

Abstract
Familial hemophagocytic lymphohistiocytosis (FHL) is a genetically heterogeneous autosomal recessive immune disorder characterized by the occurrence of uncontrolled activation of lymphocytes and macrophages infiltrating multiple organs. Disease-causing mutations in the perforin (PRF1; also known as FHL2), Munc13-4 (UNC13D; also known as FHL3), and syntaxin-11 (STX11; also known as FHL4) genes have been identified in individuals with FHL. These genes all encode proteins involved in the cytotoxic activity of lymphocytes. Here, we show that the gene encoding syntaxin-binding protein 2 (Munc18-2; official gene symbol STXBP2) is mutated in another subset of patients with FHL (designated by us as “FHL5”). Lymphoblasts isolated from these patients had strongly decreased STXBP2 protein expression, and NK cells exhibited impaired cytotoxic granule exocytosis, a defect that could be overcome by ectopic expression of wild-type STXBP2. Furthermore, we provide evidence that syntaxin-11 is the main partner of STXBP2 in lymphocytes, as its expression required the presence of STXBP2. Our work shows that STXBP2 deficiency causes FHL5. These data indicate that STXBP2 is required at a late step of the secretory pathway for the release of cytotoxic granules by binding syntaxin 11, another component of the intracellular membrane fusion machinery.
Introduction
Familial hemophagocytic lymphohistiocytosis (FHL; MIM 267700) is a genetically heterogeneous immune disorder of autosomal recessive inheritance. A hallmark of this condition consists in the occurrence of uncontrolled proliferation and activation of polyclonal T/NK lymphocytes and macrophages that infiltrate multiple organs including liver and spleen and the CNS. It causes cytopenia and hyperferritinemia and has a fatal outcome (1).
Disease-causing mutations in the genes encoding perforin (PRF1; FHL2) (2), Munc13-4 (UNC13D; FHL3) (3), and syntaxin-11 (STX11; FHL4) (4) have been previously identified in FHL. They account for approximately 80% of FHL cases. These genes encode proteins that are involved in the cytotoxic activity of lymphocytes. Lymphocyte cytotoxic activity plays a major role in the defense against viral infection and cancer by seeking out and killing infected or tumorigenic cells. Once cytotoxic cells have recognized their targets and formed a conjugate, cytotoxic activity is delivered in a multi-step process. This includes the polarization of the cytotoxic granules containing perforin and granzymes toward the immunologic synapse (IS) formed with the target, docking, priming, and fusion of the cytotoxic granules with the plasma membrane so that release of their content induces target cell destruction by apoptosis (5–7). The inability of activated cytotoxic cells to clear antigen-presenting targets results in sustained immune stimulation, likely accounting for the unremitting polyclonal CD8+ T cell activation and hyperimmune reaction that characterizes FHL. Along with perforin, which is critical for granzyme delivery to target cells, Munc13-4 is essential in priming cytotoxic granules docked at the IS, whereas STX11 regulates membrane fusion events. In other patients however, the underlying molecular cause remains unknown. Here, we report that biallelic mutations in the gene encoding syntaxin binding protein 2 (STXBP2) account for what we believe is a new genetic form of FHL. We have identified 2 mutations that result in an absence or markedly decreased expression of STXBP2 in leukocytes from 8 patients. Remarkably, the latter mutation is associated with late-onset FHL. When STXBP2 is defective, patients’ NK cells conjugate with targets and polarize their cytotoxic granule toward the IS but are unable to release cytotoxic granule contents.
Results
STXBP2 mutations cause FHL.
