

Abstract
The mechanisms underlying the timing of the laminar fate decisions during cortical neurogenesis remain poorly understood. Here we show that β-catenin signaling in cortical neural precursors can regulate the laminar fate of their daughters. In ventricular zone neural precursors, β-catenin signaling is higher when deep-layer neurons are being generated and lower when upper-layer neurons are being generated. Overactivation of β-catenin in cortical precursors midway through corticogenesis increased the relative production of deep-layer neurons, while inhibition of signaling increased the relative production of upper-layer neurons. Furthermore, in late-gestation upper-layer precursors, overactive β-catenin signaling was able to partially restore production of deep-layer neurons. These observations suggest that increased β-catenin signaling can reset the timing of cortical precursors to promote the production of deep-layer neurons, while inhibition of β-catenin signaling advances the timing to promote upper-layer production.
Introduction
The mammalian neocortex is composed of six morphologically and functionally distinct neuronal layers generated from progenitors in a proliferative neuroepithelium called the ventricular zone (VZ). The time point at which progenitors exit the cell cycle to generate a postmitotic neuron (its “birthdate”) is closely tied to the neuron's final laminar position (Angevine and Sidman, 1961; Rakic, 1971; Takahashi et al., 1999). Radial progenitor cells in the cortical VZ ultimately generate all the excitatory projection neurons of the mature cortex. With the exception of the earliest-born Cajal–Retzius neurons found in layer 1, neurons that are born early in cortical development comprise the deepest layers of the cortex, whereas later-born neurons migrate past their earlier-born counterparts and compose the more superficial layers of the cortex.
As cortical neurogenesis progresses, the subsets of cortical cell types that progenitors are able to produce is progressively restricted (Frantz and McConnell, 1996; Desai and McConnell, 2000; Shen et al., 2006). Transplantation studies suggest that at the beginning of neurogenesis, progenitors are capable of generating both superficial and deep-layer neurons, whereas later progenitors become committed to producing upper-layer neurons (Desai and McConnell, 2000).
Both external and cell-intrinsic factors contribute to laminar fate restriction. Early cortical progenitors blocked from undergoing neurogenesis by transient expression of constitutive active Notch receptor produced upper-layer neurons when allowed to undergo neurogenesis later in cortical development (Mizutani and Saito, 2005). These experiments suggested that changes in extrinsic signals contributed to the progressive restriction of laminar fate potential. In contrast, isolated neural stem/progenitor cells and embryonic stem cells in culture are able to sequentially generate neurons expressing markers of different cortical layers, suggestive of an intrinsic clock within precursors that regulates cell cycle exit and laminar fate choice (Shen et al., 2006; Gaspard et al., 2008).
Recent studies suggest that some aspects of laminar fate restriction may be reversible. The FOXG1 transcription factor represses the production of the earliest-born Cajal–Retzius neurons (Hanashima et al., 2004). Inactivation of Foxg1 later in development reinitiates Cajal–Retzius production (Hanashima et al., 2004), leading to the model that FOXG1 actively suppresses the competence of later precursors to make Cajal–Retzius neurons. Reduction of Foxg1 expression by shRNA in cultured progenitors reset the timing of midgestation (but not late-gestation) progenitors to allow them to generate earlier cortical cell types (Shen et al., 2006).
The relationship of neuronal birthday and laminar fate determination suggests that factors that regulate cell cycle exit and re-entry might affect the relative production of neurons in different layers. Overactivity of β-catenin decreases precursor cell cycle exit (Chenn and Walsh, 2002) and interferes with the production of progenitors responsible for the production of upper-layer cortical neurons (Wrobel et al., 2007). While these observations suggest that increased β-catenin activity may regulate laminar fate by influencing neural precursor maturation, whether varying levels of β-catenin signaling regulate laminar identity remains unclear.
Materials and Methods
Animals.
Electroporation studies, with the exception of Ctnnb studies, were conducted on timed-pregnant Swiss Webster mice (Charles River Laboratories). BAT-gal reporter mice (a gift from Stefano Piccolo, University of Padova, Padova, Italy) have been previously described (Maretto et al., 2003). β-Cateninflox/flox mice (B6.129-Ctnnb1tm2Kem/KnwJ) were obtained from The Jackson Laboratory. Mice were genotyped according to The Jackson Laboratory protocols available at http://jaxmice.jax.org/pub-cgi/protocols/protocols.sh?objtype=protocol&protocol_id=471. Allmice in this study were treated according to protocols reviewed and approved by the institutional animal care and use committees of Northwestern University.
Statistical analysis.
All statistics were computed using GraphPad Prism 3.0 (GraphPad Software).
Plasmid DNA.
The N-terminally truncated β-catenin construct (Δ90β-catenin-GFP) used has been described previously (Chenn and Walsh, 2002). It was subcloned into a pCAG electroporation vector designed to allow expression in both neural precursors and mature cell types (Niwa et al., 1991). EGFP-tagged dominant-negative TCF4 (pCAG-DNTCF4-GFP) and Cre-IRES2-EGFP were created as previously described (Woodhead et al., 2006). pCAG-GFP and pCAG-mCherry were used in control experiments. The TOP-dGFP/pCAG-Cherry dual reporter construct was created by inserting TOP-dGFP (dGFP is destabilized GFP) into the pCAG-mCherry plasmid [both previously described (Woodhead et al., 2006)].
In utero electroporation studies.
Our protocol for in utero electroporation of plasmid DNA constructs has been described previously (Woodhead et al., 2006). Briefly, timed pregnant mice were anesthetized with a ketamine–xylazine mixture (10 parts to 1, respectively). Plasmid DNA solution (2.5 μl at 0.5 μg/μl) was injected into the lateral ventricle of the embryonic brain through the uterine wall by using a pulled capillary pipette. After injection, for embryonic day 13.5 (E13.5) embryos, five 50 ms square pulses of 40 mV at 950 ms intervals were delivered by an electroporator (BTX 830). For E15.5 embryos, five 50 ms square pulses of 50 mV at 950 ms intervals were used. Afterward, the timed pregnant females were sutured and allowed to recover. Mice were then killed at predetermined intervals and embryonic brains were recovered and fixed with 4% PFA. Postnatal samples were perfused with 4% PFA before removal.
Immunofluorescence and X-gal histochemistry.
After fixation, embryonic brains were either sunk in 30% sucrose solution and then flash frozen in O.C.T. compound (Tissue-Tek) for cryosections (14 μm) or embedded in 2% agarose for vibratome sections (100 μm). Sections were incubated with the following primary antibodies: anti-GFP (Invitrogen, rabbit polyclonal, and Abcam, chicken polyclonal), anti-DsRed (Clontech, rabbit polyclonal), anti-BRN1 (Santa Cruz Biotechnology, goat polyclonal), anti-FOXP2 (Abcam, rabbit polyclonal), anti-TUJ1 (Covance, mouse monoclonal), anti-CTIP2 (Abcam, rat monoclonal), anti-SATB2 (Abcam, mouse monoclonal), anti-cleaved caspase 3(Cell Signaling Technology, rabbit polyclonal), and anti-BrdU (Serotec, rat monoclonal). Appropriate secondary antibodies (all from Invitrogen) were used to detect staining. For BrdU antigen retrieval, sections were exposed to 3 N HCl for 15 min, followed by three washes each of 0.5× TBE and 1× PBS. DNA was counterstained with DAPI. Optical sections (1 μm) were obtained using the 40× objective on a Zeiss confocal microscope.
