

Abstract
Herpes simplex virus type 1 (HSV-1)-induced cell fusion is mediated by viral glycoproteins and other membrane proteins expressed on infected cell surfaces. Certain mutations in the carboxyl terminus of HSV-1 glycoprotein B (gB) and in the amino terminus of gK cause extensive virus-induced cell fusion. Although gB is known to be a fusogenic glycoprotein, the mechanism by which gK is involved in virus-induced cell fusion remains elusive. To delineate the amino-terminal domains of gK involved in virus-induced cell fusion, the recombinant viruses gKΔ31-47, gKΔ31-68, and gKΔ31-117, expressing gK carrying in-frame deletions spanning the amino terminus of gK immediately after the gK signal sequence (amino acids [aa] 1 to 30), were constructed. Mutant viruses gKΔ31-47 and gKΔ31-117 exhibited a gK-null (ΔgK) phenotype characterized by the formation of very small viral plaques and up to a 2-log reduction in the production of infectious virus in comparison to that for the parental HSV-1(F) wild-type virus. The gKΔ31-68 mutant virus formed substantially larger plaques and produced 1-log-higher titers than the gKΔ31-47 and gKΔ31-117 mutant virions at low multiplicities of infection. Deletion of 28 aa from the carboxyl terminus of gB (gBΔ28syn) caused extensive virus-induced cell fusion. However, the gBΔ28syn mutation was unable to cause virus-induced cell fusion in the presence of the gKΔ31-68 mutation. Transient expression of a peptide composed of the amino-terminal 82 aa of gK (gKa) produced a glycosylated peptide that was efficiently expressed on cell surfaces only after infection with the HSV-1(F), gKΔ31-68, ΔgK, or UL20-null virus. The gKa peptide complemented the gKΔ31-47 and gKΔ31-68 mutant viruses for infectious-virus production and for gKΔ31-68/gBΔ28syn-mediated cell fusion. These data show that the amino terminus of gK modulates gB-mediated virus-induced cell fusion and virion egress.
Herpes simplex virus type 1 (HSV-1) specifies at least 11 virally encoded glycoproteins, as well as several nonglycosylated and lipid-anchored membrane-associated proteins, which serve important functions in virion infectivity and virus spread. Although cell-free enveloped virions can efficiently spread viral infection, virions can also spread by causing cell fusion of adjacent cellular membranes. Virus-induced cell fusion, which is caused by viral glycoproteins expressed on infected cell surfaces, enables transmission of virions from one cell to another, avoiding extracellular spaces and exposure of free virions to neutralizing antibodies (reviewed in reference 56). Most mutations that cause extensive virus-induced cell-to-cell fusion (syncytial or syn mutations) have been mapped to at least four regions of the viral genome: the UL20 gene (5, 42, 44); the UL24 gene (37, 58); the UL27 gene, encoding glycoprotein B (gB) (9, 51); and the UL53 gene, coding for gK (7, 15, 35, 53, 54, 57).
Increasing evidence suggests that virus-induced cell fusion is mediated by the concerted action of glycoproteins gD, gB, and gH/gL. Recent studies have shown that gD interacts with both gB and gH/gL (1, 2). Binding of gD to its cognate receptors, including Nectin-1, HVEM, and others (12, 29, 48, 59, 60, 62, 63), is thought to trigger conformation changes in gH/gL and gB that cause fusion of the viral envelope with cellular membranes during virus entry and virus-induced cell fusion (32, 34). Transient coexpression of gB, gD, and gH/gL causes cell-to-cell fusion (49, 68). However, this phenomenon does not accurately model viral fusion, because other viral glycoproteins and membrane proteins known to be important for virus-induced cell fusion are not required (6, 14, 31). Specifically, gK and UL20 were shown to be absolutely required for virus-induced cell fusion (21, 46). Moreover, syncytial mutations within gK (7, 15, 35, 53, 54, 57) or UL20 (5, 42, 44) promote extensive virus-induced cell fusion, and viruses lacking gK enter more slowly than wild-type virus into susceptible cells (25). Furthermore, transient coexpression of gK carrying a syncytial mutation with gB, gD, and gH/gL did not enhance cell fusion, while coexpression of the wild-type gK with gB, gD, and gH/gL inhibited cell fusion (3).
Glycoproteins gB and gH are highly conserved across all subfamilies of herpesviruses. gB forms a homotrimeric type I integral membrane protein, which is N glycosylated at multiple sites within the polypeptide. An unusual feature of gB is that syncytial mutations that enhance virus-induced cell fusion are located exclusively in the carboxyl terminus of gB, which is predicted to be located intracellularly (51). Single-amino-acid substitutions within two regions of the intracellular cytoplasmic domain of gB were shown to cause syncytium formation and were designated region I (amino acid [aa] positions 816 and 817) and region II (aa positions 853, 854, and 857) (9, 10, 28, 69). Furthermore, deletion of 28 aa from the carboxyl terminus of gB, disrupting the small predicted alpha-helical domain H17b, causes extensive virus-induced cell fusion as well as extensive glycoprotein-mediated cell fusion in the gB, gD, and gH/gL transient-coexpression system (22, 49, 68). The X-ray structure of the ectodomain of gB has been determined and is predicted to assume at least two major conformations, one of which may be necessary for the fusogenic properties of gB. Therefore, perturbation of the carboxyl terminus of gB may alter the conformation of the amino terminus of gB, thus favoring one of the two predicted conformational structures that causes membrane fusion (34).
The UL53 (gK) and UL20 genes encode multipass transmembrane proteins of 338 and 222 aa, respectively, which are conserved in all alphaherpesviruses (15, 42, 55). Both proteins have multiple sites where posttranslational modification can occur; however, only gK is posttranslationally modified by N-linked carbohydrate addition (15, 35, 55). The specific membrane topologies of both gK and UL20 protein (UL20p) have been predicted and experimentally confirmed using epitope tags inserted within predicted intracellular and extracellular domains (18, 21, 44). Syncytial mutations in gK map predominantly within extracellular domains of gK and particularly within the amino-terminal portion of gK (domain I) (18), while syncytial mutations of UL20 are located within the amino terminus of UL20p, shown to be located intracellularly (44). A series of recent studies have shown that HSV-1 gK and UL20 functionally and physically interact and that these interactions are necessary for their coordinate intracellular transport and cell surface expression (16, 18, 21, 26, 45). Specifically, direct protein-protein interactions between the amino terminus of HSV-1 UL20 and gK domain III, both of which are localized intracellularly, were recently demonstrated by two-way coimmunoprecipitation experiments (19).
According to the most prevalent model for herpesvirus intracellular morphogenesis, capsids initially assemble within the nuclei and acquire a primary envelope by budding into the perinuclear spaces. Subsequently, these virions lose their envelope through fusion with the outer nuclear lamellae. Within the cytoplasm, tegument proteins associate with the viral nucleocapsid and final envelopment occurs by budding of cytoplasmic capsids into specific trans-Golgi network (TGN)-associated membranes (8, 30, 47, 70). Mature virions traffic to cell surfaces, presumably following the cellular secretory pathway (33, 47, 61). In addition to their significant roles in virus-induced cell fusion, gK and UL20 are required for cytoplasmic virion envelopment. Viruses with deletions in either the gK or the UL20 gene are unable to translocate from the cytoplasm to extracellular spaces and accumulated as unenveloped virions in the cytoplasm (5, 15, 20, 21, 26, 35, 36, 38, 44, 55). Current evidence suggests that the functions of gK and UL20 in cytoplasmic virion envelopment and virus-induced cell fusion are carried out by different, genetically separable domains of UL20p. Specifically, UL20 mutations within the amino and carboxyl termini of UL20p allowed cotransport of gK and UL20p to cell surfaces, virus-induced cell fusion, and TGN localization, while effectively inhibiting cytoplasmic virion envelopment (44, 45).