We studied a cohort of 6 consanguineous families in which 10 individuals had been diagnosed with FHL. Known causes of FHL (2–4) were excluded by genetic analyses. In order to determine the molecular genetic basis of the FHL in these families, we screened for homozygous chromosomal regions using genome-wide SNP analysis with DNA obtained from 8 available patients (Figure 1A). This analysis revealed a common region on chromosome 19p13.2–p13.3 in which none of the markers showed evidence of heterozygosity. This region was flanked by EMR1 on the telomeric side of the critical interval and ELAVL1 on the centromeric side. Of the 40 genes contained in the interval, STXBP2 (Munc18-2 gene) appeared to be a plausible candidate, since STXBP2 is known to be expressed in the hematopoietic system, interact with soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) syntaxins, and regulate intracellular vesicle trafficking (8–10).
Figure 1. STXBP2 mutations in FHL5.

(A) Pedigrees from the FHL families with STXBP2 mutations. F3P1 and F3P2 belonged to the same tribe in Saudi Arabia. Consanguinity (double horizontal bars), affected individuals (black boxes and circles), carriers (half-filled boxes and circles), deceased individuals (diagonal bars) and subjects not available to participate in the study (asterisks) are indicated. (B) Positions of the 2 STXBP2 mutations. The splice mutation in intron 14 introduced 56 bp from the intronic sequence into the end of exon 15. The proline transition (P477L) occurred at a residue that had been conserved over evolution and within the members of the Munc18 family.
DNA sequencing of the STXBP2 gene in the 8 expressive probands from these families revealed 2 different homozygous missense mutations (Table 1 and Figure 1B). Five individuals from 3 families originating from Saudi Arabia (F1P1, F2P1, F2P2, F3P1, and F3P2, where “F” indicates the family number and “P” indicates the patient number) carried homozygous missense mutations in exon 16, prompting a 1430C>T nucleotide change and a P477L aa substitution. This proline is highly conserved in all members of the Munc18 family and in all species (Figure 1B). The 3 other affected individuals from 3 different families of Turkish, Arabic, or Iranian origin were found to carry a homozygous splice mutation resulting in the use of cryptic acceptor and donor splice sites at intron 14 (1247-1G>C indicated as IVS14-1G>C) (Table 1 and Figure 1B). In addition, we identified the same homozygous mutation in F5P2 (the 2-year-old brother of F5P1), who was asymptomatic at the age of 32 months (Table 1). In all cases, the parents were found to be heterozygous for the mutated STXBP2 allele. With the exception of F5P2, the tested healthy siblings were heterozygous or homozygous for the WT allele. None of these mutations were present in 80 French, 50 Turkish, or 80 Arabian or Iranian healthy subjects. The mutations did not show any influence on STXBP2 steady-state mRNA levels, which was normally abundant (Figure 2, A and B). Sequencing of the RT-PCR products confirmed the mutations and enabled characterization of the new splice product for the individuals with the IVS14-1G>C mutation. Rather than leading to a premature stop codon, this splice mutation resulted in exchange of the first 17 aa of exon 15 by 19 new aa from the intronic sequence, in frame with the last 20 aa of the exon 15 WT sequence (Figure 1B).
Table 1 .
Phenotypes and genotypes observed in the 9 individuals carrying STXBP2 mutations

Figure 2. STXBP2 and STX11 expression in FHL5.

(A) Lymphoblasts from affected individuals showed normal levels of STXPB2 transcript. (B) STXBP2 transcript levels were quantified as the fold difference of mRNA levels for STXBP2 normalized to 18S, a housekeeping gene, in the lymphoblasts from control subjects and patients. Analyses were performed in triplicate. Data are mean ± SD. (C) Left: Western blot showing the expression of STXBP2, STX3, and STX11 proteins in lymphocytes from a healthy control, 3 STXBP2-deficient patients (F2P1, F6P1, and F5P1), and 2 STX11-deficient patients with nonsense mutations in STX11 located in the 5ι (P2) or 3ι (P1) part of the gene. Right: The abundance of STX3 and STX11 (relative to PI3K) via signal intensity measurement. Results are presented as the syntaxin/PI3K ratio, normalized against that obtained with a control (in arbitrary units).
Defective expression of STXBP2.
