurralde, and K. L. Beemon. 2000. Rous sarcoma virus DR posttranscriptional elements use a novel RNA export pathway. J. Virol. 74:9507-9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palmarini, M., N. Maeda, C. Murgia, C. De-Fraja, A. Hofacre, and H. Fan. 2001. A phosphatidylinositol 3-kinase docking site in the cytoplasmic tail of the Jaagsiekte sheep retrovirus transmembrane protein is essential for envelope-induced transformation of NIH 3T3 cells. J. Virol. 75:11002-11009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palmarini, M., C. Murgia, and H. Fan. 2002. Spliced and prematurely polyadenylated Jaagsiekte sheep retrovirus-specific RNAs from infected or transfected cells. Virology 294:180-188. [DOI] [PubMed] [Google Scholar]
- 50.Palmarini, M., J. M. Sharp, M. de las Heras, and H. Fan. 1999. Jaagsiekte sheep retrovirus is necessary and sufficient to induce a contagious lung cancer in sheep. J. Virol. 73:6964-6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palmarini, M., J. M. Sharp, C. Lee, and H. Fan. 1999. In vitro infection of ovine cell lines by Jaagsiekte sheep retrovirus. J. Virol. 73:10070-10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peng, S. S., C. Y. Chen, N. Xu, and A. B. Shyu. 1998. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 17:3461-3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perales, C., L. Carrasco, and M. E. Gonzalez. 2005. Regulation of HIV-1 env mRNA translation by Rev protein. Biochim. Biophys. Acta 1743:169-175. [DOI] [PubMed] [Google Scholar]
- 54.Rosen, C. A., E. Terwilliger, A. Dayton, J. G. Sodroski, and W. A. Haseltine. 1988. Intragenic cis-acting art gene-responsive sequences of the human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 85:2071-2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharp, J. M., and A. J. Herring. 1983. Sheep pulmonary adenomatosis: demonstration of a protein which cross-reacts with the major core proteins of Mason-Pfizer monkey virus and mouse mammary tumour virus. J. Gen. Virol. 64:2323-2327. [DOI] [PubMed] [Google Scholar]
- 56.Siomi, H., H. Shida, S. H. Nam, T. Nosaka, M. Maki, and M. Hatanaka. 1988. Sequence requirements for nucleolar localization of human T cell leukemia virus type I pX protein, which regulates viral RNA processing. Cell 55:197-209. [DOI] [PubMed] [Google Scholar]
- 57.Walter, P., and A. E. Johnson. 1994. Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane. Annu. Rev. Cell Biol. 10:87-119. [DOI] [PubMed] [Google Scholar]
- 58.Wang, W., H. Furneaux, H. Cheng, M. C. Caldwell, D. Hutter, Y. Liu, N. Holbrook, and M. Gorospe. 2000. HuR regulates p21 mRNA stabilization by UV light. Mol. Cell. Biol. 20:760-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weihofen, A., and B. Martoglio. 2003. Intramembrane-cleaving proteases: controlled liberation of proteins and bioactive peptides. Trends Cell Biol. 13:71-78. [DOI] [PubMed] [Google Scholar]
- 60.York, D. F., R. Vigne, D. W. Verwoerd, and G. Querat. 1992. Nucleotide sequence of the Jaagsiekte retrovirus, an exogenous and endogenous type D and B retrovirus of sheep and goats. J. Virol. 66:4930-4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zapp, M. L., and M. R. Green. 1989. Sequence-specific RNA binding by the HIV-1 Rev protein. Nature 342:714-716. [DOI] [PubMed] [Google Scholar]