

Abstract
Nearly all livers transplanted into hepatitis C virus (HCV)-positive patients become infected with HCV, and 10 to 25% of reinfected livers develop cirrhosis within 5 years. Neutralizing monoclonal antibody could be an effective therapy for the prevention of infection in a transplant setting. To pursue this treatment modality, we developed human monoclonal antibodies (HuMAbs) directed against the HCV E2 envelope glycoprotein and assessed the capacity of these HuMAbs to neutralize a broad panel of HCV genotypes. HuMAb antibodies were generated by immunizing transgenic mice containing human antibody genes (HuMAb mice; Medarex Inc.) with soluble E2 envelope glycoprotein derived from a genotype 1a virus (H77). Two HuMAbs, HCV1 and 95-2, were selected for further study based on initial cross-reactivity with soluble E2 glycoproteins derived from genotypes 1a and 1b, as well as neutralization of lentivirus pseudotyped with HCV 1a and 1b envelope glycoproteins. Additionally, HuMAbs HCV1 and 95-2 potently neutralized pseudoviruses from all genotypes tested (1a, 1b, 2b, 3a, and 4a). Epitope mapping with mammalian and bacterially expressed proteins, as well as synthetic peptides, revealed that HuMAbs HCV1 and 95-2 recognize a highly conserved linear epitope spanning amino acids 412 to 423 of the E2 glycoprotein. The capacity to recognize and neutralize a broad range of genotypes, the highly conserved E2 epitope, and the fully human nature of the antibodies make HuMAbs HCV1 and 95-2 excellent candidates for treatment of HCV-positive individuals undergoing liver transplantation.
Hepatitis C virus (HCV) is a major cause of liver failure and infects more than 170 million people worldwide. HCV is a member of the Flaviviridae family and contains a 9.6-kb positive-strand RNA genome. The genome is translated into a single polypeptide that is cleaved by viral and cellular proteases into at least nine different proteins. The major HCV surface glycoproteins, E1 and E2, form a noncovalent heterodimer on the virion surface (23) and are believed to mediate viral entry via a complex set of poorly understood interactions with cellular coreceptors, including CD81 (28), claudin-1 (8), occludin (29), scavenger receptor class B type I (30), and others (38). The E2 glycoprotein has been shown to interact directly with receptors (38); currently, no function has been assigned to E1, although it is known to be required for viral infection. These viral glycoproteins provide an obvious target for neutralizing monoclonal antibodies (MAbs).
Isolation of potently neutralizing HCV-specific MAbs has been complicated by the lack of an in vitro cell culture system to study the full infection cycle of the virus. Recently, systems have been developed that allow for the generation of infectious viral particles, highlighting the importance of E1 and E2 in viral binding and entry. A novel in vitro infection system employs HCV pseudotyped viral particles (HCVpp) generated from a lentivirus that are devoid of native glycoproteins and engineered to contain HCV glycoproteins E1 and E2 (4, 15). HCVpp specifically infect cell lines derived from human liver cells and can be neutralized by polyclonal and MAbs directed against the HCV envelope glycoproteins.
HCVpp have allowed the identification of antibodies that can neutralize HCV infection in cell culture. E1 has proven to be a difficult target for MAb-mediated neutralization, possibly because it appears to have low immunogenicity (32), has no identified binding proteins on the cell surface, and has an undefined role in cell entry. Despite this challenge, two groups have identified HCV neutralizing MAbs directed to E1: these MAbs are H-111, which has moderate neutralizing activity (17), and the recently isolated IGH505 and IGH526, which neutralize numerous HCV genotypes (1a, 1b, 2a, 4a, 5a, and 6a but not 2b and 3a) (22). Although they are predicted to inhibit viral binding or fusion, the mechanism by which these E1-directed MAbs neutralize HCV infection is unclear.
A diverse group of mouse anti-E2 antibodies, recognizing both linear and discontinuous epitopes, has been generated. Many of these MAbs showed broad neutralization of multiple HCV genotypes, but not surprisingly, several HCV isolates were refractory to neutralization. In contrast, AP33, a mouse MAb that largely recognizes a highly conserved linear epitope in the N terminus of E2 (amino acids 412 to 423), was identified as a broadly cross-reactive antibody that neutralized strains from all genotypes tested (1a, 1b, 2a, 2b, 3a, 4, 5, and 6), with the exception of one genotype 5 virus (UKN5.14.4; GenBank accession no. AY894682) (24). Recently, several cross-reactive neutralizing MAbs have been identified that are of human origin and have the capacity to neutralize a significant fraction of the genotypes tested (1, 5, 12, 13, 27, 31) or to neutralize all genotypes tested (16, 20, 25). As with the vast majority of previously described human MAbs (HuMAbs), these MAbs recognize conformation-dependent epitopes of E2. One broadly neutralizing human antibody, AR3B, was tested in a mouse model of infection and showed significant protection from viremia (20). Given the known function of the E2 envelope glycoprotein, the high level of immunogenicity, the surface vulnerability, and the abundance of data pertaining to E2 and HCV neutralization, E2 provides a promising target for the development of fully human neutralizing antibodies.
Liver deterioration due to HCV infection is the leading reason for liver transplantation in the United States. Unfortunately, it is highly likely that the transplanted liver will also become infected with HCV, and 10 to 25% of these patients develop cirrhosis within 5 years of transplant (9, 40). Here we describe the characterization of HuMAbs directed against the HCV E2 envelope glycoprotein, generated using transgenic mice. Based on epitope conservation and broad neutralization capacity, HuMAbs HCV1 and 95-2 provide excellent candidates for prevention of graft reinfection of HCV-infected individuals undergoing liver transplantation.
MATERIALS AND METHODS
Cells and cell culture.
Hep3B, HEK-293T/17, and P3X-AG8.653 cells were obtained from the American Type Culture Collection. Hep3B and HEK-293T/17 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 IU penicillin-streptomycin (PS). P3X-AG8.653 cells were grown in RPMI supplemented with 10% FBS, 4 mM glutamine, 1 mM pyruvate, 5 mM HEPES, 100 IU PS, and 55 μM 2-mercaptoethanol. Hybridomas were cultured in DMEM supplemented with 10% FBS, nonessential amino acids, 1× oxaloacetate-pyruvate-insulin (Sigma), 4 mM glutamine, 1× hypoxanthine-aminopterin-thymidine (Sigma), 100 IU PS, 1× hybridoma cloning factor (Bioveris), 55 μM 2-mercaptoethanol, and 10% NCTC-109 (Invitrogen). All cells were grown at 37°C in air supplemented with 5% CO2.
Cloning of soluble E2 envelope glycoprotein and full-length E1/E2 glycoprotein.
The amino acid sequences of the genotype 1a H77 core protein and E1 and E2 envelope glycoproteins were obtained from GenBank (accession no. NC004102). A codon-optimized gene encoding the structural proteins of HCV was constructed using overlapping oligonucleotides in conjunction with PCR (3), and the sequence was confirmed. To generate a codon-optimized gene encoding a soluble version of the E2 envelope glycoprotein (CO-E2661-1a), nucleotides encoding amino acids 364 to 661 of the codon-optimized HCV structural region were amplified to contain a 5′ HindIII restriction site followed by a Kozak sequence (GCCGCCACC) and a 3′ XbaI restriction site. The PCR product was digested with HindIII/XbaI and ligated into pcDNA3.1 (Invitrogen) containing a sequence encoding a His6 epitope tag and containing a stop codon immediately 3′ of the XbaI restriction site. The coding sequence of pcDNA3.1-CO-E2661-1a-His was subsequently confirmed. Sequences encoding HCV genotype 1b soluble E2 envelope glycoprotein (E2661-1b) were amplified from RNA extracted from the serum of an infected patient and cloned into pcDNA3.1 as described above. This construct contained the native HCV 1b gene and was not codon optimized (GenBank accession no. GQ259488).