We assessed the expression of STXBP2 in lymphoblasts obtained from F2P1, F2P2, F5P1, and F6P1. In a protein blot analysis, STXBP2 protein was not found in lymphoblasts from F2P1 (Figure 2C) or F2P2 (data not shown) but was detected in lymphoblasts from F5P1 and F6P1, albeit at markedly lower levels than in control lymphoblasts (Figure 2C). These data indicate that FHL5 is associated with loss-of-function mutations in the STXBP2 gene.
The phenotypic expression of FHL varied considerably as a function of STXBP2 mutations, as shown in Table 1. In all patients with the P477L transition, FHL had a very early onset and rapidly led to death in most cases. In contrast, FHL manifestations occurred several years later in all patients with the in-frame splice site mutation in STXBP2. This strong genotype/phenotype correlation probably accounts for the absence of disease symptoms in F5P2 at the age of 32 months but also predicts that this individual runs a high risk of developing FHL later on.
STXBP2, like other Sec1/Munc18 family members, has an arch-shaped structure consisting of 3 domains (Figure 3A) (11, 12). The central cavity (formed by domains 1 and 3a) provides the binding surface for syntaxin. Both the mutant STXBP2 and Munc18-2 proteins bear aa replacements in the hydrophobic core (Figure 3A). The position of proline 477 within the core of the protein — at the border between domains 3b and 2 (Figure 1B and Figure 3A) — predicts that replacement of this aa will create a kink likely to affect the positioning of neighboring secondary structure units and destabilize the folding of STXBP2, thus leading to degradation. Remarkably, the spliced mutant protein in which the 19 new aa introduced by the splice mutation replace the WT 17 aa is predicted to have a relatively unperturbed structure (Figure 3, A and B). As emphasized by the sequence alignment shown in Figure 3B, there is strong conservation of hydrophobic residues when comparing the WT and mutant STXBP2/Munc18-2. The region exhibits an amphipathic α helix with an alternating hydrophobic/nonhydrophobic residue pattern every 3–4 residues (since the pitch of an α helix is 3.6 residues). As a result, the helix is generally hydrophobic on one side and hydrophilic on the other. Thus, the spliced mutant protein is also expected to feature an α helix at this location (Figure 3, A and B) and may therefore retain the arch-shaped structure and residual syntaxin binding.
Figure 3. Model structure of STXBP2.

(A) A ribbon representation of the STXBP2 protein. By homology with STXBP1, the 3 domains are colored as follows: domain 1 in blue, domain 2 in green (with light and dark green indicating the 2 non-contiguous segments of the polypeptide chain that form this domain), domain 3a in yellow, and domain 3b in brown. The central cavity (formed by domains 1 and 3a) provides the binding surface for syntaxin. The mutated proline (P477L) and the α helix affected by the splice are highlighted in red. The IVS14-1G>C mutant induces aa replacements in the hydrophobic core. (B) Splice mutation, which replaces 17 aa of the WT sequence with 19 new residues. A remarkable conservation of a hydrophobic aa pattern can be observed. Hydrophobic aa are highlighted in green; the alternation of hydrophobic and hydrophilic aa every 3–4 residues is typical for amphipathic α helices.
STXBP2 interacts with STX11.