In this paper, we demonstrate that the amino terminus of gK expressed as a free peptide of 82 aa (gKa) is transported to infected cell surfaces by viral proteins other than gK or UL20p and facilitates virus-induced cell fusion caused by syncytial mutations in the carboxyl terminus of gB. Thus, functional domains of gK can be genetically separated, as we have shown previously (44, 45), as well as physically separated into different peptide portions that retain functional activities of gK. These results are consistent with the hypothesis that the amino terminus of gK directly or indirectly interacts with and modulates the fusogenic properties of gB.
MATERIALS AND METHODS
Cells.
African green monkey kidney (Vero) cells and human embryonic kidney (HEK293T) cells were obtained from the American Type Culture Collection (Rockville, MD). Cells were maintained in Dulbecco's modified Eagle's medium (Gibco-BRL, Grand Island, NY) supplemented with 10% fetal calf serum and antibiotics. The gK-transformed cell line VK302 was originally obtained from D. C. Johnson, Oregon Health Sciences University, and maintained as described earlier (20, 36).
Plasmids.
A gene cassette encoding the gKa peptide was constructed to include a 3× FLAG epitope inserted in frame after gK aa 82 and cloned into transient expression vector p3XFLAG-CMV14 (Invitrogen, Inc.). gKa-A40V and gKa-A40T plasmids were constructed by PCR-assisted mutagenesis using the gKa plasmid as the template, as described previously (13). The gKa-R plasmid contained the same 82-aa sequence cloned in reverse orientation into the pcDNA 3.1 plasmid (Invitrogen). Plasmids expressing T7 polymerase, the firefly luciferase gene under T7 promoter control, and plasmid control vector pCAGGS were obtained from Richard Longnecker (Northwestern University).
Recombinant-virus construction.
The mutant viruses gKΔ31-47 (encoding gK carrying an in-frame deletion of aa 31 to 47), gKΔ31-68, gKΔ31-117, gK-null (ΔgK), gBΔ28syn (deletion of 28 aa from the carboxyl terminus of gB), and gKΔ31-68/gBΔ28syn were created as described earlier (40), using a markerless two-step red recombination mutagenesis system (67) implemented on the bacterial artificial chromosome plasmid pYEbac102 carrying the HSV-1(F) genome (a kind gift from Y. Kawaguchi, Japan). All ΔgK recombinant viruses specified gK carrying in-frame deletions spanning the amino terminus of gK immediately after the gK signal sequence (aa 1 to 30). The ΔgK recombinant virus was created by inserting a kanamycin resistance cassette after aa position 17 within the gK signal sequence. The recombinant virus gBΔ28syn was constructed by deleting the carboxyl-terminal 28 aa of gB and inserting one stop codon at the site of the deletion. This viral genome was utilized for construction of the double mutant virus gKΔ31-68/gBΔ28syn.
Plaque morphology and one-step viral growth kinetics.
Visual analysis of plaque morphology of mutant viruses was performed essentially as we have described previously (27, 44, 46). Specifically, photographs of viral plaques were taken at a ×25 magnification on a Leica model DM IRB inverted wide-angle microscope. Seventy randomly selected plaques were imaged for each virus. The resulting images were analyzed using Zeiss AxioVision SP1 software, release 4.7.1.0 (Carl Zeiss MicroImaging GmbH, Jena, Germany), to determine the area of each plaque in square pixels. The plaque area measurements were analyzed using the SAS statistical package, version 9.1.3. Distributions were examined for normality using the univariate procedure with a Shapiro-Wilks test of normality. The general linear-model procedure was used to conduct a one-way analysis of variance on the data. When overall analyses determined significance (P ≤ 0.05), Tukey's honestly significant difference test was used to examine pairwise differences between the means for each of the five mutants.
One-step growth kinetics was performed as we have described previously (18, 21, 25). Briefly, viruses were adsorbed on wells of a six-well plate of nearly confluent Vero cell monolayers at 4°C for 1 h at a multiplicity of infection (MOI) of 0.1 (low MOI) and an MOI of 2 (high MOI). Thereafter, plates were incubated at 37°C and 5% CO2 and virus was allowed to penetrate for 1 h at 37°C. Any remaining extracellular virus was inactivated by low-pH treatment (pH 3.0), and cells were incubated at 37°C and 5% CO2. Supernatants and cell pellets (in the same volume of cell medium as supernatants) were collected at different times postinfection and stored at −80°C. Virus titers were determined (in triplicate) by endpoint titration of virus stocks on VK302 cells.
Transfections and Western immunoblot analyses.
Subconfluent Vero cells in six-well plates were transfected with 5 μg of plasmid gKa using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's directions. Twenty-four hours posttransfection, cells were infected with the gBΔ28syn virus (MOI of 2). Twenty-four hours postinfection (hpi), cells were processed for Western blot analysis. Cells were collected by low-speed centrifugation, washed with phosphate-buffered saline (PBS), and lysed at room temperature for 15 min in mammalian protein extraction reagent supplemented with a cocktail of protease inhibitors (Invitrogen-Life Technologies, Carlsbad, CA). Lysed Vero cell extracts were treated with endoglycosidase H (endo H) or peptide N-glycosidase F (PNGase F) as recommended by manufacturer instructions. Samples were further processed for sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting as detailed previously (19, 25, 40).
Complementation of the replication of gK mutant viruses by the gKa peptide.
Nearly confluent monolayers of Vero cells (triplicates) were transfected with either gKa or control gKa-R plasmid containing the gKa coding sequence cloned in the reverse orientation. Twenty-four hours posttransfection, cells were infected with either the gKΔ31-47 or the gKΔ31-68 virus. Virus stocks were prepared at 9 and 21 hpi, and virus titers were determined on VK302 cell monolayers as described above.
Immunohistochemistry/immunocytochemistry.
Vero cell monolayers in 12-well plates were transfected with gKa and gKa-R. Twenty-four hours posttransfection, cells were infected at an MOI of 0.2 with gKΔ31-68/gBΔ28syn. Thirty hpi, monolayers were washed with Tris-buffered saline (TBS)-Ca-Mg and either fixed with 100% methanol for 10 min or left unfixed (live). Immunohistochemistry was performed by utilizing a Vector Laboratories Vectastain Elite ABC kit essentially as described in the manufacturer's directions. Briefly, cells were washed with TBS-Ca-Mg and incubated in TBS blocking buffer supplemented with normal horse serum-normal goat serum at room temperature for 1 h. After the blocking step, cells were incubated with anti-FLAG antibody (1:500) (Sigma Chemical) or polyclonal rabbit anti-HSV-1 antibody (1:500) (Dako) in TBS blocking buffer for 1 h. Cells were subsequently washed three times and incubated with biotinylated horse anti-mouse antibody and goat anti-rabbit secondary antibody. Excess antibody was removed by four washes, and the cells were subsequently incubated with Vectastain Elite ABC reagent for 30 min. Finally, cells were washed three times with TBS-Ca-Mg and developed with Nova Red substrate (Vector Laboratories) according to the manufacturer's direction. Cells were visualized under a light microscope.
Flow cytometry assay.