5-Bromo-4-chloro-3-indolyl-β-d-galactoside (X-gal) histochemistry was performed as previously described with slight modifications (Currle et al., 2005). Briefly, 14 μm coronal cryosections from comparable regions of E12.5, E14.5, and E16.5 cortex were incubated in the same X-gal reaction solution for 40 min. Bright-field images were captured by Spot RT digital imaging on an upright Nikon E600 microscope. The X-gal data were taken from equivalent coronal sections at each of the three ages. The intermediate thalamus and the third ventricle were used as landmarks to ensure that equivalent regions were used.
Quantification of β-catenin signaling.
To quantify β-catenin signaling, we electroporated TOP-dGFP/pCAG-mCherry dual expression plasmid (described above) into the lateral ventricles of the developing mouse brain. Sections of fixed embryonic cortex were prepared and imaged as described above. To preserve the relationship between fluorescence intensities, care was taken not to saturate any of the images, and all images were obtained using identical settings and conditions. Using ImageJ (http://rsb.info.nih.gov/ij/) with the LSM reader plugin (http://rsb.info.nih.gov/ij/plugins/lsm-reader.html), regions were drawn around the soma of each VZ cell expressing mCherry. The mean, maximum, and minimum pixel intensities for each region were recorded. Then, using the “Set Measurement” command, the pixel intensities of the corresponding region in the dGFP channel were measured. The average background fluorescence was computed using areas of sections with no electroporated cells present and was subtracted from all measurements, which were then rescaled. Using these data, the ratio of TOP-dGFP expression to pCAG-mCherry expression was determined using Microsoft Excel 2003. Rainbow scale images were created using LSM Image Browser 3.5 (Carl Zeiss).
Cell counting and identification.
The analysis throughout the paper was performed in a semiblinded manner (because the morphology of the cells with the different constructs is usually distinctive, the observer can identify which construct is used). However, for each experiment, GFP+ electroporated cells were marked on an image containing only the channels for GFP and DAPI. The position of these marked cells was then transferred to an image containing the marker of interest and the colocalization of these markers was noted. Cells are identified by colocalization of the fluorescent construct with DAPI (nuclear stain). In the case of junctional staining (Δ90β-catenin-GFP), the fluorescence must encircle the entire DAPI-stained region to be counted as positive staining. To confirm localization of electroporated cells, in all experiments, mCherry was coelectroporated, and cell position was determined by antibody staining for mCherry (data not shown).
Results
β-Catenin signaling in cortical VZ precursors decreases during neurogenesis
To characterize the levels of β-catenin-mediated transcriptional activation in developing cortical progenitors, we first used a well characterized transgenic reporter mouse line, BAT-gal (Maretto et al., 2003). In these mice, the lacZ gene is expressed under the control of seven TCF/LEF consensus sequences, which activate transcription when β-catenin signaling is present; β-galactosidase activity faithfully reflects β-catenin signaling levels in both neural and non-neural tissues (O'Brien et al., 2004; Backman et al., 2005; Junghans et al., 2005; Lie et al., 2005; Sick et al., 2006) (Fig. 1). Consistent with prior reports, we observed a medial to lateral gradient in β-galactosidase activity in the developing cortex (Machon et al., 2007). Additionally, when comparing β-galactosidase activity in E12.5, E14.5, and E16.5 cortices fixed and processed identically for X-gal staining, we observed a reduction in staining intensity in cortical VZ progenitors as development progressed, suggesting a temporal gradient in β-catenin signaling.
Figure 1.

β-Catenin signaling levels undergo progressive reduction during cortical neurogenesis. Comparable coronal sections from E12.5 (A), E14.5 (B), and E16.5 (C) BAT-gal mouse cortex after X-gal staining reaction. BAT-gal mice express β-galactosidase in response to β-catenin signaling. Additional detail of the ventricular zone is shown in high-power insets below (A′, B′, C′). Scale bars: low-power images, 100 μm; high-power images, 20 μm.
To compare levels of β-catenin signaling at a cellular level in cortical progenitors at different developmental stages more quantitatively, we designed a bicistronic plasmid containing two fluorescent proteins whose expression is driven by two different promoters (Yokota et al., 2009). TOP, a TCF/LEF consensus site upstream from a minimal c-Fos promoter (Dorsky et al., 2002), drives the expression of dGFP, and CAG, a strong, constuitively active promoter, independently drives the expression of mCherry. Destabilized green fluorescent protein is designed to be rapidly degraded (with a half-life of ∼2 h) (Reya et al., 2003), while mCherry is used as a transfection control. To compare levels of β-catenin signaling we measured the ratio of dGFP fluorescence to mCherry fluorescence following transfection of this reporter plasmid to cells.
Using this reporter, we examined signaling in VZ progenitor cells at E13.5 and E15.5, the peaks in neuronal production for deep- and superficial-layer neurons, respectively (Takahashi et al., 1999). We delivered the TOP-dGFP/CAG-mCherry dual plasmid to progenitor cells in the developing cortex at E13.5 (Fig. 2A–C) and E15.5 (Fig. 2D–F) using in utero electroporation and examined fluorescent signal 24 h afterward. We found that the mean normalized level of β-catenin signaling was more than five times greater in E13.5 progenitors as compared to E15.5 progenitors. We used the ratio of dGFP fluorescence intensity to mCherry fluorescence intensity to quantify the average signaling intensity (p < 0.0001, t test, two tailed) in E13.5 [2.48 ± 0.393 (±SEM), n = 3 brains, 74 cells] and E15.5 (0.477 ± 0.068, n = 4 brains, 104 cells) cells, all of which were VZ progenitors expressing mCherry. Together, the observations that the progressive reduction of β-catenin signaling in cortical progenitors paralleled the progressive restrictions on laminar fate during cortical development raised the possibility that levels of β-catenin signaling might contribute to laminar fate determination.
Figure 2.

β-Catenin signaling in progenitors decreases between E13.5 and E15.5. E13.5 and E15.5 timed pregnant Swiss Webster mouse embryos were electroporated with TOP-dGFP/CAG-mCherry dual expression plasmid. This construct contains two main elements: TOP-dGFP reports β-catenin signaling and CAG-mCherry serves as a transfection control. After 24 h the embryos were killed and their brains were sectioned and imaged. All images are from the dorsolateral cerebral cortex midway through the rostral–caudal continuum of the cortex. Throughout the figure the nomenclature x refers to merged images, X' refers to dGFP only, and X“ refers to mCherry only. A–C, Representative images from the E13.5-electroporated cortices. D–F, Representative images from E15.5-electroporated cortices. The small panels (B, C, E, F) to the right of the image highlight individual cells from the larger panels. While the majority of ventricular zone cells express dGFP, the migratory intermediate zone (IZ) cells largely express only mCherry (A, D). Scale bars: large panels, 50 μm; small panels, 20 μm. All images are 1 μm optical sections. G, Schematic of TOP-dGFP/CAG-mCherry dual expression plasmid.