The Sec1/Munc18 proteins (via interaction with syntaxins) are required for the induction of secretory vesicle docking and fusion with the plasma membrane (10, 13) and also stabilize syntaxin expression (14). It has been reported that STXBP2 interacts with STX3 and, to a lesser extent, with STX2 (8, 9, 15). We therefore investigated whether STX3 expression was affected in STXBP2-deficient cells. The expression of STX3 in patients was found to be similar to that of control lymphoblasts (Figure 2C). In a striking contrast, we observed that expression of STX11, a syntaxin family member that is highly expressed in hematopoietic cells and is known to be deficient in a variant of FHL (FHL4), was dramatically impaired (Figure 2C). STX11 expression was indeed not detected in patient cells carrying the proline substitution and was also significantly lower in cells from patients carrying the STXBP2 splice mutation (Figure 2C). The level of STX11 expression thus fully correlates with that of STXBP2, suggesting that the interaction between these proteins is relevant. Accordingly, the residual STX11 expression in lymphoblasts from patients carrying the splice mutation in STXBP2 could account for the observed milder phenotype. The STXBP2-STX11 interaction was confirmed in cells coexpressing the 2 proteins. (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI40732DS1). Similarly, STXBP2 splice mutant did interact with STX11 when co-expressed (Supplemental Figure 1). In order to see whether a defect in STX11 leads to a decrease in STXBP2 stability, expression of the latter was analyzed in STX11-deficient lymphoblasts from 2 FHL4 patients. Remarkably, the STXBP2 expression level was similar in control and patient cells either completely lacking STX11 or expressing a truncated form of the protein (Figure 2C). Thus, since expression of STX11 (but not of STX3) entirely depends on the expression of STXBP2, STX11 appears to be the main syntaxin partner of STXBP2 in lymphocytes. In contrast, STXBP2 stabilization does not require STX11 or it could involve binding to other syntaxins (8, 9, 15).
STXBP2 regulates NK cytotoxic activity.
In line with the pathophysiology of FHL, defective cytotoxic NK cell degranulation is a hallmark of STX11 deficiency, which causes FHL4 (16, 17), whereas defective cytotoxic function of both T and NK lymphocytes was shown to result from defects in perforin (causing FHL2) (2) and Munc13-4 (causing FHL3) (3). We therefore tested cell degranulation of STXBP2 NK and T lymphocytes by measuring CD107 expression at the cell membrane upon activation (17, 18). Freshly isolated NK cells from F5P1 and his asymptomatic brother, F5P2, failed to degranulate in an antibody-dependent cellular cytotoxicity assay (Figure 4A) and to exert natural cytotoxicity against K562 target cells (Supplemental Figure 2). As reported for STX11-deficient cells (17), IL-2 stimulation of NK cells partially restored degranulation (Figure 4A). Impaired degranulation was also detected in F5P1, F5P2, and F6P1 NK cells that were cultured for 15 to 20 days (Figure 4B). The degranulation of STXBP2-deficient CTLs was not significantly impaired (Figure 4C), similar to that of STX11-deficient CTLs (17) (Figure 4C). In order to confirm the role of STXBP2 in the release of cytotoxic granules, NK cells were cotransfected with the cDNA encoding WT STXBP2 and an otherwise empty fluorescent vector. Degranulation was restored in the fraction of patients’ NK cells that had been cotransfected, while it remained impaired in NK cells transfected with the empty vector alone (Figure 4D). In control cells, transfection did not significantly modify the ability of the NK cells to degranulate (Figure 4D). Lymphocyte-activated killer (LAK) cells were then used to evaluate the polarization of perforin-containing granules of NK cells toward target cells. Although STXBP2-deficient NK cells were impaired in their ability to degranulate, they were nevertheless able to polarize perforin-containing granules toward cognate target cells in the same manner as control NK cells (Figure 5). Thus, STXBP2, as reported for STX11 (17), is required for the degranulation of NK cell cytotoxic granules. STXBP2 probably operates at a late step in vesicle docking or fusion with the plasma membrane.
Figure 4. Defective cytotoxic granule exocytosis of STXBP2-deficient NK lymphocytes.