Prototypical genotype 1a E1/E2 envelope glycoprotein genes were amplified from an H77 expression plasmid (p90HCVconsensuslongpU) (18) obtained through the AIDS Research and Reference Program, Division of AIDS, NIAID, NIH, from Charles M. Rice via Apath, LLC. Alternative E1/E2-encoding genes (genotypes 1a GR5, 1b, 2b, 3a, and 4a) were all amplified from HCV-infected patient sera (GenBank accession no. GQ379232, GQ259488, GQ379229, GQ379231, and GQ379230, respectively). All PCR products contained 5′ HindIII and 3′ XbaI restriction sites and were cloned into pcDNA3.1 containing a 3′ His6 epitope tag.
Expression and purification of soluble E2 envelope glycoproteins.
Constructs encoding soluble E2 envelope glycoproteins were transfected into HEK-293T/17 cells by using Lipofectamine 2000 (Invitrogen) as described by the manufacturer. Briefly, cells were grown to 80% confluence in 150-mm tissue culture dishes in 15 ml DMEM-10% FBS. Thirty micrograms of DNA mixed with 75 μl of Lipofectamine 2000 was added to the cells, and plates were incubated overnight at 37°C. Medium was removed and stored, and fresh complete DMEM was added to the cells. Cells were incubated for an additional 24 h prior to the addition of 3 mM sodium butyrate (Sigma) to the medium. Supernatants were removed from the plate after an additional 24-hour incubation. These supernatants were combined with the transfection supernatant and filtered using a 0.45-nm filter apparatus. Filtered supernatants were mixed with Ni-nitrilotriacetic acid agarose (Invitrogen) at a ratio of 0.5 ml of agarose for 40 ml of culture supernatant. Supernatant-agarose mixtures were incubated for 2 h on a rocking platform at room temperature. Supernatant was removed from the agarose by column filtration. Agarose was washed with phosphate-buffered saline (PBS) and protein eluted using 250 mM imidazole. Eluted protein was dialyzed against PBS for 2 h at room temperature and concentrated with an Amicon Centriprep YM-10 concentrator. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Coomassie blue staining were used to determine the purity of isolated proteins.
Mouse immunizations and hybridoma isolation.
HuMAb mice (Medarex Inc., Bloomsbury, NJ) are transgenic for human immunoglobulin genes; mouse heavy chain and kappa light chain immunoglobulin genes are inactivated. HuMAb mice were injected weekly with 100 μg of CO-E2661-1a, using RIBI (Corixa) as adjuvant, for a total of 6 to 16 weeks. Enzyme-linked immunosorbent assay (ELISA) was employed to measure serum responses to antigen, and animals were sacrificed when serum responses reached a plateau. Hybridomas were generated by the standard polyethylene glycol method, using P3X63-AG8.653 mouse myeloma cells as the fusion partner.
ELISA.
Culture supernatants or purified antibody was assessed for antigen binding by use of ELISA. Ninety-six-well microtiter plates were coated with 0.5 to 2 μg/ml of antigen in PBS overnight at 4°C. One hundred microliters of culture supernatant or purified antibody at various dilutions was added to the wells and incubated at 22°C for 2 h. Antibody binding was detected using an anti-human immunoglobulin G (IgG)-alkaline phosphatase (AP) conjugate (1:5,000; Jackson Immunoresearch) followed by p-nitrophenyl phosphate disodium salt (PNPP) at 1 mg/ml in 1 M diethanolamine. Absorbance (405 nm) was analyzed using a Molecular Devices Emax plate reader with Softmax software.
For peptide-specific ELISAs, peptides (New England Peptide) were covalently attached through the carboxy terminus to Covalink NH ELISA plates (Nunc) as described by the manufacturer. Covalink plates contain reactive secondary amines conjugated to the solid support, allowing covalent capture of molecules containing a free carboxylic acid group. Antibody binding to peptides was detected using identical reagents to those described above.
Antibody cloning and expression.
RNAs were extracted from hybridomas by using an RNeasy kit (Qiagen) as described by the manufacturer. For amplification of the antibody heavy chain variable regions, reverse transcription-PCR (RT-PCR) was performed using a Superscript III One-Step RT-PCR kit (Invitrogen) in combination with a cocktail of forward oligonucleotide primers complementary to all 5′ VH sequences encoded by the mouse and a reverse oligonucleotide complementary to the constant region of IgG1/IgG3. Heavy chain variable region PCR products were cloned into a mammalian expression vector in frame with the human IgG1 constant region contained in the vector. Light chain variable regions were amplified using a BD Smart RACE kit (BD Biosciences) as described by the manufacturer. Amplified light chain variable regions were cloned into a mammalian expression vector in frame with the human kappa constant region present in the vector. Heavy and light chain vectors were combined into a single vector and electroporated into CHO cells. Stable transfectants were selected and expanded.
Antibody purification.
Hybridoma cell cultures or stably transfected CHO cells were expanded, and supernatants were harvested by centrifugation. Culture supernatants were incubated with protein A or protein G Sepharose beads (GE Healthcare) for 2 h at room temperature while rocking. Beads were removed by column filtration and washed with PBS, and antibody was eluted with 100 mM glycine, pH 2.8. The eluate was dialyzed against PBS and concentrated using an Amicon YM-30 Centriprep concentrator as described by the manufacturer. Purified antibody was filter sterilized, and the protein concentration was determined by spectrophotometry. For large-scale antibody purification, HiTrap (Amersham) protein A column chromatography was employed, and purified antibody was concentrated as described above.
HCVpp neutralization assays.
Pseudovirus was generated by employing a human immunodeficiency virus backbone that contained a mutation to prevent human immunodeficiency virus envelope glycoprotein expression and a luciferase gene to direct luciferase expression in target cells (pNL4-3.Luc.R−E− [6, 14], obtained through the AIDS Research and Reference Program, Division of AIDS, NIAID, NIH, via Nathaniel Landau). HCV E1/E2 glycoproteins were provided in trans by cotransfection of HEK-293T/17 cells with pcDNA-E1/E2-H77-1a (prototypical H77 sequence) or pcDNA-genotypes-E1/E2 (E1/E2 sequences from other genotypes, obtained from patient sera) and with pNL4-3.Luc.R−E−. Supernatant containing virus particles was harvested at 48 to 72 h posttransfection, concentrated using Centricon 70 concentrators, aliquoted, and stored frozen at −80°C. Pseudovirus was preincubated with various concentrations (fivefold dilutions) of HuMAbs for 1 h at room temperature before being added to Hep3B cells. After incubation for 72 h, infection was quantitated by luciferase detection with the BrightGlo luciferase assay (Promega) and read in a Victor3 plate reader (Perkin Elmer) for light production.
Cloning, expression, and purification of N-terminal fragments of the E2 protein.
DNAs encoding N-terminal fragments of E2 that were longer than 12 amino acids were amplified from pcDNA3.1-CO-E2661-1a-His and cloned into pET-32a(+) (Novagen) in frame with the carboxy-terminal His6 epitope tag. For constructs encoding fragments of 12 amino acids in length, complementary phosphorylated oligonucleotides containing the appropriate overhangs were annealed and ligated into pET32a(+). All constructs were confirmed by DNA sequencing.
Escherichia coli BL21Star cells (Invitrogen) were transformed with pET32a(+)-E2 fragment vectors and grown overnight at 37°C in Luria-Bertani broth containing 100 μg/ml ampicillin (LB-amp). The culture was diluted 1:10 in LB-amp broth and grown for 2.5 h at 37°C, followed by the addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside), and then further grown at 37°C for 2.5 h. Bacteria were harvested by centrifugation, and pellets were frozen at −20°C. Bacteria were lysed and proteins purified as described previously (2), employing Ni-nitrilotriacetic acid agarose.