β-Catenin signaling in progenitors alters the laminar positioning of cortical neurons
To test the effects of β-catenin signaling on cortical laminar cell fate, we alternatively activated and inhibited the pathway in VZ progenitors by using in utero electroporation approaches. First, by transfecting only a small subset of cells, we observed transfected cells in a wild-type background allowing us to focus on cell-autonomous effects. Second, this technique allowed us to easily track transfected cells by the fluorescence-tagged proteins they express. Finally, we avoided the architectural disorganization in conditional knock-out and transgenic models that make analysis of laminar fate impossible (Brault et al., 2001; Chenn and Walsh, 2002; Machon et al., 2003; Zechner et al., 2003; Junghans et al., 2005).
To manipulate β-catenin signaling we used two GFP-tagged constructs (Δ90β-catenin-GFP) and dominant-negative TCF4 (DNTCF4-GFP) and a GFP control construct. In Δ90β-catenin-GFP, the regulatory elements normally used to degrade β-catenin in the absence of WNT ligand have been removed, resulting in a stabilized version of β-catenin that mediates transcriptional activation in neural precursors (Chenn and Walsh, 2002). For DNTCF4-GFP, the β-catenin binding domain has been deleted. The resultant construct efficiently blocks the transcription of WNT/β-catenin target genes (Tetsu and McCormick, 1999; Woodhead et al., 2006).
To examine the role of β-catenin signaling in laminar fate determination, we electroporated VZ precursors at E13.5 and allowed the embryos to develop normally until E19.5. Using coronal sections, the dorsolateral cortical plate of each of the embryos was divided into 10 equally sized bins ranging from the pial surface to the white matter tracts and the location of each electroporated cell body was recorded (Fig. 3A–C). To confirm neuronal differentiation following β-catenin manipulation, we examined the coexpression of the neuronal marker TUJ1 in electroporated cells (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). In each of the experimental groups, all electroporated cells in the cortical plate expressed TUJ1, indicating that the alteration of β-catenin signaling does not promote non-neuronal cell fates in progenitors. Immunostaining for cleaved caspase 3 showed that progenitors electroporated with Δ90β-catenin (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) and DNTCF4 (Woodhead et al., 2006) have very low rates of apoptosis that are not significantly different from GFP control.
Figure 3.
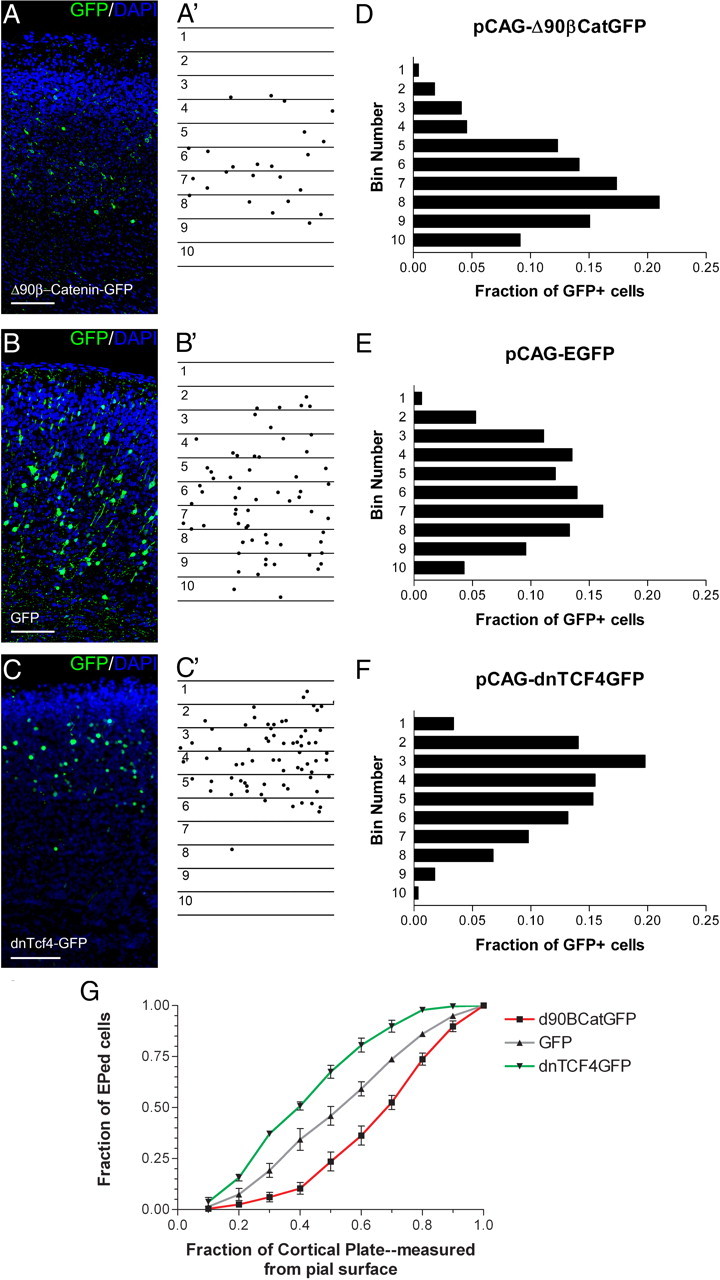
β-Catenin signaling alters laminar positions of cortical neurons. A–C, E13.5 embryos were electroporated with Δ90β-catenin-GFP (n = 4 brains) (A), DNTCF4-GFP (n = 3 brains) (B), or GFP control (n = 4) (C), along with pCAG-mCherry, and killed at E19.5. Electroporated cells were identified using antibody staining against GFP and sections counterstained with the DNA dye DAPI (A–C). To quantify changes in cortical positioning of electroporated cells, 10 equally sized bins were drawn over each image covering the complete cortical plate. Each dot corresponds with the soma of an electroporated cell (A′–C′). D–F, The fraction of the total GFP+ cells in each of the 10 bins was then graphed for the three experimental conditions. Bin 1 corresponds to the section of the cortical plate closest to the pial surface, while bin 10 is adjacent to the white matter tracts. G, Data from D–F in a cumulative view. The x axis denotes the fraction of the total area of cortical plate (measured from the pia) included for a given data point. For example, data points at 0.4 on the x axis refer to the fraction of all electroporated cells that reside in the top 40% of the cortical plate. Scale bars, 100 μm. Brackets indicate SEM. EPed, Electroporated.