Subconfluent monolayers of 293T cells in T25 flasks were transfected with 8 μg of plasmid gKa using Superfect (Qiagen). Twenty-four hours posttransfection, cells were infected with HSV-1 wild-type, ΔgK, and UL20-null viruses (MOI of 0.1). Twelve hpi, cells were treated with Accutase (Innovative Cell Technologies, Inc., San Diego, CA) to obtain a single-cell suspension, and the numbers were adjusted to 106 cells/ml. Appropriately diluted concentrations of green Live/Dead fixable dead cell stain (Invitrogen) were added to 100-μl cell suspensions and incubated for 10 min at 37°C. Cells were then washed two times with Dulbecco's PBS-bovine serum albumin wash buffer and stained with anti-FLAG-phycoerythrin antibody (clone M2; Abcam) for 30 min on ice in the dark, followed by two washes with Dulbecco's PBS-bovine serum albumin wash buffer. Cells were kept on ice, and data were acquired within 6 h of staining by use of a FACSAria flow cytometer (BD immunocytometry system). At least 200,000 events were collected by gating on live cells, and the data were analyzed using FlowJo software (TreeStar, Inc.), version 8.7.1. Percentages of FLAG-positive cells were calculated and compared between different treatment groups.
Complementation of gB-mediated virus-induced cell fusion.
Nearly confluent Vero cell monolayers were transfected with plasmids expressing gKa, gKa-R, gKa-A40V (Ala-to-Val change), or gKa-A40T (Ala-to-Thr change). Twenty-four hpi, the cells were infected with the gKΔ31-68/gBΔ28syn virus (MOI of 0.2). Thirty to 36 hpi, cells were fixed with ice-cold methanol and cell fusion was visualized by immunostaining using either anti-FLAG or anti-HSV-1 antibody, as described above.
Quantification of cell-to-cell fusion.
Quantification of cell fusion was accomplished using a chemiluminescence-based system as described previously (43, 50, 52). Briefly, subconfluent Vero cells (effector cells) in six-well plates were transfected with the plasmid expressing gKa, gKa-A40V, gKa-A40T, or gKa-R (200 ng of each) and T7 RNA polymerase (200 ng per well). Target cells in six-well plates were transfected with a plasmid vector expressing the luciferase gene under the control of the T7 promoter (200 ng). The total amount of DNA transfected per well was kept constant at 2.4 μg per well by adding the pCAGGS empty vector plasmid DNA. Lipofectamine 2000 (Invitrogen) was used for all transfections. Six hours posttransfection, fresh medium was added, and after 5 h of incubation, cells were detached using trypsin-EDTA. Cells were washed once and resuspended in 1 ml of complete medium. Effector and target cells were mixed in a 1:1 ratio (100 μl plus 100 μl) and seeded in a 24-well plate. Twelve hours postseeding, the monolayers were infected at an MOI of 0.2 with gKΔ31-68/gBΔ28syn virus. Twenty-four hpi, cells were washed with PBS and lysed with passive lysis buffer, and supernatants were collected. A luciferase reporter assay system (Promega, Madison, WI) was used to quantify the luciferase activity. Luciferin substrate was added to the supernatants, and luminescence was measured with a TD-20/20 luminometer (Turner Designs).
RESULTS
Construction of recombinant HSV-1(F) viruses carrying deletions in gK and gB.
Most syncytial mutations in gK are conserved amino acid changes located in the amino terminus of gK predicted to localize in extracellular spaces (18) (Fig. 1). To characterize the amino-terminal domains of gK involved in virus-induced cell fusion, a set of three recombinant viruses carrying in-frame deletions immediately following the signal sequence of gK were constructed using the two-step red recombination method implemented on the HSV-1(F) genome cloned into a bacterial artificial chromosome, as described in Materials and Methods. Specifically, the recombinant viruses gKΔ31-47, gKΔ31-68, and gKΔ31-117 encoded gK carrying in-frame deletions of aa 31 to 47, 31 to 68, and 31 to 117, respectively. In addition, a new ΔgK virus was generated by inserting a kanamycin gene cassette within the DNA segment encoding the gK signal sequence immediately after codon 17 (Thr).
FIG. 1.

HSV-1 gK membrane topology. (A) The experimentally validated topology of gK is shown in conjunction with the secondary predicted structure of gK (18). Features of the gK topology include four membrane-spanning hydrophobic domains (hpd1 to -4), a signal sequence of 30 aa, and two N-linked glycosylation sites (CHO). The locations of different syncytial mutations published previously (17, 18) are indicated by stars. (B) Schematic showing the in-frame truncations within the amino terminus of gK demarcated with dotted lines spanning aa 31 to 47, 31 to 68, and 31 to 117. Similar dotted markings are also included in panel A. The gene construct encoding the amino-terminal 82 aa of gK in-frame with the 3× FLAG epitope is shown.
Single-amino-acid replacements or deletions within the carboxyl terminus of gB are known to cause extensive virus-induced cell fusion (9, 10, 28, 69). We previously showed that deletion of the carboxyl-terminal 28 aa of gB causes extensive cell fusion, while larger deletions, which encompassed the entire predicted alpha-helical domain H17a, were lethal for virus replication and prevented virus-induced cell fusion (4). The recombinant virus gBΔ28syn was constructed by deleting the carboxyl-terminal 28 aa of gB and inserting one stop codon at the site of the deletion. This virus was similar to the previously characterized virus HSV-1(KOS) 1511gB(Δ28), with the exception that the gBΔ28syn virus was constructed on the HSV-1(F) genome cloned into YEbac102 by using the two-step red recombination method to ensure that all viruses used in the present study were constructed in similar manners and in the same genetic background. To investigate the potential relationships between the amino-terminal domains of gK and the carboxyl terminus of gB, a double recombinant virus was constructed by introducing the gKΔ31-68 mutation in the gBΔ28syn genetic background into the YEbac102 HSV-1(F) genome, as described in Materials and Methods. All mutant viruses were confirmed by DNA sequencing prior to recovery as infectious virus in Vero cells, as well as after virus stocks were made in Vero or VK302 cells (see Materials and Methods).
Kinetics of viral replication and plaque morphologies of recombinant viruses carrying mutations in gK and/or gB.
Recombinant viruses gKΔ31-47 and gKΔ31-117 produced viral plaques that were very similar to those produced by the ΔgK virus on Vero cells. In contrast, the recombinant virus gKΔ31-68 formed plaques of intermediate size between the ΔgK and HSV-1(F) wild-type viruses on Vero cells (Fig. 2). All gK mutant viruses were efficiently complemented in VK302 cells, which constitutively express gK, indicating that these mutant viruses did not contain any secondary mutations elsewhere in their genomes.
FIG. 2.

Plaque morphologies and relative sizes of gK mutant and wild-type HSV-1(F) viruses. Vero cells were infected with an MOI of 0.001, and viral plaques were fixed with methanol and stained with anti-HSV antibodies as described in Materials and Methods. (A) Representative viral plaques of all gK mutant viruses and the HSV-1(F) wild-type virus are shown on both Vero (A to E) and VK302 (F to J) cells (constitutively expressing gK). (B) Bar graph showing the average plaque sizes of 70 randomly chosen viral plaques for each virus; error bars indicate the standard error (see Materials and Methods).