Generation of site-directed mutants in H77 1a E1/E2 expression plasmid.
Mutagenesis of the expression plasmid for H77 1a full-length E1/E2 (pCDNA-E1/E2-H77-1a) was performed using a QuikChange II site-directed mutagenesis kit (Stratagene) following the manufacturer's instructions. Following mutagenesis, the E1/E2-encoding region was sequenced to confirm the introduction of the desired mutation as well as the integrity of the entire coding sequence.
Biacore affinity determination.
Surface plasmon resonance technology was used to determine the affinities of HuMAbs HCV1 and 95-2 for the Trx-E2412-423-His protein. Each HuMAb was separately captured at nonsaturating levels on the surface of a Biacore chip that was coated with goat anti-human Fc antibody fragment. Various concentrations of Trx-E2412-423-His in solution were flowed over the Biacore chip with the captured HuMAb. Results for association and dissociation rate constants were calculated and used to derive the dissociation constant (KD) (33).
Informed consent.
Informed consent was obtained from healthy individuals and patients with chronic HCV infection without advanced liver disease enrolled in the study. The study underwent full committee review and was approved by the Committee for the Protection of Human Subjects in Research at the University of Massachusetts Medical School (Worcester, MA).
RESULTS
Generation and selection of HuMAbs directed against the HCV E2 envelope glycoprotein.
To generate a panel of MAbs directed against the HCV E2 protein, a codon-optimized, soluble version of the E2 envelope glycoprotein, CO-E2661-1a, consisting of amino acids 384 to 661 of E2 (strain H77, genotype 1a) fused with a carboxy-terminal His6 epitope tag, was produced in HEK-293T/17 cells and purified from culture supernatants by nickel-affinity chromatography. Mice transgenic for human immunoglobulin heavy (IgM, IgG1, and IgG3) and light (kappa) chain genes (HuMAb mice; Medarex Inc.) were immunized with soluble CO-E2661-1a glycoprotein to generate a panel of fully human E2-reactive MAbs. Serum titers directed against CO-E2661-1a were measured by antigen-specific ELISA, and splenic fusions were performed based on high serum responses to CO-E2661-1a. Spleens from a total of 51 mice were fused, and antigen-specific ELISA for human IgG yielded 487 hybridomas reactive to CO-E2661-1a.
A rapid screening procedure was devised to limit the number of hybridomas that would be evaluated extensively (Fig. 1). Given the requirement for a MAb to cross-react with multiple genotypes of HCV, we first assessed the reactivity of the CO-E2661-1a-reactive HuMAbs with genotype 1b E2 envelope glycoprotein. HCV RNA was isolated from the serum of a patient infected with HCV genotype 1b, and E2661 gene sequences were amplified and cloned into a mammalian expression vector (E2661-1b). The encoded amino acid sequence of the E2661-1b protein was 79% identical to the amino acid sequence of the CO-E2661-1a protein. Soluble E2661-1b glycoprotein was expressed and purified from HEK-293T/17 cells and used in ELISA to screen the antibodies contained in hybridoma supernatants. To increase the screening process throughput, a single dilution of supernatant was tested to grossly determine if antibody was cross-reactive. Of the 487 CO-E2661-1a-reactive antibodies, only 50 (∼10%) demonstrated recognition of E2661-1b (data not shown).
FIG. 1.
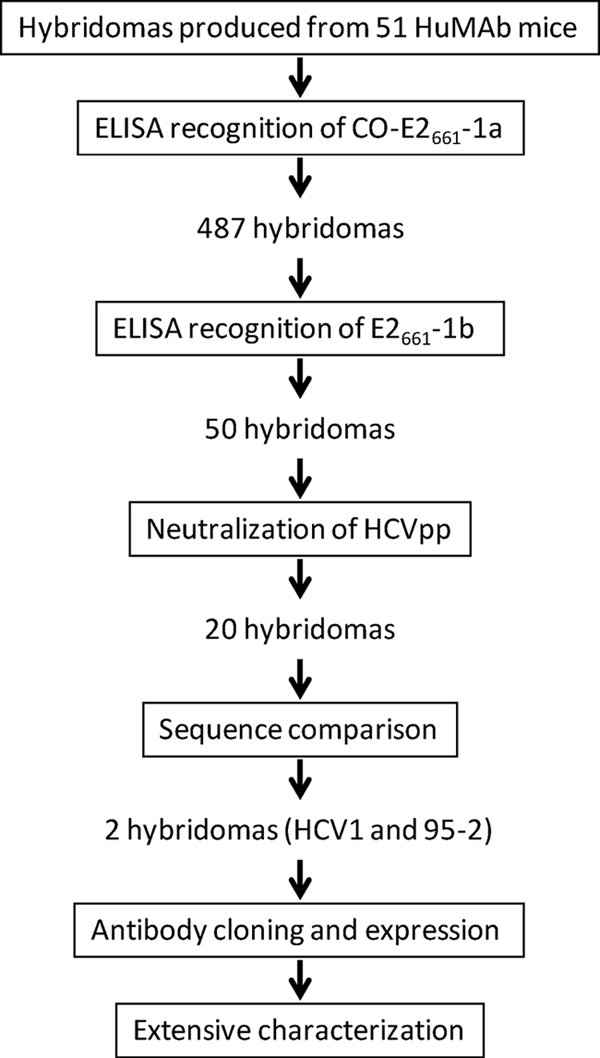
Schematic representation of rapid screening method devised to select lead-candidate hybridomas. The large panel of CO-E2661-1a-reactive hybridomas were screened for reactivity with E2661-1b as well as for neutralization of genotype 1a HCVpp. Cross-reactive, neutralizing antibodies produced by the hybridomas were sequenced and grouped according to both amino acid identity and similarity in VH gene usage. HCV1 and 95-2 were selected as lead candidates, cloned into expression vectors, and transfected into CHO cells for the production of recombinant antibody. Antibodies were purified and subsequently characterized more extensively.
To determine if E2-reactive antibodies from the selected hybridomas could prevent infection in vitro, a neutralization-of-infection assay using HCVpp was performed. Antibodies were purified from the 50 lead-candidate hybridomas and tested for the capacity to neutralize infection of genotype 1a (H77) HCVpp. Of the 50 cross-reactive antibodies, 20 were able to potently inhibit HCVpp infectivity, as measured by a reduction in light output generated from a luciferase gene engineered into the viral genome (data not shown). Of the 20 antibody-producing hybridomas that could bind HCV genotype 1a and 1b E2 soluble proteins and neutralize infection with genotype 1a HCVpp, 19 were isolated from mouse 97895 and one was from mouse 85083.
RNAs were purified from all 20 lead-candidate hybridomas, and sequence analysis of RT-PCR amplicons specific for the human IgG heavy chain determined that all 20 were >90% identical. Figure 2 represents the complementarity-determining regions (CDRs) of the three unique sequences from the 19 antibodies found from mouse 97895 (GenBank accession no. GQ423620, GQ423621, and GQ423622) and the one unique sequence from mouse 85083 (GenBank accession no. GQ423623). In addition, all possessed the VH3-33, D3-10, and JH4b gene segments. Interestingly, the 19 hybridomas from mouse 97895 contained human IgG1 constant regions, whereas the hybridoma from mouse 85083 contained a human IgG3 constant region. The lone hybridoma from mouse 85083 (HCV1) and one representative hybridoma from mouse 97895 (95-2) were chosen for further characterization.
FIG. 2.

CDRs of broadly cross-reactive lead-candidate HuMAbs. CDR1, CDR2, and CDR3 of the four unique sequences of cross-reactive anti-HCV HuMAbs are shown, with alignment scores denoted below the amino acid entries. 95-14, 95-38, and 95-2 were isolated from mouse 97895, and HCV1 was isolated from mouse 85083.