We found that activation of signaling with Δ90β-catenin-GFP increased the fraction of cells that resided in deep layers (Fig. 3D) compared with control GFP-electroporated cells (Fig. 3E). In contrast, blocking signaling with DNTCF4-GFP resulted in more superficial positioning of electroporated cells (Fig. 3F,G). Approximately 70% of DNTCF4-GFP-expressing cells were located in the superficial half of the cortex, compared to slightly <50% of GFP control and <25% of Δ90β-catenin-GFP-expressing cells (Fig. 3G). A similar distribution of cells was observed when E13.5 electroporated brains were allowed to develop until postnatal day 7, suggesting that the alterations in laminar distribution are not a result of delayed or altered or migration (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Following electroporation of Cre plasmid into embryos with the β-catenin gene flanked by loxP sites [β-cateninflox/flox mice (B6.129-Ctnnb1tm2Kem/KnwJ)] to conditionally excise both alleles of β-catenin, we observed a similar phenotype as we observed when blocking β-catenin signaling with DNTCF4-GFP with electroporated cells residing in the most superficial parts of the cortex (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Together, these results suggest that differing levels of β-catenin signaling alters the positioning of cortical neurons. Overactivity of β-catenin increases the relative number of cortical neurons in deeper layers, while inhibition of signaling decreases relative numbers of superficial-layer neurons.
β-Catenin signaling in progenitors influences the laminar identity of their daughter neurons
To examine whether the molecular identity of cells following manipulation of β-catenin signaling correlated with the alterations in positioning in the cortical plate, we stained for cortical layer molecular markers. The expression of FOXP2, a transcription factor in the winged-helix/forkhead family, is limited to the layers 5 and 6 of the dorsolateral cortex (Ferland et al., 2003), while BRN1, a POU homeodomain protein, is expressed more superficially, in cortical layers 2, 3 and 4 (Sugitani et al., 2002). Additionally, the postmitotic determinants of layer fate SATB2 and CTIP2 can be used to differentiate superficial and deep-layer neurons. CTIP2 is expressed in cortical layers 5 and 6 (immunostaining is more intense in layer 5). CTIP2 is both necessary and sufficient for the production of the subcortical projection neurons that largely populate the deep cortical layers (Arlotta et al., 2005; Chen et al., 2008). SATB2 functions by repressing CTIP2 expression and is a marker for the corticocortical projection neurons found in cortical layers 2–4 (Alcamo et al., 2008; Britanova et al., 2008).
We found that increasing β-catenin signaling increased the fraction of cells that expressed FOXP2 (58 vs 40%; >1.4-fold increase) (Fig. 4; supplemental Fig. S5, available at www.jneurosci.org as supplemental material) while decreasing the fraction that expressed BRN1 (∼26 vs ∼54% for GFP control; ∼48% of control) (Fig. 5; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). In contrast, decreasing β-catenin signaling with DNTCF4 decreased the fraction of FOXP2 cells (29 vs 40% for GFP control; ∼72% of control) while increasing the fraction of BRN1-expressing (∼74 vs ∼54% for GFP control; ∼1.4-fold increase) (Figs. 4, 5). Similarly, increasing β-catenin signaling increased the fraction of cells that expressed CTIP2 (55 vs 41%; >1.3-fold increase) (Fig. 4; supplemental Fig. S5, available at www.jneurosci.org as supplemental material) while decreasing the fraction that expressed SATB2 (∼30 vs ∼58% for GFP control; ∼52% of control) (Fig. 5; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Meanwhile, decreasing β-catenin signaling with DNTCF4 decreased the fraction of CTIP2 cells (20 vs 41% for GFP control; ∼49% of control) while increasing the fraction of SATB2-expressing cells (∼82 vs ∼58% for GFP control; >1.4-fold increase) (Figs. 4, 5).
Figure 4.
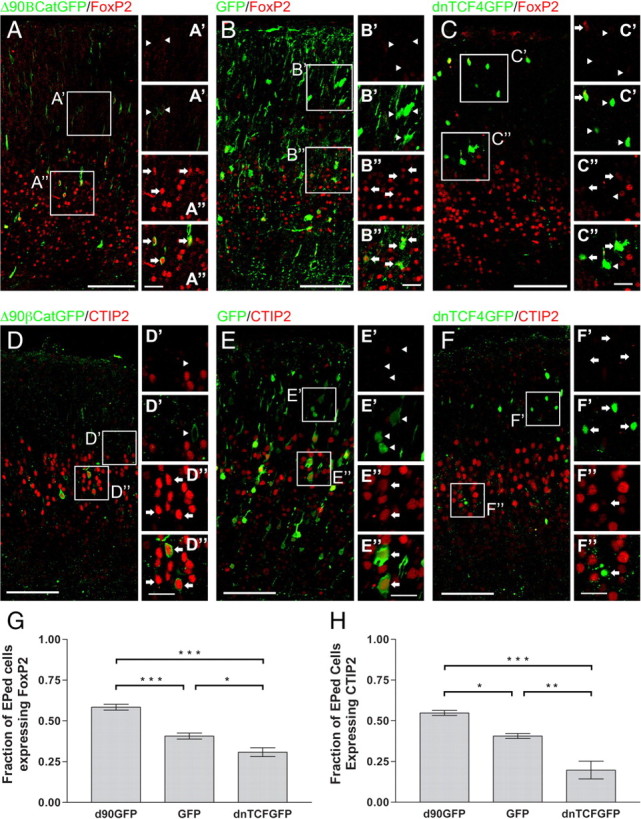
β-Catenin signaling influences the laminar cell fate of deep-layer cortical projection neurons. D–F, E13.5 embryos were electroporated with Δ90β-catenin-GFP (n = 4 brains), DNTCF4-GFP (n = 3 brains), or GFP (n = 4 brains) constructs. Embryos were killed at E19.5 and sections were stained for GFP and FOXP2 (A–C) or CTIP2 (D–F), markers for cortical layers 5 and 6. G, Fractions of GFP+ cells coexpressing FOXP2 among the three groups were significantly different (p < 0.0001; ANOVA; Newman–Keuls post-test analysis: Δ90β-catenin-GFP vs DNTCF4-GFP, p < 0.001; Δ90β-catenin-GFP vs GFP, p < 0.001, DNTCF4-GFP vs GFP, p < 0.05). H, CTIP2 expression was also significantly different among the three groups (p = 0.0010, ANOVA; Newman–Keuls post-test analysis: Δ90β-catenin-GFP vs DNTCF4-GFP, p < 0.001, Δ90β-catenin-GFP vs GFP, p < 0.05, DNTCF4-GFP vs GFP, p < 0.01). Increasing β-catenin signaling by Δ90β-catenin-GFP increased the fraction of FOXP2+ cells (0.580 ± 0.018, n = 581) and CTIP2+ cells (0.548 ± 0.016, n = 142), while blocking β-catenin signaling decreased the fraction of FOXP2 (0.287 ± 0.027, n = 376)- and CTIP2 (0.197 ± 0.054, n = 297)-positive cells, when compared to control (FOXP2: 0.370 ± 0.018, n = 504; CTIP2: 0.407 ± 0.015, n = 496). Arrows in insets (A′, A″, B′, B″, C′, C″) denote FOXP2+ cells; arrowheads denote FOXP2− cells. Arrows in insets (D′, D″, E′, E″, F′, F″) denote CTIP2+ cells; arrowheads denote CTIP2− cells. The top panel for each inset shows FOXP2 or CTIP2 immunofluorescence, while the bottom panels show merged images. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars: main panels, 100 μm; insets, 25 μm. Brackets on graphs show SEM.