(A) Exocytosis of cytotoxic granules from resting or STXBP2-deficient CD3–CD56+ NK cells cultured with IL-2 for 72 hours (F5P1, triangle and F5P2, diamond) compared with cells of healthy adult and infant donors (controls). Cytotoxic granule exocytosis (ΔCD107) represents the percentage of CD107+ NK cells stimulated with anti-CD16 and P815 cells subtracted from the percentage of CD107+ NK cells incubated with P815 cells alone. Horizontal bars represent mean values. **P < 0.01, control values compared with the median value for each patient. (B) CD107 expression of NK cells cultured with IL-2 for 15–20 days from either controls or STXBP2-deficient (F5P1, F5P2, and F6P1) individuals. Values represent mean (± SD) percentages of the CD107+ NK cells. (C) Cytotoxic granule exocytosis by T lymphoblasts. Induced CD107 surface expression on CD3+CD8+ T lymphoblasts from controls, STXBP2-deficient individuals (F2P1, F5P1, and F6P1), or STX11-deficient individuals (P1 and P2). Horizontal bars represent mean values. #P > 0.1. (D) Restoration of the cytotoxic granule exocytosis in patient cells transfected with WT STXBP2 construct. PBMCs described in B were cotransfected with the WT STXBP2-FLAG–tagged vector or empty-FLAG–tagged vector and the insertless ECFP vector. For each individual, the ECFP-positive (transfected) and ECFP-negative (nontransfected) NK cell populations were tested as described in B. Dot plots were gated on CD3–CD56+ NK cells, and gates were set individually on the basis of NK cells incubated with P815 alone. Numbers indicate the percentage of degranulating NK cells. The results shown are representative of 2 independent experiments with similar results.
Figure 5. Normal polarization of cytotoxic granules in STXBP2-deficient NK cells.
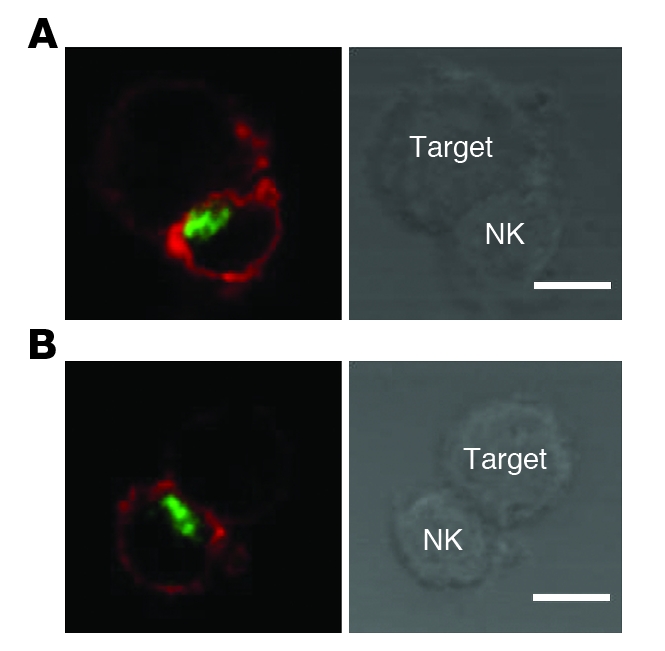
Confocal microscopy of WT (A) and STXBP2-deficient (B) LAK cells conjugated with anti-CD16 P815 target cells. Cells were stained with perforin mAb (green) and phalloidin (red). Perforin polarized in 66% of control NK cell conjugates (n = 30) and 70% of STXBP2-deficient NK cells conjugates (n = 30). Scale bars: 5 μm. Data are representative of 3 independent experiments.