To produce the quantities of antibody required for our studies, the heavy and light chain genes of HuMAbs HCV1 and 95-2 were cloned into mammalian expression vectors and expressed in CHO cells. Note that the constant region of HCV1 was replaced with an IgG1 constant region to facilitate ease of purification. The relative affinity of cloned HCV1 in the context of an IgG1 heavy chain was essentially identical to that of the IgG3 HCV1 produced from the isolated hybridoma (data not shown), suggesting that this alteration was not deleterious.
HuMAbs HCV1 and 95-2 recognize and neutralize a broad range of HCVpp genotypes.
As discussed above, it was clear that HuMAbs HCV1 and 95-2 were able to recognize E2661 from both genotypes 1a and 1b. To assess the relative binding capacities of the HuMAbs for each genotype, a more extensive binding ELISA was performed. Dilution series of HCV1 (Fig. 3A) and 95-2 (Fig. 3B) were tested against CO-E2661-1a and E2661-1b in an ELISA. To control for equivalent coating of each soluble E2 protein, a His6-directed antibody (generated in-house) was employed, and the results are shown in Fig. 3C. Both HuMAbs gave similar binding profiles for both CO-E2661-1a and E2661-1b.
FIG. 3.

HuMAbs HCV1 and 95-2 interact with HCV E2 glycoproteins derived from genotypes 1a and 1b. HuMAbs HCV1 (A) and 95-2 (B) and mouse His6-directed antibody (C) were tested for the capacity to bind to soluble E2 glycoproteins derived from genotypes 1a and 1b in ELISA. ELISA plates were coated with 2 μg/ml of CO-E2661-1a (triangles) or E2661-1b (squares), and a dilution series of each antibody was introduced to the assay, as well as the mouse MAb directed against the His6 epitope tag present in each E2 protein. Bound antibody was detected employing goat anti-human secondary antibody (HuMAbs) or goat anti-mouse secondary antibody (mouse anti-His6) conjugated to AP, followed by development with PNPP substrate, and the absorbance at 405 nm for each dilution was plotted.
To determine the capacity of HuMAbs HCV1 and 95-2 to recognize and prevent infection of a broad range of genotypes in vitro, we employed the HCVpp system to assess neutralization of the prototype genotype 1a strain (H77) as well as a panel of pseudoviruses with HCV E1/E2 sequences isolated from patient sera representing genotypes 1a, 1b, 2b, 3a, and 4a. E1/E2 glycoprotein RNAs representing the various genotypes were obtained from patient serum samples (with the exception of 1a H77) and cloned into a mammalian expression vector. Lentiviral pseudovirus was generated for each genotype isolate by cotransfection of HEK-293T/17 cells with the HCV E1/E2 expression plasmid and the lentiviral backbone with a defective native glycoprotein gene and an engineered luciferase reporter gene. HCVpp were harvested from the culture supernatants, concentrated, and stored frozen in aliquots. The volume of HCVpp-containing supernatant that generated roughly 50,000 cps light output after infection of a human-derived liver cell line (Hep3B) was determined for each virus. This constant amount of virus was incubated with a range of dilutions (0.001 to 300 nM) of HuMAbs HCV1 and 95-2 and an irrelevant isotype-matched control to test for neutralization of HCVpp derived from the various genotypes (Fig. 4A to F). HuMAbs HCV1 and 95-2 were able to potently neutralize pseudoviruses bearing E1/E2 glycoproteins from genotypes 1a, 1b, 2b, 3a, and 4a, while the irrelevant HuMAb did not neutralize any of the different genotype HCVpp, even with the largest amount of antibody tested (Fig. 4A to F). It is clear that HuMAbs HCV1 and 95-2 have the capacity to bind and neutralize a broad range of HCV genotypes in the HCVpp model of infection.
FIG. 4.

HCV1 and 95-2 neutralize HCVpp representing a diverse group of HCV genotypes. Fivefold dilutions of HuMAbs HCV1 (squares) and 95-2 (diamonds) and an irrelevant HuMAb (negative control; triangles) were incubated for 1 h at room temperature with lentiviral pseudovirus with an engineered luciferase gene. The pseudovirus (HCVpp) was generated with E1/E2 glycoproteins from the following genotypes: H77 1a (A), GR5 1a (B), 1b (C), 2b (D), 3a (E), and 4a (F). The HCVpp-antibody mixture was added to Hep3B cells, followed by incubation at 37°C for 72 h. Infection was assessed with Brightglo luciferase reagent, followed by analysis using a Victor3 multilabel reader. Light output, expressed as counts per second (CPS), was plotted.
Epitope and affinity determination of HuMAbs HCV1 and 95-2.
To grossly determine which region of the E2 envelope glycoprotein was recognized by both HCV1 and 95-2, a series of mammalian-expressed carboxy-terminal truncations of E2 was generated. ELISA was utilized to determine that both HCV1 and 95-2 were specific for the amino-terminal 80 amino acids of E2 (data not shown). To further characterize the epitope, a panel of constructs was generated to express various amino acids within the N-terminal 80 amino acids of genotype 1a E2. This panel of proteins was expressed in bacteria as a fusion of HCV sequence with an N-terminal thioredoxin domain (Trx) and a C-terminal His6 epitope tag for purification and detection (Fig. 5). A total of eight proteins representing various regions of the E2 N terminus were expressed, purified, and used in subsequent ELISAs. Appropriate and consistent coating was confirmed using the His6-directed MAb (data not shown). HCV1 and 95-2 recognized only a subset of the eight N-terminal truncation proteins (Fig. 5). Interestingly, both HuMAbs HCV1 and 95-2 recognized all bacterially expressed truncation proteins that contained amino acids 412 to 423 of the E2 glycoprotein (Fig. 5). To confirm this region as the minimal epitope, a peptide with the sequence of E2 amino acids 412 to 423 (QLINTNGSWHIN) was covalently attached to ELISA plates through the carboxy terminus, using Covalink NH ELISA plates (Nunc) as described by the manufacturer. The epitope was strongly detected with HuMAbs HCV1 and 95-2 (data not shown). The antibodies gave no signal above background for a negative control peptide (amino acids 432 to 443) (data not shown). These data identify amino acids 412 to 423 as the epitope for both HCV1 and 95-2. They also suggest that HCV1 and 95-2 binding does not depend on E2 glycosylation, given that bacterial fusion proteins as well as synthetic peptides are not glycosylated.
FIG. 5.

HuMAbs HCV1 and 95-2 recognize amino acids 412 to 423 of the E2 envelope glycoprotein. The top graphic depicts the amino-terminal 80 amino acids of the E2 envelope glycoprotein, with the amino acid numbers indicated above the schematic. Hypervariable region 1 (HVR1) is denoted with hashing. Various bacterial fusion proteins representing different truncation protein products of the E2 glycoprotein were generated. The corresponding amino acids for each truncated E2 product are indicated. The various truncations were assayed in ELISA for recognition by HuMAbs HCV1 and 95-2. Bound antibodies were detected with goat anti-human antibody conjugated to AP, followed by PNPP substrate addition. ELISA results are listed to the right of the schematic; positive recognition is indicated by a plus sign, while signals equivalent to background are indicated by a minus sign.
The affinities of HuMAbs HCV1 and 95-2 for the E2 412-423 epitope were determined by surface plasmon resonance (Biacore). HuMAbs were captured on the surface of the Biacore chip, and binding constants were determined by assaying the ability of various concentrations of bacterially expressed thioredoxin fusion protein containing amino acids 412 to 423 of E2 (Trx-E2412-423-His) to bind to the chip. Both HCV1 and 95-2 were determined to have a high affinity (KD), at 3.8 nM and 1.9 nM, respectively, for this 12-amino-acid linear epitope.
Determination of residues in the HCV1 and 95-2 epitope required for binding.