Replication kinetics of all mutant viruses were performed at both a high MOI (MOI of 2) and a low MOI (MOI of 0.1) on both Vero and VK302 cells. At an MOI of 0.1, the gKΔ31-47 and gKΔ31-117 viruses replicated in a manner similar to that of the ΔgK virus, achieving viral titers of nearly 3 logs less than those of the HSV-1(F) parental virus (reconstituted from pYEbac102bac) within infected cells (Fig. 3A). However, the number of infectious virions found extracellularly was reduced by nearly 4 logs (Fig. 3B). In contrast, the gKΔ31-68 recombinant virus replicated >1 log more than the ΔgK, gKΔ31-47, and gKΔ31-117 viruses and approximately 1 log less than the parental HSV-1(F) virus intracellularly (Fig. 3A), while viral titer differences for the same viruses produced extracellularly approached 2 logs (Fig. 3B). At an MOI of 2, all gK amino-terminal deletion mutant viruses replicated in similar manners, achieving intracellular and extracellular viral titers which were approximately 1.5 logs higher than those for the ΔgK virus and approximately 1 log lower than that for the HSV-1(F) wild-type virus (Fig. 3C and D). The ratios of viral titers obtained at 24 hpi from intracellular (cells) and extracellular spaces were calculated (Fig. 3F). These ratios revealed that all viruses egressed to extracellular spaces more efficiently at high-MOI than at low-MOI infections. However, the gK mutant virus egress from infected cells was substantially less efficient than that of the HSV-1(F) wild-type virus at low-MOI infections (Fig. 3F). All viruses replicated in similar manners in VK302 cells at an MOI of 0.1 (Fig. 3E) or an MOI of 2 (not shown), indicating that the gK mutant viruses were unlikely to contain any secondary genomic mutations.
FIG. 3.

One-step replication kinetics of HSV-1(F) wild-type (wt) and gK mutant viruses. Vero cells were infected at either a low MOI (MOI of 0.1) or a high MOI (MOI of 2), and the numbers of infectious viruses produced were determined on VK302 cells at different times postinfection. At each time point, supernatants and infected cells were collected, and virus titers were determined separately. Viral titers after low-MOI infection are shown in panels A (intracellular) and B (supernatant). High-MOI titers are shown in panels C (intracellular) and D (supernatant). Viral titers obtained after low-MOI infections on VK302 cells are shown in panel E. Ratios of intracellular to extracellular viral titers obtained at 24 hpi are shown in panel F. gKins17, ΔgK recombinant virus created by inserting a kanamycin resistance cassette after aa position 17 within the gK signal sequence.
Deletion of the carboxyl-terminal 28 aa of gB caused extensive virus-induced cell fusion, producing a distinctive syncytial virus plaque on Vero cells (Fig. 4A). In contrast, the gKΔ31-68/gBΔ28syn mutant virus produced nonsyncytial, substantially smaller plaques on Vero cells (Fig. 4B), while it produced syncytial viral plaques on the complementing VK302 cells (Fig. 4D). The gBΔ28syn virus appeared to produce more-extensive cell fusion in VK302 cells than in Vero cells (Fig. 4C).
FIG. 4.

Plaque morphologies of the gBΔ28syn and gKΔ31-68/gBΔ28syn viruses on Vero and VK302 cells. Viral plaques were obtained on both Vero and VK302 cells and visualized after methanol fixing and immunostaining with anti-HSV antibodies at 48 hpi. Magnified parts of viral plaques are shown as insets in each photograph.
Transient expression and characterization of the gKa peptide, containing the amino-terminal 82 aa of gK.
We investigated whether the amino terminus of gK could complement in trans the gKΔ31-68 defects for infectious-virus production and gBΔ28syn-mediated virus-induced cell fusion. A gene cassette encoding the gKa peptide was constructed to include a 3× FLAG epitope inserted in frame after gK aa 82 and cloned into transient expression vector p3XFLAG-CMV14 (Invitrogen, Inc.). Transient expression in Vero cells produced protein species migrating with an apparent molecular mass of approximately 12 to 22 kDa in the absence or presence of infection with the gBΔ28syn virus (Fig. 5, lanes 5 and 6, respectively). Endo H recognizes and cleaves high-mannose precursor carbohydrate chains characteristic of glycosylation occurring in the rough endoplasmic reticulum of cells, while Golgi network-dependent glycosylation causes resistance to endo H digestion. In contrast, the enzyme PNGase F cleaves mature N-linked carbohydrate chains that contain terminal sugars, such as fucose and galactose (65, 66). Treatment of transfected or transfected and subsequently infected Vero cell extracts with endo H or PNGase F revealed that the gKa-related peptide species were sensitive to both endo H and PNGase F action. Transfected cells subsequently infected with the gBΔ28syn virus appeared to produce a small portion of glycosylated gKa which was resistant to glycosidase action (Fig. 5, lanes 2 and 4).
FIG. 5.
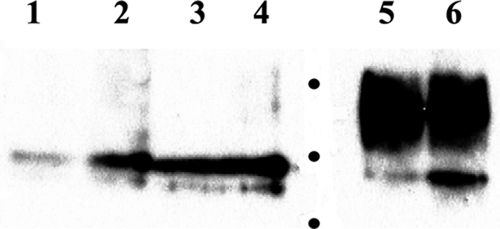
Transient expression of the gKa peptide. Vero cells were transfected with the gKa-expressing plasmid, and gKa expression was detected by a Western immunoblot assay utilizing the anti-FLAG antibody. Cellular extracts from transfected Vero cells (lane 5) and transfected cells followed by infection with the gBΔ28syn virus (lane 6) are shown. Cellular extracts were treated with either endo H (lane 1, gKa transfection alone; lane 2, gKa transfection followed by gBΔ28syn infection) or PNGase F (lane 3, gKa transfection alone; lane 4, gKa transfection followed by gBΔ28syn infection). Molecular mass markers, shown as dots, are 10, 15, and 20 kDa.
Complementation of the replication of gK mutant viruses by the gKa peptide.
To assess whether the gKa peptide complemented the replication defect of mutant viruses gKΔ31-47 and gKΔ31-68, Vero cells were transfected either with the gK-expressing plasmid or with the control gKa-R plasmid containing the gKa coding sequence cloned in the reverse orientation. This gene cassette did not code for a peptide or the FLAG epitope, since it lacked an initiation codon and contained two internal stop codons (not shown). All transfected Vero cells (in triplicate) were subsequently infected with either the gKΔ31-47 or the gKΔ31-68 virus. Virus stocks were prepared at 9 or 21 hpi, and virus titers were determined on VK302 cell monolayers. Cell-free viral titers were increased, on average, twofold by the gKa peptide compared with gKa-R (Fig. 6A). Intracellular titers of infected cultures were increased approximately threefold for the gKΔ31-47 virus and 50% for the gKΔ31-68 virus in the gKa- versus the gKa-R-transfected cells (Fig. 6B). Overall viral titers at 9 hpi were approximately twofold higher for the gKa- than for the gK-R-transfected cells. Expression of the full gK gene produced approximately sixfold-higher levels of infectious viruses than did expression of the gKa peptide for the gKΔ3147 virus (Fig. 6C), and virus titers were twofold higher for the ΔgK31-68 virus than for the gKa peptide (not shown).
FIG. 6.

Complementation of viral replication of gK mutant viruses by the gKa peptide. Vero cells were transfected with the gKa-expressing plasmid or a control plasmid containing the gKa coding sequence cloned in reverse (see Materials and Methods), and 24 h posttransfection, cells were infected with either the gKΔ31-47 or the gKΔ31-68 virus at an MOI of 1. To better visualize the ∼2-fold complementation for viral replication effect, the numbers of viral plaques obtained at the 10−3 dilution for extracellular (A) and combined extracellular and intracellular (B) virus stocks obtained at 21 hpi are shown. Complementation by wild-type gK for total virus (extracellular plus intracellular) at 9 hpi in comparison to complementation by gKa is shown in panel C. Error bars indicate the standard error.
Virus-dependent cell surface expression of the gKa peptide.