Discussion
We report here that recessive mutations of the STXBP2 gene are responsible for a new genetic form of FHL, designated FHL5. Mutations in already known genes (perforin, Munc13-4, and STX11) were identified in 80% of a cohort of 80 FHL patients. STXBP2 mutations accounted for an additional 10% of the FHL cohort, and the causes remain unknown for the remaining 10% of FHL cases. The complete absence of STXBP2 resulted in early-onset severe disease, whereas a hypomorphic mutation was associated with delayed disease onset (as observed with hypomorphic mutations of the perforin encoding gene, for example) (6). In addition, our data show that STXBP2 is required for the release of cytotoxic granules by NK cells by promoting granule exocytosis after their polarization to the IS. Both CD16-mediated and natural cytotoxicity are shown to depend on STXBP2 function. It is unlikely that a difference in STXBP2 or STX11 expression between NK and T lymphocytes accounts for the functional difference observed, since STX11 was not detected in the F2P1 lymphocyte population that contained 75% T cells and efficiently degranulated. Defective cytotoxic function of both T and NK lymphocytes in FHL2 and FHL3 was shown to result from defects in perforin and Munc13-4, respectively (2, 3). However, when we tested the degranulation of STXBP2-deficient CTLs, there was no significant difference between patients and control groups. These data are consistent with those observed in FHL4 resulting from a STX11 deficiency (17) and differ from those observed in Munc13-4 deficiency (3). Thus, the same defective cytotoxicity phenotype characterizes both STX11 and STXBP2 deficiencies, an observation that further suggests functional interaction between these proteins in the degranulation process. The fact that FHL associated with a partial NK cell cytotoxic defect (FHL4 and FHL5) and FHL associated with severe impairment of both NK and T lymphocyte cytotoxicity (FHL2 and FHL3) exhibit indistinguishable phenotypes strongly suggests that the in vitro cytotoxic assays do not faithfully reflect the in vivo function of cytotoxic cells. The potent in vitro stimulation of activated T lymphocytes before CTL testing might bypass the STXBP2 STX11-dependent in vivo function. It may promote cytotoxic granule exocytosis, perhaps by causing the docking step to be skipped, as has been observed for other cells under sustained stimulation conditions, a process that has been termed “crash fusion” (13). Alternatively, this may indicate that NK cells could play a more important role in the hemophagocytic lymphohistiocytosis (HLH) disease phenotype. However, we should stress that studies performed in the perforin knockout mice that were depleted of either the CD8+ or the NK cell population indicate that CTLs, rather than the NK1.1+ cell population, play a critical role in the occurrence of HLH (19). Finally, additional mechanisms may operate in FHL4 and FHL5, in addition to the impairment of NK cell cytotoxic activity.
STXBP2 belongs to the Sec1/Mun18 family of fusion accessory proteins, which clasp SNARE’s 4-helix bundle and play complementary roles in membrane fusion (10, 20). The role of Sec1/Munc18 proteins in vesicle exocytosis has mainly been deduced by studying STXBP1 in neurons, where its deletion leads to a complete loss of neurotransmitter secretion in mice (14, 21). As in the process of cytotoxic granule exocytosis, a sequential docking/priming/fusion pathway is thought to orchestrate synaptic vesicle exocytosis at the neurologic synapse. STXBP1 was shown in a first step to bind to the closed conformation of its specific syntaxin (STX1) and thus create a docking platform that gates initiation of the reaction (20). Docked vesicles are then primed for fusion by Munc13-1 (equivalent to Munc13-4 in cytotoxic cells). Completion of the fusion reaction occurs when STXBP1 clasps across the zippering 4-helix assembled trans SNARE complex (20). Our present data strongly suggest that at the IS of a cytotoxic cell conjugated with a target, the STXBP2 STX11 complex could exert a role similar to that of the STXBP1 STX1 complex at the neurologic synapse by regulating granule docking and initiation of SNARE complex formation upstream of the priming step.
In summary, what we believe is a newly recognized genetic form of FHL, FHL5, is caused by mutation in STXBP2, resulting in defective cytotoxic activity of NK cells. The same defective NK cytotoxic function characterizes FHL4, caused by deficiency of STX11, a protein herein shown to be the main partner of STXBP2 in lymphocytes. Together with the previously reported findings of defective cytotoxicity of T and NK lymphocytes in FHL2 and FHL3 (2, 3), a critical role of the cytotoxic function in the regulation of lymphocyte responses is further pinpointed. Following the identification of 4 causative gene mutations, the frequency of unknown FHL cases is now down to 10%. Nevertheless, further delineation of still unknown causes of FHL is likely to provide important information on how the various components of this machinery are physiologically orchestrated.