The 412-423 region of the HCV E2 envelope glycoprotein has previously been recognized as having a high degree of conservation between a broad range of HCV genotypes (34). We analyzed the sequences of amino acids 412 to 423 of the E2 envelope glycoproteins from the panel of different genotypes of HCVpp that were neutralized by HuMAbs HCV1 and 95-2, shown in Fig. 4. The analysis revealed a high degree of conservation in the epitope, with conservative changes at only three amino acid positions, namely, 412, 414, and 416 (Fig. 6A). Clearly, these alterations in the 412-423 epitope do not abrogate HCV1 or 95-2 binding or neutralization. To further confirm that this epitope is refractory to mutation, we performed an extensive analysis of the amino acid sequences of residues 412 to 423 in E2 proteins found in the NIH HCV database (19). Of the sequences in the HCV database, 56% have an amino acid sequence at residues 412 to 423 that is identical to that of the H77-derived 1a glycoprotein that was used in this study. Sixteen percent of the sequences in the database are identical to the E2 412-423 sequence of the 1b and 3a E2 glycoproteins used to generate HCVpp described herein. The sequence for amino acids 412 to 423 of the 4a HCVpp E2 is identical to 8% of the sequences in the database, and the sequence for 2b HCVpp is identical to 0.4% of the sequences. The sequence diversity found at amino acids 412 to 423 in our panel of HCV1- and 95-2-neutralized HCVpp genotypes encompasses 80% of the E2 sequences reported in the NIH HCV sequence database, suggesting that HCV1 and 95-2 may have the capacity to neutralize a high percentage of HCV variants.
FIG. 6.

Determination of critical residues required for HCV1 and 95-2 binding. (A) The sequence of the 412-423 region of E2 is listed in one-letter amino acid code for the genotype 1a H77 strain that was used for HCVpp generation. Listed below that sequence are the sequences for all strains of HCVpp that were tested in Fig. 4. Positions identical to the position in H77 are indicated by dashes, while differences are indicated in one-letter amino acid code. (B) Bacterially expressed fusion proteins containing E2 amino acids 412 to 423 with the indicated amino acid mutated to alanine were assayed in ELISA with 0.052 nM HuMAbs HCV1 and 95-2. Bound antibodies were detected with goat anti-human antibody conjugated to AP. The data were plotted as percentages of the absorbance obtained with the wild-type Trx-E2412-423-His protein derived from native H77.
To define critical residues for both HCV1 and 95-2 binding, alanine scanning mutagenesis was performed with amino acids 412 to 423 in the Trx-E2412-423-His protein. The capacity of HuMAbs HCV1 and 95-2 to recognize each of the purified alanine substitution mutants was assessed by ELISA. Antibody specific for the His6 epitope tag was used as a control to ensure the use of similar amounts of input protein in the assay (data not shown). In this experiment, HCV1 and 95-2 were tested at a concentration representing half of that required to saturate binding (0.052 nM). The concentration was chosen in this manner to ensure that subtle binding differences would be detected. HuMAb HCV1 and 95-2 binding was reduced 75% to 90% by both the L413A and W420A mutations (Fig. 6B). Interestingly, the G418A mutation partially reduced HCV1 binding capacity but had no effect on the ability of 95-2 to bind. Increasing the concentration of HCV1 in the assay restored full binding capacity to the G418A mutant, indicating that the affinity of HCV1 for this mutation was reduced and not abolished (data not shown). These results demonstrate that HuMAbs HCV1 and 95-2 bind to the same epitope of the E2 glycoprotein and suggest that the molecular interactions between the E2 glycoprotein and the antibodies have modest differences.
The capacity of HuMAbs HCV1 and 95-2 to recognize diverse sequences that naturally occur in HCV infection was assessed in ELISA by selecting a panel of sequences from the NIH HCV database that contained changes in the 412-423 epitope (Fig. 7A). Additionally, a genotype 5 sequence (amino acids 415 to 418, QNGS) that was not neutralized by a mouse MAb (AP33) that also interacts with the 412-423 epitope was included (UKN5.14.4; GenBank accession no. AY894682) (24). Recognition with the His6 epitope tag antibody was used to control for differences in protein coating (data not shown). When used at a less-than-saturating concentration (0.026 nM), HuMAbs showed reduced binding to three of the mutant epitopes in the ELISA (Fig. 7A). HuMAbs HCV1 and 95-2 showed dramatically reduced binding to proteins with amino acids 415 to 418 changed to either KNGS or QNGS (Fig. 7A). Also, HCV1, but not 95-2, demonstrated reduced binding to a mutant protein containing changes at amino acids 413 and 414 (LI to FV).
FIG. 7.

HCV1 and 95-2 bind E2 amino acids 412 to 423 isolated from viruses of infected patients, representing a diverse group of genotypes. (A) Bacterially expressed fusion proteins containing E2 amino acids 412 to 423 of isolates identified from the NIH HCV database (y axis) were used to coat ELISA plates and probed with 0.026 nM HuMAbs HCV1 (black) and 95-2 (gray). The top-most sequence represents the H77 genotype 1a 412-423 sequence. In subsequent listings, positions identical to the position in H77 are indicated by dashes, and differences are listed in one-letter amino acid code. Absorbance was plotted on the x axis. Various dilutions of HuMAbs HCV1 (B) and 95-2 (C) were measured in ELISA against the E2 412-423 fusion proteins that gave suboptimal absorbance in panel A. Each assayed protein, with the corresponding amino acid sequence and symbol, is listed in the legend. As a control for maximal antibody binding kinetics, the fusion protein containing amino acids 412 to 423 of the H77 1a sequence (squares) was included. Absorbance was plotted as a function of antibody concentration.
To determine the extent to which these mutations affected HuMAb binding, more extensive dilution series of antibodies were tested in ELISA. HCV1 recognized the QNGS and KNGS mutants poorly at all concentrations tested (Fig. 7B). However, at higher concentrations, HCV1 could bind to the FV mutant (Fig. 7B), suggesting that this mutant has a reduced affinity for HCV1. Similar to HCV1, 95-2 interacted poorly with the KNGS mutation in ELISA (Fig. 7C). As expected from Fig. 7A, 95-2 recognition of the FV mutant was essentially equivalent to that of the native 412-423 sequence. Also, unlike the case for HCV1, the QNGS mutant could be detected by 95-2 at higher concentrations of antibody (Fig. 7C). These data reinforce the conclusion that although HCV1 and 95-2 bind to the same epitope, the mechanisms by which this binding occurs have subtle differences.
HuMAb HCV1 and 95-2 neutralization of HCVpp containing mutations in the critical epitope.
To determine if changes in the 412-423 epitope would alter HuMAb HCV1 or 95-2 neutralization of HCVpp, mutations deemed relevant from the alanine scanning data, as well as mutations selected from the HCV database, were engineered into the expression construct for full-length H77 genotype 1a E1 and E2. Additionally, any changes found in the NIH HCV database for E2 amino acids 413, 418, and 420, critical residues according to alanine scanning mutagenesis, were also engineered into the full-length 1a E1 and E2 expression construct (Fig. 8A). Two additional changes were engineered at amino acid 415, based on recent reports that identified alterations within the 412-423 epitope (11, 39). When transfected into HEK-293T/17 cells, all of the mutant proteins were expressed at levels equivalent to that of the wild type, as determined by Western blot analysis of cell lysates by use of an antibody specific for an epitope distinct from amino acids 412 to 423 (data not shown). A total of 28 different mutations were made throughout the 412-423 epitope (Fig. 8A), and pseudovirus was generated with each of the mutated full-length 1a E1/E2 proteins to determine which constructs could produce infectious pseudovirus. Of the 28 mutants, only 5 produced infectious pseudovirus (Fig. 8A, residues shown in bold and underlined), demonstrating the importance of the 412-423 region for HCV infection. No changes at amino acid 420 produced infectious virus, and the only change at amino acid 413 that produced a detectable amount of infectious virus in our assay was the conservative leucine-to-isoleucine change, suggesting that these two residues are critical for viral attachment and/or entry (also described previously [26]).