The expression of the gKa peptide on cell surfaces was examined using immunocytochemistry under both fixed (methanol) and live-cell conditions. Vero cells were transfected with plasmids expressing either the gKa peptide or the control plasmid gKa-R. Twenty-four hours posttransfection with either the gKa or the gKa-R plasmid, transfected cells were infected with the gKΔ31-68/gBΔ28syn virus. Positive reactions were visualized after staining with anti-FLAG and anti-HSV antibodies, as described in Materials and Methods. The anti-FLAG antibody efficiently detected expression of the gKa peptide in methanol-fixed Vero cells transfected with the gKa-expressing plasmid and subsequently infected with the gKΔ31-68/gBΔ28syn virus. FLAG-positive cells appeared to form small syncytia throughout the Vero cell culture. As expected, the anti-FLAG antibody did not react with Vero cells transfected with the gKa-R plasmid and subsequently infected with the gKΔ31-68/gBΔ28syn virus (Fig. 7A, panels c and d). The anti-HSV antibody detected HSV proteins in cells transfected with either the gKa or the gKa-R plasmid followed by infection with the gKΔ31-68/gBΔ28syn virus. However, an increased amount of labeling was noted for cells transfected with the gKa peptide in comparison to that for cells transfected with the gKa-R plasmid. Furthermore, cells expressing the gKa peptide appeared to form large syncytia that were positive for HSV gene products (Fig. 7A, panel a). Similar experiments were performed, with the exception that Vero cell monolayers were labeled with anti-FLAG or anti-HSV antibody under live conditions. Previous work has shown that under live reaction conditions, antibodies detect only cell surface-expressed proteins (18, 23). The anti-FLAG antibody detected gKa on Vero cell surfaces but did not react with the control Vero cell monolayer, which was transfected with the gKa-R plasmid (Fig. 7B, panels c and d). The anti-HSV antibody detected HSV proteins in Vero cells transfected with either the gKa or the gKa-R plasmid and subsequently infected with the gKΔ31-68/gBΔ28syn virus. However, Vero cells expressing the gKa peptide appeared to form relatively large syncytia, which were stained with the anti-HSV antibody (Fig. 7B, panels a and b).
FIG. 7.

Cell surface expression of the gKa peptide. Vero cells were transfected with the gKa plasmid or the control plasmid gKa-R, and transfected cells were infected with the gKΔ31-68/gBΔ28syn virus. Cells were stained with either the anti-FLAG (α-FLAG) or the anti-HSV-1 (α-HSV) antibody at 30 hpi under either methanol-fixed (A) or live (B) conditions. The percentages of FLAG-positive live cells in gKa-transfected cells infected with either wild-type HSV-1(F) or the ΔgK or ΔUL20 mutant are shown as contour plots (C) and as bar graphs (D). SSC, side scatter.
To quantify the relative amounts of gKa peptide that were expressed on cell surfaces in the presence or absence of virus infection and to determine whether gK or UL20p was required for gKa cell surface expression, 293T cells were transfected with the gKa plasmid alone or with the gKa plasmid followed by infection with wild-type HSV-1(F), ΔgK, or ΔUL20 virus. 293T cells were used in these experiments instead of Vero cells, because they could readily be prepared as single-cell populations for cytometry analysis. Single-cell suspensions were generated by using Accutase treatment as described in Materials and Methods. Cells were gated on live cells, and the portion of this population of cells that stained positive for the anti-FLAG-phycoerythrin antibody was measured. Infection with any of these three viruses increased the basal level of FLAG epitope detected in cells transfected with the gKa plasmid by more than threefold (relative to that with gKa alone), indicating that viral infection substantially increased cell surface expression of the gKa peptide independently of gK or UL20p (Fig. 7C and D).
Complementation of gB-mediated virus-induced cell fusion by the gKa peptide, but not by gKa peptides containing syncytial mutations.
A number of syncytial mutations have been mapped to the amino terminus of gK. One of these types of mutation is a single-amino-acid change at Ala to either Val or Thr at aa position 40 (17, 54). To test for the ability of the gKa peptide or gKa-derived peptides containing either the Ala-to-Val or the Ala-to-Thr mutation to complement gKΔ31-68/gBΔ28syn virus-induced cell fusion, Vero cells were transfected with plasmids encoding gKa, gKa-A40V, or gKa-A40T. Subsequently, transfected cells were infected with the gKΔ31-68/gBΔ28syn virus, and the amount of virus-induced cell fusion was monitored using phase-contrast microscopy. Expression of the gKa, gKa-A40V, and gKa-A40T peptides was detected efficiently using the anti-FLAG antibody in methanol-fixed Vero cells, while there was no reaction with cells transfected with the control plasmid gKa-R (Fig. 8A, panel e). The anti-HSV antibody detected expression of viral proteins in all infected cell monolayers; however, cells expressing the gKa peptide appeared to form relatively large syncytia, while cells expressing either the gKa-A40V or the gKa-A40T peptide did not exhibit any visible signs of virus-induced cell fusion (Fig. 8A, panels a to d). Cell fusion was also evident in cells expressing the gKa peptide stained with the anti-FLAG antibody (Fig. 8A, panel f). To quantify the relative amount of virus-induced cell fusion produced by transient expression of the gKa peptide, a previously described chemiluminescence assay was employed (43) (see Materials and Methods). Control samples were prepared from Vero cells transfected with either the T7-driven luciferase plasmid alone mixed with untransfected Vero cells or two populations of Vero cells transfected with the T7-driven luciferase and the T7 polymerase, respectively. These two cell populations emitted background chemiluminescence after infection with the gKΔ31-68/gBΔ28syn virus. Vero cells expressing the gKa peptide and subsequently infected with the gKΔ31-68/gBΔ28syn virus emitted 17 times more chemiluminescence than control cells that did not express the gKa peptide. Vero cells expressing the gKa-A40V or the gKa-A40T mutant peptide produced background chemiluminescence at levels comparable to those for negative controls (Fig. 8B).
FIG. 8.

Ability of the gKa peptide to complement gKΔ31-68/gBΔ28syn-induced cell fusion. (A) Vero cells were transfected with the gKa, gKa-R, gKa-A40V, or gKa-A40T plasmid, and cells were subsequently infected with the gKΔ31-68/gBΔ28syn virus at an MOI of 0.2. Cells were reacted with the anti-FLAG (α-FLAG) or the anti-HSV (α-HSV) antibody under methanol-fixed conditions and visualized by phase-contrast microscopy after immunostaining. (B) Vero cells were transfected and infected as detailed above, with the exception that two different populations of cells were transfected at the same time, with each of the gK-expressing plasmids with a plasmid expressing T7 polymerase (pol) or a plasmid expressing the luciferase gene under the T7 promoter. These equal populations of cells were mixed together prior to infection with the gKΔ31-68/gBΔ28syn virus. The amount of relative fusion was obtained by measuring the relative level of luminescence (RLU) emitted by cellular extracts at 24 hpi. Error bars indicate the standard error.
DISCUSSION
HSV-1-induced cell fusion is a complex process that involves the concerted actions of multiple viral glycoproteins and membrane proteins. Generally, it is thought that glycoproteins gD, gB, gH, and gL form a functional complex that is required for both virus entry and virus-induced cell fusion. Specifically, it has been hypothesized that initial binding of gD to its cognate host receptors triggers conformational and possibly sequential changes to gH/gL and then gB to promote membrane fusion (11, 64). However, in viral infections gK and UL20p are absolutely required for virus-induced cell fusion, and syncytial mutations in either gK or UL20p cause extensive cell fusion. Here, we provide evidence in support of a model in which the gK/UL20p complex regulates infectious-virus production and gB-mediated cell fusion via the amino terminus of gK.