Figure 5.

β-Catenin signaling influences the laminar cell fate of upper-layer cortical projection neurons. E13.5 embryos were electroporated with Δ90β-catenin-GFP (n = 4 brains), DNTCF4-GFP (n = 3 brains) or GFP (n = 4 brains) constructs. A–F, Embryos were killed at E19.5 and sections were stained for GFP and BRN1 (A–C) or SATB2 (D–F), markers for cortical layers 2–4. G, Fractions of GFP+ cells that coexpressed BRN1 in the three experimental groups were significantly different (p = 0.0004, ANOVA; Newman–Keuls post-test analysis: Δ90β-catenin-GFP vs DNTCF4-GFP, p < 0.001, Δ90β-catenin-GFP vs GFP, p < 0.01, DNTCF4-GFP vs GFP, p < 0.05). H, SATB2 expression was also significantly different among the three groups (p = 0.0013, ANOVA; Newman–Keuls post-test analysis: Δ90β-catenin-GFP vs DNTCF4-GFP, p < 0.01, Δ90β-catenin-GFP vs GFP, p < 0.05, DNTCF4-GFP vs GFP, p < 0.05). Increasing β-catenin signaling by Δ90β-catenin-GFP decreased the fraction of BRN1+ cells (0.267 ± 0.034, n = 255) and SATB2+ cells (0.304 ± 0.013, n = 117), while blocking β-catenin signaling increased the fraction of BRN1 (0.740 ± 0.017, n = 351) and SATB2 (0.817 ± 0.041, n = 232), when compared to GFP control (BRN1: 0.534 ± 0.065, n = 634; SATB2: 0.577 ± 0.079, n = 453). Arrows in insets (A′, A″, B′, B″, C′, C″) denote BRN1+ cells; arrowheads denote BRN1-. Arrows in insets (D′, D″, E′, E″, F′, F″) denote SATB2+ cells; arrowheads denote SATB2− cells. The top panel for each inset shows BRN1 or SATB2 immunofluorescence, while the bottom panels show merged images. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars: main panels, 100 μm; insets, 25 μm. Brackets on graphs show SEM. EPed, Electroporated.
To determine whether β-catenin signaling could alter the projection patterns of cortical neurons, we coelectroporated E13.5 embryos with pCAG-mCherry and either pCAG-GFP, pCAG-Δ90β-catenin-GFP, or pCAG-dnTCFGFP and analyzed the embryos at E19.5 (Fig. 6; supplemental Fig. S7, available at www.jneurosci.org as supplemental material). Coronal sections through the level of the anterior commissure were produced for all three groups. At this section level, both corticocortical projection pathways (through the corpus callosum and the anterior commissure) and subcortical projection pathways (through the internal capsule, whose fibers are perpendicular to the plain of the section) are visible. Cells coelectroporated with GFP control plasmid and mCherry projected many mCherry-positive processes through both subcortical (internal capsule) and corticocortical (anterior commissure and corpus callosum) projection pathways. Cells electroporated with Δ90β-catenin-GFP exhibit many mCherry-positive processes through the internal capsule but few through the corpus callosum or the anterior commissure. Meanwhile, cortical cells electroporated with dnTCF4GFP have very few mCherry-positive processes in the internal capsule but numerous mCherry-positive processes through the anterior commissure and corpus callosum.
Figure 6.

Manipulation of β-catenin signaling alters the axonal projection patterns of electroporated cortical neurons. E13.5 embryos were electroporated with pCAG-mCherry and either pCAG-GFP, pCAG-Δ90β-catenin-GFP, or pCAG-dnTCFGFP, and axonal projection patterns were analyzed at E19.5. Coronal sections through the level of the anterior commissure were produced for all three groups. At this section level, both corticocortical projection pathways (through the corpus callosum and the anterior commissure) and subcortical projection pathways (through the internal capsule whose fibers are perpendicular to the plain of the section) are visible. Cells coelectroporated with GFP control plasmid and mCherry projected many mCherry-positive processes through both subcortical (internal capsule) and corticocortical (anterior commissure and corpus callosum) projection pathways. Cells electroporated with Δ90β-catenin-GFP exhibit many mCherry-positive processes through the internal capsule but few through the corpus callosum or the anterior commissure. Meanwhile, cortical cells electroporated with dnTCF4GFP have very few mCherry-positive processes in the internal capsule but numerous mCherry-positive processes through the anterior commissure and corpus callosum. mCherry staining is pseudocolored red and DNA is pseudocolored blue. Scale bars, 125 μm (high-power images).
To examine whether the upper-layer neurons generated by DNTCF4-electroporated cells are born at the time when other upper-layer neurons are normally generated, we exposed E13.5-electroporated embryos to BrdU at E15.5 (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). We found that few DNTCF4-electroporated neurons were labeled by BrdU (∼1.6%) compared with GFP-electroporated neurons (∼11.2%). Consistent with previous findings that DNTCF4 causes premature cell cycle exit (Woodhead et al., 2006), this observation suggests that progenitors electroporated with DNTCF4-GFP generate upper-layer cortical neurons despite leaving the cell cycle at a time when deep-layer neurons are normally being generated.
β-Catenin signaling during late neurogenesis
As neurogenesis proceeds, the subtypes of neurons that can be produced by progenitors are restricted (Frantz and McConnell, 1996; Desai and McConnell, 2000; Shen et al., 2006). To examine whether β-catenin signaling could alter the cell fates of progenitors during late neurogenesis, we electroporated embryos at E15.5, the peak period of superficial-layer neurogenesis, with Δ90β-catenin and GFP control plasmids at E15.5 and killed them at E19.5. We found that expression of Δ90β-catenin was able to partially rescue early progenitor cell fate (Fig. 7). A significantly smaller proportion of cells electroporated with Δ90β-catenin expressed the superficial-layer marker BRN1 when compared to control (Fig. 7) (0.8983 ± 0.01558, n = 206) when compared to control (0.9785 ± 0.002237, n = 247; p = 0.0022, t test, two tailed), while the proportion of Δ90β-catenin-electroporated cells expressing the deep-layer marker FOXP2 is nearly three times greater than the proportion of control cells (0.07780 ± 0.01518, n = 363) when compared to control (0.02693 ± 0.01214, n = 148; p = 0.0397, t test, two tailed). Correspondingly, cells expressing Δ90β-catenin were found in deeper cortical positions and a greater proportion of Δ90β-catenin electroporated neurons expressed the deep-layer marker CTIP2 versus control GFP cells (supplemental Fig. S8, available at www.jneurosci.org as supplemental material). These findings suggest that β-catenin activation may partially reset the timing of laminar fate restriction of late-gestation cortical progenitors and allow them to produce deep-layer neurons.