FIG. 8.

Generation and neutralization of HCVpp containing E2 envelope glycoproteins with mutations in amino acids 412 to 423. (A) A total of 28 mutations were introduced into amino acids 412 to 423 of the genotype 1a (H77) full-length E2 envelope glycoprotein (listed at the top), using site-directed mutagenesis. Directly below are listed two mutants that had four amino acid changes each from the H77 sequence. The other 26 mutants had single amino acid changes and are listed in the lower part of the panel. The five bold, underlined amino acid changes represent mutations that yielded infectious HCVpp. Unchanged amino acids are indicated as dashes, and the altered positions are indicated with the one-letter amino acid code. (B) Fivefold dilutions of HuMAbs HCV1 and 95-2 were incubated for 1 h at room temperature with HCVpp generated with H77 1a E1/E2 glycoproteins or H77 1a E1/E2 glycoproteins containing the desired changes in the E2 protein, indicated to the left. The neutralization assays were performed and developed as described in the legend to Fig. 4. The concentration of antibody required to reduce the light output to 50% of that of the negative control antibody is indicated for both HCV1 and 95-2.
To determine the capacity of our lead HuMAbs to neutralize mutant pseudovirus, infectious pseudovirus produced from each of the five different mutants of the genotype 1a E1/E2 envelope glycoproteins was preincubated with various concentrations of either HCV1 or 95-2 for 1 hour at room temperature before being added to Hep3B cells. An isotype-matched, irrelevant human antibody was used as a negative control (data not shown). The concentration of HuMAb that was required to reduce the light output generated from HCVpp infection to 50% of that of the negative control antibody sample was determined for each mutant and wild-type pseudovirus. The results were recorded as the HuMAb concentration required for 50% neutralization (Fig. 8B). Both HuMAbs HCV1 and 95-2 were found to potently neutralize four of the five infectious pseudoviruses (Fig. 8B). Interestingly, E1/E2 with an N415K mutation was found to produce infectious HCVpp but was not neutralized by either HuMAb (Fig. 8B). This result was striking given that the alanine scanning mutagenesis did not identify the asparagine at position 415 as a critical amino acid for binding (Fig. 6B). In addition, the N415K mutation in the Trx-E2412-423-His protein did not affect binding by either HuMAb in ELISA (Fig. 7A). Furthermore, cell surface staining with HuMAbs HCV1 and 95-2 for E1/E2 proteins expressed on the surfaces of CHO cells gave identical binding profiles for dilution series of the HuMAbs with both native H77 1a protein and the H77 1a N415K mutant (data not shown). Clearly, changes at position 415 failed to disrupt binding in the ELISA and cell surface assays, but the lysine change at this position abolished neutralization in the HCVpp assay. These data taken together demonstrate that the N415K mutation is the only identified alteration that generates HCVpp resistant to neutralization by HuMAbs HCV1 and 95-2. In the NIH HCV sequence database, the N415K mutation is represented in approximately 0.7% of the total sequences.
DISCUSSION
We have identified two HuMAbs that recognize, with high affinity, a conserved, linear epitope (amino acids 412 to 423) present in the HCV E2 envelope glycoprotein. These HuMAbs show broad neutralization capacity against all HCV genotypes tested (1a, 1b, 2b, 3a, and 4a), as determined with HCVpp. A diverse group of mutants in the 412-423 epitope identified in the Los Alamos HCV database were recognized by both HCV1 and 95-2, demonstrating the broad reactivity of these HuMAbs.
Essentially all HCV-neutralizing human antibodies reported to date recognize discontinuous epitopes, predominantly in the E2 envelope glycoprotein. This fact demonstrates that HCV1 and 95-2 are distinct in that they bind to a linear epitope in the E2 envelope glycoprotein. Previously described human antibodies were isolated from either phage display analysis (1, 5, 16, 20, 27, 31) or Epstein-Barr virus immortalization of B cells (12, 13, 25) derived from HCV-positive patients. It is possible that the immune response in patients naturally favors the production of conformation-dependent antibodies, thereby limiting the diversity in antibodies that can be isolated using these techniques. In fact, screening patient sera for antibodies reactive to amino acids 412 to 423 of the E2 envelope glycoprotein demonstrated a very low response to this epitope (34). The utilization of HuMAb mouse technology offers the advantage of relying on mouse-directed antigen recognition for the generation of an immune response. In this system, antigen presentation by T cells and macrophages and rearrangement and somatic hypermutation of antibody genes are directed by mouse proteins and enzymes. Using this technology, we isolated HCV1 and 95-2, human antibodies directed against the linear 412-423 E2 epitope.
It is important that the 412-423 epitope appears to be weakly immunogenic in the HuMAb mouse. Of 51 mice immunized with CO-E2661-1a, hybridomas producing antibody reactive to E2 amino acids 412 to 423 were isolated from only two mice. Following the initial isolation of HuMAbs HCV1 and 95-2, multiple HuMAb mice were immunized with a keyhole limpet hemocyanin-modified 412-423 peptide as well as with bacterial fusion proteins containing the 412-423 epitope. In all cases, serum responses to immunogen were low and hybridomas reactive to the immunogen could not be isolated (data not shown). It is unclear if this was due to the antigenic properties of the epitope or because HuMAb mice are immunocompromised and have limited diversity in human antibody genes.
Interestingly, antibodies that interact with amino acids 412 to 423 of the E2 envelope glycoprotein have been isolated from wild-type rodents. We (data not shown) and other groups have developed mouse and rat antibodies that interact with this epitope. Mouse MAb AP33 (24, 35) and rat MAb 3/11 (10, 24, 36, 39) have previously been described to bind E2 amino acids 412 to 423; MAb 3/11 has broad neutralization ability but has a low affinity for the epitope, while AP33 has generated excitement for its ability to potently neutralize nearly all HCV genotypes tested and has a high affinity for the E2 envelope glycoprotein. HCV1 and 95-2 resemble MAb AP33, given their potent neutralization capacity and shared recognition of the conserved E2 412-423 epitope. In addition, HCV1, 95-2, and MAb AP33 are all sensitive to alterations at amino acids L413 and W420. However, there are, in fact, distinct differences between HCV1, 95-2, and MAb AP33. HCV1 and MAb AP33, but not 95-2, are affected by alterations at amino acid G418, yet HCV1 appears less impacted by this mutation than MAb AP33. In addition, unlike the case for MAb AP33, HCV1 and 95-2 binding was not reduced when the MAbs were tested against the N415A protein. MAb AP33 was unable to bind or neutralize HCVpp derived from a UKN5.14.4 E1/E2 sequence that contained a four-amino-acid change of NTNG to QNGS at positions 415 to 418. Unfortunately, we were unable to generate H77 HCVpp containing this sequence of mutations, and thus we were unable to test neutralization with HCV1 or 95-2. These HuMAbs did interact with a protein containing this specific mutation, albeit with lower affinity than that for the native genotype 1a H77 sequence (Fig. 7B and C). Given these data, it seems clear that the E2 residues required for HCV1 and 95-2 binding to the E2 412-423 epitope are more restricted than those required for MAb AP33. In addition, HuMAbs HCV-1 and 95-2 are human rather than mouse antibodies and therefore are more promising human therapeutics than the mouse-derived MAb AP33.