Most of the syncytial mutations in gK are contained within the amino-terminal segment, which has been predicted and experimentally verified to lie extracellularly (17, 18). Generally, these mutations cause conservative amino acid changes, which are not predicted to alter the secondary structure of the amino terminus of gK. One of these loci is syn20, located at aa position 40, close to the gK signal sequence and immediately before the first N-glycosylation site (Fig. 1). syn20 mutations are known to cause extensive virus-induced cell fusion of all cell types, unlike gB syncytial mutations, which can cause weaker fusion in a more limited repertoire of cells (41). The experiments described herein have revealed that the amino terminus of gK is involved in infectious-virus production and virus-induced cell fusion. Specifically, mutant virus gKΔ31-68 appeared to replicate more efficiently than mutant virus gKΔ31-47 or gKΔ31-117. This result suggests that aa 48 to 67 contain a gK domain that regulates infectious-virus production. Furthermore, the ratio of intracellular to extracellular gKΔ31-68 virions was more than fivefold higher than that for the HSV-1(F) virus at a low MOI, indicating a defect in virion egress. Consequently, viral plaques produced by the gKΔ31-68 virus were on average half the size of wild-type viral plaques, and the quantity of infectious virus produced was approximately 1 log less.
We have shown previously that the gK/UL20 complex has genetically separable functions in virion envelopment at the TGN and in virus-induced cell fusion at the plasma membrane, presumably through extracellular domains of gK and UL20 that modulate other viral glycoproteins involved in virus-induced cell fusion (44, 45). It is possible that aa 48 to 67 alter the gK structure and its interactions with UL20p, causing defects in cytoplasmic virion envelopment and egress. This potential steric hindrance may be relieved by the slightly larger gK deletion of aa 31 to 68, allowing for improved cytoplasmic envelopment and egress. At high-MOI infections, all gK mutant viruses appeared to egress substantially more efficiently than at low-MOI infections. In this regard, higher expression of several viral proteins may compensate for the egress defects of the gK mutant viruses because of the increased stability of the gK mutant proteins through interactions with other viral proteins. In support of this hypothesis, gK mutant viruses appear to replicate more efficiently in Vero cells constitutively expressing UL20p, suggesting that overexpression of UL20p may partially complement gK defects (not shown).
UL20p and gK are known to be essential for virion envelopment. However, UL20p and gK domains that function in cytoplasmic virion envelopment can be genetically segregated from domains that function in virus-induced cell fusion (44, 45). UL20p and gK domains that function in virion envelopment are located exclusively within their intracellular domains, which are suspected to interact with one or more tegument and capsid proteins during the process of cytoplasmic virion envelopment. Therefore, it is possible that perturbation of the amino terminus of gK may alter the overall structure of gK and UL20p, causing the observed decrease in infectious-virus production and virion egress. Alternatively, mutant gK carrying deleted portions of the gK amino terminus may affect unknown cytoplasmic events that create a hostile environment for viral replication. In this regard, it is noted that overexpression of gK in certain cell lines reduces viral titers in part by blocking viral replication and nuclear egress from gK-transformed cells (24).
One of the most interesting aspects of this work is the finding that the gBΔsyn28 mutation, which is known to cause extensive virus-induced cell fusion, was drastically inhibited in the presence of the gKΔ31-68 mutation at both low and high MOIs. Similar findings were observed for the gKΔ31-47 mutation (not shown). However, expression of the gKa peptide, composed of the amino-terminal 82 aa of gK, efficiently complemented the gKΔ31-47 and gKΔ31-68 amino-terminal deletion mutants of gK for infectious-virus production as well as the gKΔ31-68/gBΔ28syn for virus-induced cell fusion. The gKa peptide was efficiently synthesized in cells under transient-expression conditions, and it was N glycosylated, as evidenced by its sensitivity to both endo H and PNGase enzymes. Importantly, the gKa peptide was transported to cell surfaces only after infection with the wild-type virus or the gK or UL20-null mutant virus. This result indicates that the gKa peptide did not require gK or UL20p for cell surface expression, suggesting the involvement of other viral proteins. Attempts to detect the gKΔ31-68 and gKΔ31-47 mutant gKs tagged with the V5 epitope tag at their carboxyl termini failed to detect significant amounts of these gKs on cell surfaces (not shown), suggesting that transport of the gKa peptide to cell surfaces and its ability to modulate gB fusion may be independent of the gKΔ31-68 or gKΔ31-47 gK molecule. Apparently, the majority of the gKa peptide contained high-mannose carbohydrates known to be added in the rough endoplasmic reticulum. High-mannose-containing HSV-1 precursor glycoproteins can be transported to cell surfaces under conditions where Golgi network-dependent glycosylation is inhibited (39). Therefore, it is possible that the gKa peptide is transported to cell surfaces by non-Golgi network- and -TGN-dependent pathways or, alternatively, that it cannot be glycosylated efficiently while traversing these intracellular compartments. A small amount of gK peptide appeared to be resistant to endo H treatment, suggesting that some gKa peptides may contain peripheral carbohydrates added in the Golgi and TGN compartments.
Transfection of the gKa-expressing plasmid typically produced 50% of the 293T cells expressing the gKa peptide (not shown). Viral infections of the gK-expressing cells were performed at a low MOI (0.2). Therefore, it can be calculated that a maximum of 10% of cells would be both transfected and infected (0.2 × 50%). The percentages of cells expressing the gKa peptide on their cell surfaces were 4.48% after infection with HSV-1(F) and 4.21% after infection with the ΔgK virus. Therefore, virus-dependent pathways appeared to efficiently transport gKa to cell surfaces, since a high proportion of the infected cells expressed the gKa peptide on their cell surfaces.
Transient expression of gKa peptides that carried syncytial mutations (Ala to Val or Ala to Thr at aa 40) failed to complement gKΔ31-68/gBΔ28syn-induced cell fusion. This result suggests that the gKa-A40V or gKa-A40T peptide was unable to facilitate the necessary conformational changes of the amino terminus of gB required for virus-induced cell fusion normally exerted by the intact gK. It is worth noting that expression of full-length gK appeared to inhibit cell fusion caused by transient expression of gB, gD, gH, and gL, whereas gK containing the syn1 mutation did not (3), indicating that the syn1 mutation in gK alters the functional, and possibly physical, relationships among gK, gB, gD, and gH/gL.
Overall, these results suggest a mechanistic model by which gK may regulate gB-mediated virus-induced cell fusion. According to this model, the amino terminus of gK is required for gB-mediated virus-induced cell fusion, suggesting that gB cannot assume a fully functional fusogenic conformation in the absence of the amino terminus of gK. It is possible that the gKa peptide interacts with the remaining portion of gK (gKΔ31-68), restoring gK functions. However, the gKa peptide was transported to cell surfaces after infection with the ΔgK or UL20-null virus. This indicates that interactions with gKΔ31-68 are probably not necessary for trafficking to the cell surfaces, thereby suggesting potential interactions of the gKa peptide with other viral or cellular proteins. Previous work has demonstrated that gK syn1 mutations do not cause virus-induced cell fusion in the absence of gB and that gB syn3 mutations do not cause fusion in the absence of UL20p (21). Moreover, certain mutations within the amino terminus of the UL20p protein (located intracellularly) cause extensive virus-induced cell fusion, while others inhibit gB-mediated cell fusion (44, 45). We favor the hypothesis that the carboxyl terminus of gB directly or indirectly interacts with the amino terminus of UL20p. In this model, gB syncytial mutations affect interactions between the carboxyl terminus of gB and the amino terminus of UL20p intracellularly, causing changes in the extracellular interactions of the amino terminus of gK with gB. Additional experiments are needed to further clarify the roles of both gK and UL20p in infectious-virus production and virus-induced cell fusion and to discern the biological significance of UL20p/gK regulation of virus-induced cell fusion.