Figure 7.

Alteration of β-catenin signaling during late neurogenesis influences cell positioning and fate. A–F, E15.5 embryos were electroporated with GFP (A–C) (n = 4 brains) or Δ90β-catenin-GFP (D–F) (n = 4 brains) constructs. Embryos were analyzed at E19.5 and sections were stained for GFP and BRN1, a marker for cortical layers 2–4 (A, D) or FOXP2, a marker for cortical layers 5 and 6 (B, E). The fractions of GFP+ cells that coexpressed BRN1 were significantly different (p = 0.0022, t test, two tailed). Increasing β-catenin signaling by Δ90β-catenin-GFP decreased the fraction of BRN1+ cells (0.8983 ± 0.01558, n = 206) when compared to control (0.9785 ± 0.002237, n = 247). The fractions of GFP+ cells coexpressing FOXP2 were also significantly different (p = 0.0397, t test, two tailed). Increasing β-catenin signaling by Δ90β-catenin-GFP increased the fraction of FOXP2+ cells (0.07780 ± 0.01518, n = 363) when compared to control (0.02693 ± 0.01214, n = 148). The somal position of each electroporated cell within 10 equally sized bins covering the complete cortical plate was also noted. The fraction of the total GFP+ cells in each of the 10 bins was then graphed (C, F) for both experimental conditions. Bin 1 corresponds to the section of the cortical plate closest to the pial surface, while bin 10 is adjacent to the white matter tracts. Overall, cells electroporated with Δ90β-catenin-GFP (n = 447) resided in slightly deeper cortical positions than GFP (n = 280) control cells. G, When these results are shown in a cumulative view, the shift in positioning is more evident. Arrows highlight examples of FOXP2/GFP double-positive cells. Arrowheads indicate GFP+/BRN1− cells. Scale bars, 100 μm. EPed, Electroporated.
Together these results provide evidence that β-catenin signaling regulates the laminar identity of cortical neurons. Overactivating β-catenin signaling increases the fraction of neurons located in deep cortical layers; these neurons express the deep-layer markers CTIP2 and FOXP2. In contrast, inhibition of β-catenin signaling increases the fraction of neurons located in superficial cortical layers; these neurons express the superficial cortical markers SATB2 and BRN1.
Discussion
Neurons destined for distinct laminar cell fates are produced sequentially at characteristic time points in neurogenesis. Here we show β-catenin signaling regulates laminar cell fate. By measuring relative β-catenin signaling during the peak periods for production of deep and superficial-layer neurons, we found β-catenin signaling in VZ precursors to be higher in cortical progenitors during deep-layer production versus later during upper-layer generation. Experimentally increasing the level of β-catenin signaling in midgestation VZ precursors increased the proportion of deep-layer neurons generated. Conversely, blocking β-catenin signaling caused an increase in the proportion of superficial level neurons. Moreover, we find that β-catenin overexpression can influence even late-gestation precursors to make deep-layer cortical neurons. These observations suggest a model in which relative levels of β-catenin signaling in VZ precursors determines their laminar fate decision, in which high levels of β-catenin signaling promote the production of deep-layer neurons, and reduced levels lead to upper-layer identity.
Quantification of β-catenin signaling levels
Previous studies using various reporter methods have demonstrated the presence of β-catenin signaling in the mouse ventricular zone (Maretto et al., 2003; Backman et al., 2005; Woodhead et al., 2006; Kadowaki et al., 2007). In this study we have shown that the level of β-catenin signaling decreases during the neurogenic period through two independent methods. BAT-gal transgenic mice, which have been used as a reporter of WNT/β-catenin signaling in many previous studies (Maretto et al., 2003; Backman et al., 2005; Lie et al., 2005) showed a decrease in reporter activity from early to late neurogenesis.
One of the drawbacks of lacZ reporter systems, such as the BAT-gal transgenic mouse line, is that the β-galactosidase protein product of may persist even after its reporter-driven transcription decreases. In an effort to quantify signaling more accurately we designed a novel reporter construct which contains several unique characteristics. First, we incorporated a dGFP that is rapidly degraded, allowing for more-accurate measurements. Second, electroporation of DNA injected into the lateral ventricles specifically targets only VZ precursors. Third, the use of fluorescent markers permits analysis of signaling at a cellular resolution. Finally, the incorporation of a second, constitutive promoter (CAG) driving the expression of mCherry allows for the detection of nonsignaling cells and provides a transfection control. Together these qualities allow for a more quantifiable snapshot of β-catenin signaling during different periods of neurogenesis.
Our technique for quantifying relative β-catenin signaling is not without limitations, however. While both the BAT-gal transgenic mice and the TOP consensus sequence on our plasmid reporter construct have been used extensively to measure Wnt/β-catenin signaling in a variety of systems, there is the possibility that they lack regulatory elements and modifications present on the chromatin of endogenous target genes. However, the simplicity of these reporter systems likely confers greater specificity for β-catenin signaling. While the expression of endogenous target genes are very likely regulated by inputs from multiple signaling pathways, reporter constructs lack response elements for potentially confounding non-β-catenin signaling pathways. Additionally, the BAT-gal and TOP-dGFP use the minimal promoter sequences of the distinct WNT/β-catenin signaling target genes siamois and c-fos, respectively (DasGupta and Fuchs, 1999; Maretto et al., 2003). Given the convergent results obtained from both reporter systems, it appears unlikely that our results could be due to reporter artifact.
A temporal gradient of β-catenin signaling in cortical timing
Our findings suggest a model in which one aspect of cortical progenitor timing, laminar fate determination, is regulated by β-catenin signaling levels. Early in cortical neurogenesis, β-catenin signaling levels are high and progenitors produce deep-layer cortical neurons. As cortical development continues, β-catenin signaling decreases, and progenitors become progressively restricted, giving rise to more superficial neurons (Fig. 8A). Overactivation of β-catenin signaling apparently can partially reset VZ progenitors to a younger “state” (Fig. 8B), while inhibiting signaling advances their timing to cause them to resemble older progenitors that produce upper-layer neurons (Fig. 8C).
Figure 8.