An extensive search of the HCV database identified a total of 28 mutations in the critical residues of the HCV1 and 95-2 epitope of the E2 glycoprotein. Interestingly, HCVpp could be generated for only five of these mutations when they were engineered into the H77 E1/E2 glycoprotein sequence. It is important that these mutations were identified from a wide variety of HCV genotypes. It is possible that even though these mutations did not result in infectious HCVpp in an H77 genotype 1a background, the viruses in which these sequences occur are still infectious. Compensatory mutations in other regions of the E1/E2 glycoprotein could counteract the negative effects on viral fitness that we observe in the H77 background. Given the linear nature of our antibodies, we would not expect these compensatory, distant mutations to affect antibody binding and/or neutralization. Although we consider it highly unlikely, it is possible that these mutations (for which H77 HCVpp could not be generated) may alter HCV1 and 95-2 neutralization capacity, but we are unable to assess this in our current system.
HCV1 and 95-2 could neutralize four of the five mutant infectious HCVpp (Fig. 8B). Surprisingly, we identified an asparagine (N)-to-lysine (K) mutation at amino acid 415 that blocked the ability of both HCV1 and 95-2 to neutralize HCVpp. This was an unexpected result given that these HuMAbs could bind to both bacterially expressed E2 fragments (Fig. 7A) and full-length E1/E2 protein expressed on cell surfaces (data not shown) that contained this N415K mutation. In fact, we saw absolutely no reduction in the affinity of either HuMAb for this epitope when comparing native sequence to the N415K mutation. Also, alanine substitution at N415 had no effect on HuMAb binding in the context of bacterial fusion proteins (Fig. 6B) or full-length mammal-expressed E1/E2 (data not shown). Currently, we have no explanation for the discrepancy in the data, but it is possible that a conformational change in the envelope glycoprotein heterodimer could allow antibody binding but not neutralization. It is also possible that the N415K mutant E1/E2 heterodimer assumes an alternative conformation in HCVpp compared to protein expressed on the surfaces of cultured cells and that this alternative conformation is not bound by either HCV1 or 95-2. Regardless, we believe that the N415K mutation represents an escape mutant for these HuMAbs. Analysis of the HCV database revealed that this mutation is present in 0.7% of isolated sequences, suggesting that viruses containing this mutation may be less fit in the context of human infection than those having other E1/E2 sequences.
Recent work identified one escape mutant for MAb AP33, in which the E2 envelope glycoprotein adopted an N415Y mutation (11). Although this virus was clearly an escape variant induced by MAb AP33, it was significantly compromised in replication capacity in culture. HCV1 and 95-2 were not tested for the ability to neutralize N415Y mutant HCVpp due to an inability to generate infectious H77 HCVpp containing this mutation (Fig. 8). The lack of infectious N415Y HCVpp was not unexpected, given the reduced infectivity that this mutant demonstrated in the in vitro culture system employed in the previous study. These results underscore the importance of the conserved E2 412-423 sequence in infection. Although it may be possible to generate mutations in this epitope, these mutant viruses are significantly less infectious.
The development of the HCVpp in vitro infection system has greatly improved our ability to identify antibodies that neutralize HCV. However, it remains unclear if this system is truly indicative of HCV neutralization in vivo. Confounding factors such as enhancement of HCV infection by apolipoprotein (7, 21) and epitope masking by high-density lipoprotein found in serum (37) are not present in this in vitro system. Unfortunately, the currently employed in vivo models have many drawbacks as well. Multiple mouse models have been developed that introduce human liver cells either peripherally or into mouse liver, which allows large numbers of mice to be tested for protection from infection with HCV, but it is difficult to determine if the infection represents the natural HCV infection in humans. Although costly, the preferred and only true animal model of HCV infection is the chimpanzee. Chimpanzees develop viremia shortly after exposure to HCV, and in some cases, they develop long-term infection. We are currently evaluating the efficacy of HCV1 in preventing HCV infection in the chimpanzee disease model.
Acknowledgments
We thank Asja Asceric, Katherine Baptista, Brandy Giacomozzi, Hector Hernandez, Casey Kilgus, Kirk Rowley, and Christine St. Pierre for technical assistance. We also thank Deborah Molrine, Barbra Blair, Brett Leav, Yang Wang, and Trevor Morin for thoughtful scientific discussions and manuscript review.
Footnotes
Published ahead of print on 16 September 2009.
REFERENCES
- 1.Allander, T., K. Drakenberg, A. Beyene, D. Rosa, S. Abrignani, M. Houghton, A. Widell, L. Grillner, and M. A. Persson. 2000. Recombinant human monoclonal antibodies against different conformational epitopes of the E2 envelope glycoprotein of hepatitis C virus that inhibit its interaction with CD81. J. Gen. Virol. 81:2451-2459. [DOI] [PubMed] [Google Scholar]
- 2.Babcock, G. J., T. J. Broering, H. J. Hernandez, R. B. Mandell, K. Donahue, N. Boatright, A. M. Stack, I. Lowy, R. Graziano, D. Molrine, D. M. Ambrosino, and W. D. Thomas, Jr. 2006. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 74:6339-6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babcock, G. J., D. J. Esshaki, W. D. Thomas, Jr., and D. M. Ambrosino. 2004. Amino acids 270 to 510 of the severe acute respiratory syndrome coronavirus spike protein are required for interaction with receptor. J. Virol. 78:4552-4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartosch, B., J. Dubuisson, and F. L. Cosset. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bugli, F., N. Mancini, C. Y. Kang, C. Di Campli, A. Grieco, A. Manzin, A. Gabrielli, A. Gasbarrini, G. Fadda, P. E. Varaldo, M. Clementi, and R. Burioni. 2001. Mapping B-cell epitopes of hepatitis C virus E2 glycoprotein using human monoclonal antibodies from phage display libraries. J. Virol. 75:9986-9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935-944. [DOI] [PubMed] [Google Scholar]
- 7.Dreux, M., B. Boson, S. Ricard-Blum, J. Molle, D. Lavillette, B. Bartosch, E. I. Pecheur, and F. L. Cosset. 2007. The exchangeable apolipoprotein ApoC-I promotes membrane fusion of hepatitis C virus. J. Biol. Chem. 282:32357-32369. [DOI] [PubMed] [Google Scholar]
- 8.Evans, M. J., T. von Hahn, D. M. Tscherne, A. J. Syder, M. Panis, B. Wolk, T. Hatziioannou, J. A. McKeating, P. D. Bieniasz, and C. M. Rice. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801-805. [DOI] [PubMed] [Google Scholar]
- 9.Everson, G. T. 2002. Impact of immunosuppressive therapy on recurrence of hepatitis C. Liver Transpl. 8:S19-S27. [DOI] [PubMed] [Google Scholar]
- 10.Flint, M., C. Maidens, L. D. Loomis-Price, C. Shotton, J. Dubuisson, P. Monk, A. Higginbottom, S. Levy, and J. A. McKeating. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73:6235-6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gal-Tanamy, M., Z. Y. Keck, M. Yi, J. A. McKeating, A. H. Patel, S. K. Foung, and S. M. Lemon. 2008. In vitro selection of a neutralization-resistant hepatitis C virus escape mutant. Proc. Natl. Acad. Sci. USA 105:19450-19455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Habersetzer, F., A. Fournillier, J. Dubuisson, D. Rosa, S. Abrignani, C. Wychowski, I. Nakano, C. Trepo, C. Desgranges, and G. Inchauspe. 1998. Characterization of human monoclonal antibodies specific to the hepatitis C virus glycoprotein E2 with in vitro binding neutralization properties. Virology 249:32-41. [DOI] [PubMed] [Google Scholar]