Acknowledgments
This work was supported by grant AI43000 from the National Institute of Allergy and Infectious Diseases to K.G.K. We acknowledge financial support by the LSU School of Veterinary Medicine to BIOMMED. D. V. Chouljenko was supported by an Louisiana Board of Regents Economic Development graduate student fellowship.
We acknowledge J. Walker for proofreading the manuscript.
Footnotes
Published ahead of print on 30 September 2009.
REFERENCES
- 1.Atanasiu, D., J. C. Whitbeck, T. M. Cairns, B. Reilly, G. H. Cohen, and R. J. Eisenberg. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. USA 104:18718-18723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avitabile, E., C. Forghieri, and G. Campadelli-Fiume. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 81:11532-11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avitabile, E., G. Lombardi, and G. Campadelli-Fiume. 2003. Herpes simplex virus glycoprotein K, but not its syncytial allele, inhibits cell-cell fusion mediated by the four fusogenic glycoproteins, gD, gB, gH, and gL. J. Virol. 77:6836-6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baghian, A., L. Huang, S. Newman, S. Jayachandra, and K. G. Kousoulas. 1993. Truncation of the carboxy-terminal 28 amino acids of glycoprotein B specified by herpes simplex virus type 1 mutant amb1511-7 causes extensive cell fusion. J. Virol. 67:2396-2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baines, J. D., P. L. Ward, G. Campadelli-Fiume, and B. Roizman. 1991. The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress. J. Virol. 65:6414-6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balan, P., N. Davis-Poynter, S. Bell, H. Atkinson, H. Browne, and T. Minson. 1994. An analysis of the in vitro and in vivo phenotypes of mutants of herpes simplex virus type 1 lacking glycoproteins gG, gE, gI or the putative gJ. J. Gen. Virol. 75:1245-1258. [DOI] [PubMed] [Google Scholar]
- 7.Bond, V. C., and S. Person. 1984. Fine structure physical map locations of alterations that affect cell fusion in herpes simplex virus type 1. Virology 132:368-376. [DOI] [PubMed] [Google Scholar]
- 8.Browne, H., S. Bell, T. Minson, and D. W. Wilson. 1996. An endoplasmic reticulum-retained herpes simplex virus glycoprotein H is absent from secreted virions: evidence for reenvelopment during egress. J. Virol. 70:4311-4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bzik, D. J., B. A. Fox, N. A. DeLuca, and S. Person. 1984. Nucleotide sequence of a region of the herpes simplex virus type 1 gB glycoprotein gene: mutations affecting rate of virus entry and cell fusion. Virology 137:185-190. [DOI] [PubMed] [Google Scholar]
- 10.Cai, W. Z., S. Person, C. DebRoy, and B. H. Gu. 1988. Functional regions and structural features of the gB glycoprotein of herpes simplex virus type 1. An analysis of linker insertion mutants. J. Mol. Biol. 201:575-588. [DOI] [PubMed] [Google Scholar]
- 11.Campadelli-Fiume, G., M. Amasio, E. Avitabile, A. Cerretani, C. Forghieri, T. Gianni, and L. Menotti. 2007. The multipartite system that mediates entry of herpes simplex virus into the cell. Rev. Med. Virol. 17:313-326. [DOI] [PubMed] [Google Scholar]
- 12.Campadelli-Fiume, G., F. Cocchi, L. Menotti, and M. Lopez. 2000. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 10:305-319. [DOI] [PubMed] [Google Scholar]
- 13.Chouljenko, V., S. Jayachandra, G. Rybachuck, and K. G. Kousoulas. 1996. Efficient long PCR site directed mutagenesis of a high GC-template. BioTechniques 21:472-480. [DOI] [PubMed] [Google Scholar]
- 14.Davis-Poynter, N., S. Bell, T. Minson, and H. Browne. 1994. Analysis of the contributions of herpes simplex virus type 1 membrane proteins to the induction of cell-cell fusion. J. Virol. 68:7586-7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Debroy, C., N. Pederson, and S. Person. 1985. Nucleotide sequence of a herpes simplex virus type 1 gene that causes cell fusion. Virology 145:36-48. [DOI] [PubMed] [Google Scholar]
- 16.Dietz, P., B. G. Klupp, W. Fuchs, B. Kollner, E. Weiland, and T. C. Mettenleiter. 2000. Pseudorabies virus glycoprotein K requires the UL20 gene product for processing. J. Virol. 74:5083-5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolter, K. E., R. Ramaswamy, and T. C. Holland. 1994. Syncytial mutations in the herpes simplex virus type 1 gK (UL53) gene occur in two distinct domains. J. Virol. 68:8277-8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foster, T. P., X. Alvarez, and K. G. Kousoulas. 2003. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): the UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J. Virol. 77:499-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foster, T. P., V. N. Chouljenko, and K. G. Kousoulas. 2008. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J. Virol. 82:6310-6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foster, T. P., and K. G. Kousoulas. 1999. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J. Virol. 73:8457-8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foster, T. P., J. M. Melancon, J. D. Baines, and K. G. Kousoulas. 2004. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 78:5347-5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foster, T. P., J. M. Melancon, and K. G. Kousoulas. 2001. An alpha-helical domain within the carboxyl terminus of herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) is associated with cell fusion and resistance to heparin inhibition of cell fusion. Virology 287:18-29. [DOI] [PubMed] [Google Scholar]
- 23.Foster, T. P., J. M. Melancon, T. L. Olivier, and K. G. Kousoulas. 2004. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J. Virol. 78:13262-13277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foster, T. P., G. V. Rybachuk, X. Alvarez, O. Borkhsenious, and K. G. Kousoulas. 2003. Overexpression of gK in gK-transformed cells collapses the Golgi apparatus into the endoplasmic reticulum inhibiting virion egress, glycoprotein transport, and virus-induced cell fusion. Virology 317:237-252. [DOI] [PubMed] [Google Scholar]
- 25.Foster, T. P., G. V. Rybachuk, and K. G. Kousoulas. 2001. Glycoprotein K specified by herpes simplex virus type 1 is expressed on virions as a Golgi complex-dependent glycosylated species and functions in virion entry. J. Virol. 75:12431-12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuchs, W., B. G. Klupp, H. Granzow, and T. C. Mettenleiter. 1997. The UL20 gene product of pseudorabies virus functions in virus egress. J. Virol. 71:5639-5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fulmer, P. A., J. M. Melancon, J. D. Baines, and K. G. Kousoulas. 2007. UL20 protein functions precede and are required for the UL11 functions of herpes simplex virus type 1 cytoplasmic virion envelopment. J. Virol. 81:3097-3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gage, P. J., M. Levine, and J. C. Glorioso. 1993. Syncytium-inducing mutations localize to two discrete regions within the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B. J. Virol. 67:2191-2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geraghty, R. J., C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618-1620. [DOI] [PubMed] [Google Scholar]
- 30.Granzow, H., B. G. Klupp, W. Fuchs, J. Veits, N. Osterrieder, and T. C. Mettenleiter. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 75:3675-3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haanes, E. J., C. M. Nelson, C. L. Soule, and J. L. Goodman. 1994. The UL45 gene product is required for herpes simplex virus type 1 glycoprotein B-induced fusion. J. Virol. 68:5825-5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hannah, B. P., E. E. Heldwein, F. C. Bender, G. H. Cohen, and R. J. Eisenberg. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 81:4858-4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harley, C. A., A. Dasgupta, and D. W. Wilson. 2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J. Virol. 75:1236-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heldwein, E. E., H. Lou, F. C. Bender, G. H. Cohen, R. J. Eisenberg, and S. C. Harrison. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217-220. [DOI] [PubMed] [Google Scholar]