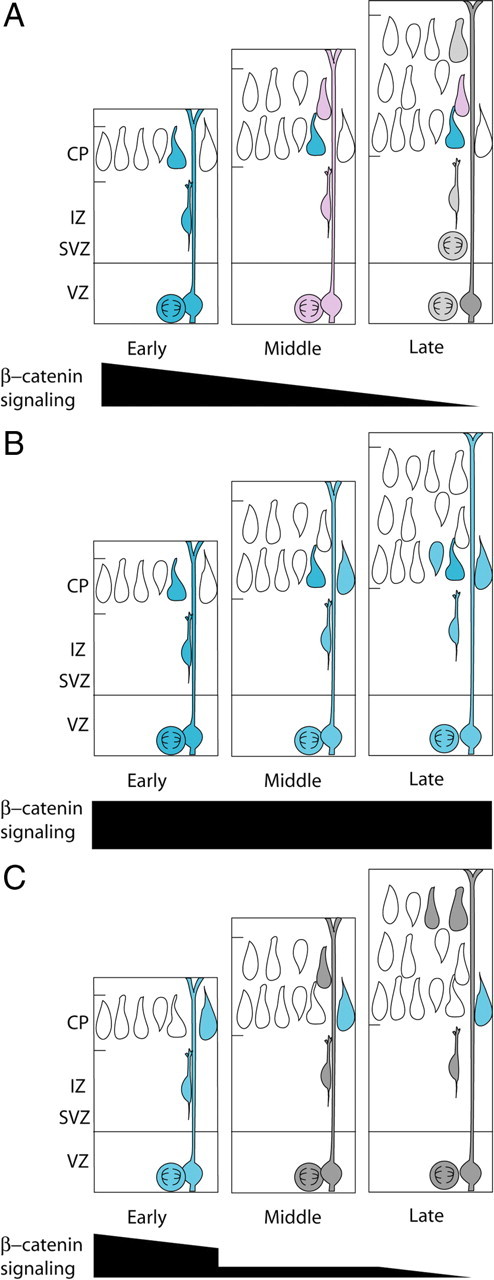
Model for the role of β-catenin signaling in determining laminar fate. In wild-type VZ progenitors (A), β-catenin signaling levels decrease throughout neurogenesis allowing for the production of progressively more superficial layers of projection neurons (depicted as blue = high, pink = medium, and gray = low signaling precursors and daughters. The progressive reduction in β-catenin signaling parallels the gradual restriction in the potential for later gestation precursors to generate deep-layer neurons. B, When β-catenin signaling is enhanced in VZ precursors (blue), the timing of precursor maturity is shifted so that they resemble more immature, deep-layer progenitors. C, When β-catenin signaling is inhibited in VZ precursors (gray cells), precursors resemble later precursors, and upper-layer neurons are produced earlier in neurogenesis.
Numerous studies have established that as neurogenesis progresses neuronal progenitors become less plastic (Frantz and McConnell, 1996; Mizutani and Saito, 2005; Shen et al., 2006; Gaspard et al., 2008). This restriction of cell fate appears to derive from both intrinsic and extrinsic cues. Despite this, under certain conditions late-progenitors have been shown to produce early neuronal fates (Molyneaux et al., 2005; Fukumitsu et al., 2006; Shen et al., 2006). In this study we were able to alter the cell fate of a small, but significant, fraction of late-neuronal progenitors. By enhancing β-catenin signaling during late neurogenesis, we were able to triple the fraction of deep-layer neurons produced. Despite this finding, it appears that even with increased β-catenin signaling, the majority of late-progenitors may require additional alterations to reset their ability to generate early cell fates. Alternatively, these observations suggest the possibility that a declining fraction of precursors may remain responsive to β-catenin signaling as neurogenesis progresses. This could represent an intrinsic process by which progenitors become less able to respond to β-catenin signaling or it could represent the absence of a yet undetermined synergistic extrinsic factor during late neurogenesis. Understanding the mechanism responsible for this observation is the target of future studies.
Recent reports identified genes involved in the production of classes of deep-layer neurons (Fezl, Ctip2, Tbr1 and Sox5) (Hevner et al., 2001; Arlotta et al., 2005; Cancer Genome Atlas Research Network, 2008) and upper-layer neurons (Satb2, Brn1, and Brn2) that that are involved in the specification of upper-layer neurons (Sugitani et al., 2002; Alcamo et al., 2008; Britanova et al., 2008). Although the relationship between these factors and β-catenin signaling is currently unknown, the expression of many of these genes (Fezl, Ctip2, Sox5, Tbr1, Satb2) in postmitotic cells raises the possibility that they function downstream of β-catenin signaling. In addition to Brn2, which is directly regulated by β-catenin in melanoma (Goodall et al., 2004), other possible direct targets of β-catenin in laminar fate decisions include the neurogenin family of bHLH transcription factors. Ngn genes have been proposed to regulate the production of early-born, deep-layer neurons (Schuurmans et al., 2004), and β-catenin has been shown to directly regulate the production of Ngn1 in progenitor cells (Israsena et al., 2004).
Distinct roles of β-catenin in proliferation and cell fate decisions
While our previous studies have shown that β-catenin signaling in neural precursors regulates cell cycle exit (Chenn and Walsh, 2002; Woodhead et al., 2006), our current findings suggest that β-catenin also functions in the determination of laminar cell fate. The correlations between birthdate and laminar fate in normal cortical development would suggest a simple model in which β-catenin activity delays cell cycle exit and predict that increased β-catenin would increase later-born (and therefore upper-layer) cortical neurons. In contrast, our results showing that increased β-catenin decreases the relative production of upper-layer cortical neurons while inhibiting β-catenin increases upper-layer neurons, suggest that neuronal birthdate and laminar fate can be dissociated. While the role of β-catenin in regulating laminar cell fate appears to be distinct from the role of β-catenin in regulating cell cycle exit, the precise downstream factors that regulate these processes remain the topic of ongoing studies.
Further studies are required to clarify how β-catenin signaling regulates laminar fate distinctly from cell cycle re-entry. Determining whether the observed relative changes in upper- and lower-layer neurons result from increased or decreased production or differential survival of distinct populations may require extended continual live imaging of cell behavior and laminar fate. Live imaging will also provide additional insight onto the dynamics of the lamination process, for example, whether the cells destined to become upper layers as a result of β-catenin inhibition migrate earlier than normal upper-layer neurons.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grants R01NS047191 (A.C.) and 1F30NS051864 (C.A.M.), a Sontag Foundation Distinguished Scholar Award (A.C.), and March of Dimes Grant 6-FY07-401 (A.C.). We thank Dr. Jack Kessler (Northwestern) and members of the Chenn lab for helpful comments on this manuscript.