- 13.Hadlock, K. G., R. E. Lanford, S. Perkins, J. Rowe, Q. Yang, S. Levy, P. Pileri, S. Abrignani, and S. K. Foung. 2000. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J. Virol. 74:10407-10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He, J., S. Choe, R. Walker, P. Di Marzio, D. O. Morgan, and N. R. Landau. 1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69:6705-6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu, M., J. Zhang, M. Flint, C. Logvinoff, C. Cheng-Mayer, C. M. Rice, and J. A. McKeating. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 100:7271-7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansson, D. X., C. Voisset, A. W. Tarr, M. Aung, J. K. Ball, J. Dubuisson, and M. A. Persson. 2007. Human combinatorial libraries yield rare antibodies that broadly neutralize hepatitis C virus. Proc. Natl. Acad. Sci. USA 104:16269-16274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keck, Z. Y., V. M. Sung, S. Perkins, J. Rowe, S. Paul, T. J. Liang, M. M. Lai, and S. K. Foung. 2004. Human monoclonal antibody to hepatitis C virus E1 glycoprotein that blocks virus attachment and viral infectivity. J. Virol. 78:7257-7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolykhalov, A. A., E. V. Agapov, K. J. Blight, K. Mihalik, S. M. Feinstone, and C. M. Rice. 1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277:570-574. [DOI] [PubMed] [Google Scholar]
- 19.Kuiken, C., M. Mizokami, G. Deleage, K. Yusim, F. Penin, I. T. Shin, C. Charavay, N. Tao, D. Crisan, D. Grando, A. Dalwani, C. Geourjon, A. Agrawal, and C. Combet. 2006. Hepatitis C databases, principles and utility to researchers. Hepatology 43:1157-1165. [DOI] [PubMed] [Google Scholar]
- 20.Law, M., T. Maruyama, J. Lewis, E. Giang, A. W. Tarr, Z. Stamataki, P. Gastaminza, F. V. Chisari, I. M. Jones, R. I. Fox, J. K. Ball, J. A. McKeating, N. M. Kneteman, and D. R. Burton. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25-27. [DOI] [PubMed] [Google Scholar]
- 21.Meunier, J. C., R. E. Engle, K. Faulk, M. Zhao, B. Bartosch, H. Alter, S. U. Emerson, F. L. Cosset, R. H. Purcell, and J. Bukh. 2005. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl. Acad. Sci. USA 102:4560-4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meunier, J. C., R. S. Russell, V. Goossens, S. Priem, H. Walter, E. Depla, A. Union, K. N. Faulk, J. Bukh, S. U. Emerson, and R. H. Purcell. 2008. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the E1 glycoprotein of hepatitis C virus. J. Virol. 82:966-973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Op De Beeck, A., L. Cocquerel, and J. Dubuisson. 2001. Biogenesis of hepatitis C virus envelope glycoproteins. J. Gen. Virol. 82:2589-2595. [DOI] [PubMed] [Google Scholar]
- 24.Owsianka, A., A. W. Tarr, V. S. Juttla, D. Lavillette, B. Bartosch, F. L. Cosset, J. K. Ball, and A. H. Patel. 2005. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J. Virol. 79:11095-11104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Owsianka, A. M., A. W. Tarr, Z. Y. Keck, T. K. Li, J. Witteveldt, R. Adair, S. K. Foung, J. K. Ball, and A. H. Patel. 2008. Broadly neutralizing human monoclonal antibodies to the hepatitis C virus E2 glycoprotein. J. Gen. Virol. 89:653-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owsianka, A. M., J. M. Timms, A. W. Tarr, R. J. Brown, T. P. Hickling, A. Szwejk, K. Bienkowska-Szewczyk, B. J. Thomson, A. H. Patel, and J. K. Ball. 2006. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol. 80:8695-8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perotti, M., N. Mancini, R. A. Diotti, A. W. Tarr, J. K. Ball, A. Owsianka, R. Adair, A. H. Patel, M. Clementi, and R. Burioni. 2008. Identification of a broadly cross-reacting and neutralizing human monoclonal antibody directed against the hepatitis C virus E2 protein. J. Virol. 82:1047-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pileri, P., Y. Uematsu, S. Campagnoli, G. Galli, F. Falugi, R. Petracca, A. J. Weiner, M. Houghton, D. Rosa, G. Grandi, and S. Abrignani. 1998. Binding of hepatitis C virus to CD81. Science 282:938-941. [DOI] [PubMed] [Google Scholar]
- 29.Ploss, A., M. J. Evans, V. A. Gaysinskaya, M. Panis, H. You, Y. P. de Jong, and C. M. Rice. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882-886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scarselli, E., H. Ansuini, R. Cerino, R. M. Roccasecca, S. Acali, G. Filocamo, C. Traboni, A. Nicosia, R. Cortese, and A. Vitelli. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017-5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schofield, D. J., B. Bartosch, Y. K. Shimizu, T. Allander, H. J. Alter, S. U. Emerson, F. L. Cosset, and R. H. Purcell. 2005. Human monoclonal antibodies that react with the E2 glycoprotein of hepatitis C virus and possess neutralizing activity. Hepatology 42:1055-1062. [DOI] [PubMed] [Google Scholar]
- 32.Seong, Y. R., S. Choi, J. S. Lim, C. H. Lee, C. K. Lee, and D. S. Im. 2001. Immunogenicity of the E1E2 proteins of hepatitis C virus expressed by recombinant adenoviruses. Vaccine 19:2955-2964. [DOI] [PubMed] [Google Scholar]
- 33.Sloan, S. E., C. Hanlon, W. Weldon, M. Niezgoda, J. Blanton, J. Self, K. J. Rowley, R. B. Mandell, G. J. Babcock, W. D. Thomas, Jr., C. E. Rupprecht, and D. M. Ambrosino. 2007. Identification and characterization of a human monoclonal antibody that potently neutralizes a broad panel of rabies virus isolates. Vaccine 25:2800-2810. [DOI] [PubMed] [Google Scholar]
- 34.Tarr, A. W., A. M. Owsianka, D. Jayaraj, R. J. Brown, T. P. Hickling, W. L. Irving, A. H. Patel, and J. K. Ball. 2007. Determination of the human antibody response to the epitope defined by the hepatitis C virus-neutralizing monoclonal antibody AP33. J. Gen. Virol. 88:2991-3001. [DOI] [PubMed] [Google Scholar]
- 35.Tarr, A. W., A. M. Owsianka, J. M. Timms, C. P. McClure, R. J. Brown, T. P. Hickling, T. Pietschmann, R. Bartenschlager, A. H. Patel, and J. K. Ball. 2006. Characterization of the hepatitis C virus E2 epitope defined by the broadly neutralizing monoclonal antibody AP33. Hepatology 43:592-601. [DOI] [PubMed] [Google Scholar]
- 36.Triyatni, M., J. Vergalla, A. R. Davis, K. G. Hadlock, S. K. Foung, and T. J. Liang. 2002. Structural features of envelope proteins on hepatitis C virus-like particles as determined by anti-envelope monoclonal antibodies and CD81 binding. Virology 298:124-132. [DOI] [PubMed] [Google Scholar]
- 37.Voisset, C., N. Callens, E. Blanchard, A. Op De Beeck, J. Dubuisson, and N. Vu-Dac. 2005. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 280:7793-7799. [DOI] [PubMed] [Google Scholar]
- 38.von Hahn, T., and C. M. Rice. 2008. Hepatitis C virus entry. J. Biol. Chem. 283:3689-3693. [DOI] [PubMed] [Google Scholar]
- 39.von Hahn, T., J. C. Yoon, H. Alter, C. M. Rice, B. Rehermann, P. Balfe, and J. A. McKeating. 2007. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 132:667-678. [DOI] [PubMed] [Google Scholar]
- 40.Wright, T. L., E. Donegan, H. H. Hsu, L. Ferrell, J. R. Lake, M. Kim, C. Combs, S. Fennessy, J. P. Roberts, N. L. Ascher, et al. 1992. Recurrent and acquired hepatitis C viral infection in liver transplant recipients. Gastroenterology 103:317-322. [DOI] [PubMed] [Google Scholar]