- 35.Hutchinson, L., K. Goldsmith, D. Snoddy, H. Ghosh, F. L. Graham, and D. C. Johnson. 1992. Identification and characterization of a novel herpes simplex virus glycoprotein, gK, involved in cell fusion. J. Virol. 66:5603-5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hutchinson, L., and D. C. Johnson. 1995. Herpes simplex virus glycoprotein K promotes egress of virus particles. J. Virol. 69:5401-5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobson, J. G., S. H. Chen, W. J. Cook, M. F. Kramer, and D. M. Coen. 1998. Importance of the herpes simplex virus UL24 gene for productive ganglionic infection in mice. Virology 242:161-169. [DOI] [PubMed] [Google Scholar]
- 38.Jayachandra, S., A. Baghian, and K. G. Kousoulas. 1997. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J. Virol. 71:5012-5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kousoulas, K. G., S. Person, and T. C. Holland. 1982. Herpes simplex virus type 1 cell fusion occurs in the presence of ammonium chloride-inhibited glycoproteins. Virology 123:257-263. [DOI] [PubMed] [Google Scholar]
- 40.Lee, H. C., V. N. Chouljenko, D. V. Chouljenko, M. J. Boudreaux, and K. G. Kousoulas. 2009. The herpes simplex virus type 1 glycoprotein D (gD) cytoplasmic terminus and full-length gE are not essential and do not function in a redundant manner for cytoplasmic virion envelopment and egress. J. Virol. 83:6115-6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manservigi, R., P. G. Spear, and A. Buchan. 1977. Cell fusion induced by herpes simplex virus is promoted and suppressed by different viral glycoproteins. Proc. Natl. Acad. Sci. USA 74:3913-3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McLean, G., F. Rixon, N. Langeland, L. Haarr, and H. Marsden. 1990. Identification and characterization of the virion protein products of herpes simplex virus type 1 gene UL47. J. Gen. Virol. 71:2953-2960. [DOI] [PubMed] [Google Scholar]
- 43.McShane, M. P., and R. Longnecker. 2005. Analysis of fusion using a virus-free cell fusion assay. Methods Mol. Biol. 292:187-196. [DOI] [PubMed] [Google Scholar]
- 44.Melancon, J. M., T. P. Foster, and K. G. Kousoulas. 2004. Genetic analysis of the herpes simplex virus type 1 UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J. Virol. 78:7329-7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melancon, J. M., P. A. Fulmer, and K. G. Kousoulas. 2007. The herpes simplex virus UL20 protein functions in glycoprotein K (gK) intracellular transport and virus-induced cell fusion are independent of UL20 functions in cytoplasmic virion envelopment. Virol. J. 4:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Melancon, J. M., R. E. Luna, T. P. Foster, and K. G. Kousoulas. 2005. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J. Virol. 79:299-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427-436. [DOI] [PubMed] [Google Scholar]
- 49.Muggeridge, M. I. 2000. Characterization of cell-cell fusion mediated by herpes simplex virus 2 glycoproteins gB, gD, gH and gL in transfected cells. J. Gen. Virol. 81:2017-2027. [DOI] [PubMed] [Google Scholar]
- 50.Okuma, K., M. Nakamura, S. Nakano, Y. Niho, and Y. Matsuura. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254:235-244. [DOI] [PubMed] [Google Scholar]
- 51.Pellett, P. E., K. G. Kousoulas, L. Pereira, and B. Roizman. 1985. Anatomy of the herpes simplex virus 1 strain F glycoprotein B gene: primary sequence and predicted protein structure of the wild type and of monoclonal antibody-resistant mutants. J. Virol. 53:243-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pertel, P. E., A. Fridberg, M. L. Parish, and P. G. Spear. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313-324. [DOI] [PubMed] [Google Scholar]
- 53.Pogue-Geile, K. L., G. T. Lee, S. K. Shapira, and P. G. Spear. 1984. Fine mapping of mutations in the fusion-inducing MP strain of herpes simplex virus type 1. Virology 136:100-109. [DOI] [PubMed] [Google Scholar]
- 54.Pogue-Geile, K. L., and P. G. Spear. 1987. The single base pair substitution responsible for the syn phenotype of herpes simplex virus type 1, strain MP. Virology 157:67-74. [DOI] [PubMed] [Google Scholar]
- 55.Ramaswamy, R., and T. C. Holland. 1992. In vitro characterization of the HSV-1 UL53 gene product. Virology 186:579-587. [DOI] [PubMed] [Google Scholar]
- 56.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 3rd ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 57.Ruyechan, W. T., L. S. Morse, D. M. Knipe, and B. Roizman. 1979. Molecular genetics of herpes simplex virus. II. Mapping of the major viral glycoproteins and of the genetic loci specifying the social behavior of infected cells. J. Virol. 29:677-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sanders, P. G., N. M. Wilkie, and A. J. Davison. 1982. Thymidine kinase deletion mutants of herpes simplex virus type 1. J. Gen. Virol. 63:277-295. [DOI] [PubMed] [Google Scholar]
- 59.Satoh, T., J. Arii, T. Suenaga, J. Wang, A. Kogure, J. Uehori, N. Arase, I. Shiratori, S. Tanaka, Y. Kawaguchi, P. G. Spear, L. L. Lanier, and H. Arase. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shukla, D., J. Liu, P. Blaiklock, N. W. Shworak, X. Bai, J. D. Esko, G. H. Cohen, R. J. Eisenberg, R. D. Rosenberg, and P. G. Spear. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13-22. [DOI] [PubMed] [Google Scholar]
- 61.Skepper, J. N., A. Whiteley, H. Browne, and A. Minson. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment → deenvelopment → reenvelopment pathway. J. Virol. 75:5697-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spear, P. G., R. J. Eisenberg, and G. H. Cohen. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1-8. [DOI] [PubMed] [Google Scholar]
- 63.Spear, P. G., and R. Longnecker. 2003. Herpesvirus entry: an update. J. Virol. 77:10179-10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Subramanian, R. P., and R. J. Geraghty. 2007. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. USA 104:2903-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tarentino, A. L., C. M. Gomez, and T. H. Plummer, Jr. 1985. Deglycosylation of asparagine-linked glycans by peptide:N-glycosidase F. Biochemistry 24:4665-4671. [DOI] [PubMed] [Google Scholar]
- 66.Tarentino, A. L., and F. Maley. 1974. Purification and properties of an endo-beta-N-acetylglucosaminidase from Streptomyces griseus. J. Biol. Chem. 249:811-817. [PubMed] [Google Scholar]
- 67.Tischer, B. K., J. von Einem, B. Kaufer, and N. Osterrieder. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. BioTechniques 40:191-197. [DOI] [PubMed] [Google Scholar]
- 68.Turner, A., B. Bruun, T. Minson, and H. Browne. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 72:873-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Walev, I., M. Lingen, M. Lazzaro, K. Weise, and D. Falke. 1994. Cyclosporin A resistance of herpes simplex virus-induced “fusion from within” as a phenotypical marker of mutations in the Syn 3 locus of the glycoprotein B gene. Virus Genes 8:83-86. [DOI] [PubMed] [Google Scholar]
- 70.Zhu, Z., M. D. Gershon, Y. Hao, R. T. Ambron, C. A. Gabel, and A. A. Gershon. 1995. Envelopment of varicella-zoster virus: targeting of viral glycoproteins to the trans-Golgi network. J. Virol. 69:7951-7959. [DOI] [PMC free article] [PubMed] [Google Scholar]