References
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Fariñas I, Grosschedl R, McConnell SK. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 2008;57:364–377. doi: 10.1016/j.neuron.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Angevine JB, Jr, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Backman M, Machon O, Mygland L, van den Bout CJ, Zhong W, Taketo MM, Krauss S. Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev Biol. 2005;279:155–168. doi: 10.1016/j.ydbio.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Britanova O, de Juan Romero C, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D, Sestan N, Molnár Z, Tarabykin V. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron. 2008;57:378–392. doi: 10.1016/j.neuron.2007.12.028. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Wang SS, Hattox AM, Rayburn H, Nelson SB, McConnell SK. The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc Natl Acad Sci U S A. 2008;105:11382–11387. doi: 10.1073/pnas.0804918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Currle DS, Cheng X, Hsu CM, Monuki ES. Direct and indirect roles of CNS dorsal midline cells in choroid plexus epithelia formation. Development. 2005;132:3549–3559. doi: 10.1242/dev.01915. [DOI] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Desai AR, McConnell SK. Progressive restriction in fate potential by neural progenitors during cerebral cortical development. Development. 2000;127:2863–2872. doi: 10.1242/dev.127.13.2863. [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Sheldahl LC, Moon RT. A transgenic Lef1/beta-catenin-dependent reporter is expressed in spatially restricted domains throughout zebrafish development. Dev Biol. 2002;241:229–237. doi: 10.1006/dbio.2001.0515. [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol. 2003;460:266–279. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- Frantz GD, McConnell SK. Restriction of late cerebral cortical progenitors to an upper-layer fate. Neuron. 1996;17:55–61. doi: 10.1016/s0896-6273(00)80280-9. [DOI] [PubMed] [Google Scholar]
- Fukumitsu H, Ohtsuka M, Murai R, Nakamura H, Itoh K, Furukawa S. Brain-derived neurotrophic factor participates in determination of neuronal laminar fate in the developing mouse cerebral cortex. J Neurosci. 2006;26:13218–13230. doi: 10.1523/JNEUROSCI.4251-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspard N, Bouschet T, Hourez R, Dimidschstein J, Naeije G, van den Ameele J, Espuny-Camacho I, Herpoel A, Passante L, Schiffmann SN, Gaillard A, Vanderhaeghen P. An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature. 2008;455:351–357. doi: 10.1038/nature07287. [DOI] [PubMed] [Google Scholar]
- Goodall J, Martinozzi S, Dexter TJ, Champeval D, Carreira S, Larue L, Goding CR. Brn-2 expression controls melanoma proliferation and is directly regulated by beta-catenin. Mol Cell Biol. 2004;24:2915–2922. doi: 10.1128/MCB.24.7.2915-2922.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanashima C, Li SC, Shen L, Lai E, Fishell G. Foxg1 suppresses early cortical cell fate. Science. 2004;303:56–59. doi: 10.1126/science.1090674. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Israsena N, Hu M, Fu W, Kan L, Kessler JA. The presence of FGF2 signaling determines whether beta-catenin exerts effects on proliferation or neuronal differentiation of neural stem cells. Dev Biol. 2004;268:220–231. doi: 10.1016/j.ydbio.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Junghans D, Hack I, Frotscher M, Taylor V, Kemler R. Beta-catenin-mediated cell-adhesion is vital for embryonic forebrain development. Dev Dyn. 2005;233:528–539. doi: 10.1002/dvdy.20365. [DOI] [PubMed] [Google Scholar]
- Kadowaki M, Nakamura S, Machon O, Krauss S, Radice GL, Takeichi M. N-cadherin mediates cortical organization in the mouse brain. Dev Biol. 2007;304:22–33. doi: 10.1016/j.ydbio.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Lie DC, Colamarino SA, Song HJ, Désiré L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Machon O, van den Bout CJ, Backman M, Kemler R, Krauss S. Role of beta-catenin in the developing cortical and hippocampal neuroepithelium. Neuroscience. 2003;122:129–143. doi: 10.1016/s0306-4522(03)00519-0. [DOI] [PubMed] [Google Scholar]
- Machon O, Backman M, Machonova O, Kozmik Z, Vacik T, Andersen L, Krauss S. A dynamic gradient of Wnt signaling controls initiation of neurogenesis in the mammalian cortex and cellular specification in the hippocampus. Dev Biol. 2007;311:223–237. doi: 10.1016/j.ydbio.2007.08.038. [DOI] [PubMed] [Google Scholar]
- Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S. Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci U S A. 2003;100:3299–3304. doi: 10.1073/pnas.0434590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani K, Saito T. Progenitors resume generating neurons after temporary inhibition of neurogenesis by Notch activation in the mammalian cerebral cortex. Development. 2005;132:1295–1304. doi: 10.1242/dev.01693. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 2005;47:817–831. doi: 10.1016/j.neuron.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- O'Brien WT, Harper AD, Jové F, Woodgett JR, Maretto S, Piccolo S, Klein PS. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. Neuron-glia relationship during granule cell migration in developing cerebellar cortex. A golgi and electronmicroscopic study in Macacus rhesus. J Comp Neurol. 1971;141:283–312. doi: 10.1002/cne.901410303. [DOI] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- Schuurmans C, Armant O, Nieto M, Stenman JM, Britz O, Klenin N, Brown C, Langevin LM, Seibt J, Tang H, Cunningham JM, Dyck R, Walsh C, Campbell K, Polleux F, Guillemot F. Sequential phases of cortical specification involve neurogenin-dependent and -independent pathways. EMBO J. 2004;23:2892–2902. doi: 10.1038/sj.emboj.7600278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Wang Y, Dimos JT, Fasano CA, Phoenix TN, Lemischka IR, Ivanova NB, Stifani S, Morrisey EE, Temple S. The timing of cortical neurogenesis is encoded within lineages of individual progenitor cells. Nat Neurosci. 2006;9:743–751. doi: 10.1038/nn1694. [DOI] [PubMed] [Google Scholar]
- Sick S, Reinker S, Timmer J, Schlake T. WNT and DKK determine hair follicle spacing through a reaction-diffusion mechanism. Science. 2006;314:1447–1450. doi: 10.1126/science.1130088. [DOI] [PubMed] [Google Scholar]
- Sugitani Y, Nakai S, Minowa O, Nishi M, Jishage K, Kawano H, Mori K, Ogawa M, Noda T. Brn-1 and Brn-2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Dev. 2002;16:1760–1765. doi: 10.1101/gad.978002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Goto T, Miyama S, Nowakowski RS, Caviness VS., Jr Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J Neurosci. 1999;19:10357–10371. doi: 10.1523/JNEUROSCI.19-23-10357.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Woodhead GJ, Mutch CA, Olson EC, Chenn A. Cell-autonomous β-catenin signaling regulates cortical precursor proliferation. J Neurosci. 2006;26:12620–12630. doi: 10.1523/JNEUROSCI.3180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel CN, Mutch CA, Swaminathan S, Taketo MM, Chenn A. Persistent expression of stabilized beta-catenin delays maturation of radial glial cells into intermediate progenitors. Dev Biol. 2007;309:285–297. doi: 10.1016/j.ydbio.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota Y, Kim WY, Chen Y, Wang X, Stanco A, Komuro Y, Snider W, Anton ES. The adenomatous polyposis coli protein is an essential regulator of radial glial polarity and construction of the cerebral cortex. Neuron. 2009;61:42–56. doi: 10.1016/j.neuron.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner D, Fujita Y, Hülsken J, Müller T, Walther I, Taketo MM, Crenshaw EB, 3rd, Birchmeier W, Birchmeier C. β-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev Biol. 2003;258:406–418. doi: 10.1016/s0012-1606(03)00123-4. [DOI] [PubMed] [Google Scholar]