Methods
Subjects.
All patients specified in this study fulfilled the diagnostic criteria for FHL according to the Histiocyte Society’s diagnostic criteria (1). Patients’ specific characteristics are given in Table 1, and the patients who presented with a milder phenotype are described below in more detail.
F4P1 was born to Palestinian Arab parents. There was no relevant family history. He first experienced typical HLH manifestations at the age 8 years associated with EBV viremia. He rapidly responded to treatment with cyclosporin A (CSA), which was then discontinued. A relapse occurred 5 months later at a time when erythrophagocytosis was detected in cerebrospinal fluid (CSF). He responded to treatment with antithymocyte globulin, high-dose corticosteroid (CS), CSA, and intrathecal methotrexate, then underwent a successful HLA identical HSC transplantation (HSCT) following cytoreductive chemotherapy. The patient was doing well at a follow-up of 40 months.
F5P1 was born to consanguineous Turkish parents. He experienced the first HLH manifestations, without neurological involvement, at the age of 10.5 years. Treatment by CS, CSA, and rituximab (because of EBV viremia) led to complete remission. Because of persistent asthenia and mild isolated thrombocytopenia, CSA therapy was very recently reinitiated.
F6P1 was born to a consanguineous Iranian family. A sister had died at the age of 22 months with a retrospective diagnosis of FHL. The F6P1 patient had first mild but typical HLH manifestations at the age of 12 years 6 months, including CSF pleiocytosis. Remission was achieved using CS, CSA, and intrathecal injection of methotrexate. Three relapses occurred within 1 year when CS dose was reduced. A more stable remission was then obtained until the age of 16, when a fourth relapse occurred. Again, CS and CSA therapy achieved a complete remission. Three years later, a fifth relapse was marked by CNS involvement with meningitis. Disease control was obtained by VP16 therapy, but the patient died from multivisceral failure at the age of 20 in the follow-up to a HSCT.
Clinical information and blood samples were collected from FHL patients and relatives as well as control individuals after informed consent had been obtained. All protocols were approved by the institutional review board of INSERM, the Newcastle upon Tyne Hospitals NHS Foundation Trust Clinical Governance and Risk, and the King Fahad Medical City institutional review board.
SNP analysis and sequence analysis.
Genomic DNA was isolated by phenol/chloroform extraction. RNA was isolated using an RNeasy Mini Kit (Qiagen). We carried out genotyping by homozygosity mapping on an Affymetrix platform using the 250K Nsp array from the Affymetrix 500K system, which consists of 262,000 SNPs (Affymetrix), according to the manufacturer’s instructions. Genotyping was performed with the Genotyping Console 2.1 Software (Affymetrix) using the BRLMM genotype calling algorithm. We detected homozygous regions using a parametric, SNP-based linkage analysis performed with the MERLIN program (version 1.1.1) (22). Genomic DNA and cDNA were amplified, sequenced, and analyzed on an ABI Prism 3700 apparatus (BigDye Terminator sequencing kit; Applied Biosystems). See Supplemental Table 1 for primers.
Gene expression analysis.
Total RNA was reverse transcribed using a Quantitec Reverse Transcriptase kit (Qiagen). RT-PCR was performed on each subject sample using STXBP2- and actin-specific primers (Supplemental Table 1). We carried out comparative real-time RT-PCR assays for each sample in triplicate and in a final reaction volume of 50 μl. The endogenous housekeeping gene (18S) and the STXBP2 gene were amplified by using about 20 ng of cDNA and STXBP2 (Hs00199557) or 18S (Hs99999901) probes labeled with 6-carboxy-fluorescein (FAM) dye (Applied Biosystems). Fluorescence measurement during PCRs was performed on an ABI Prism 7900 cycler sequence detection system (Applied Biosystems). The data were analyzed using the comparative threshold cycle method and presented as the relative change in gene expression normalized against the calibrator sample corresponding to control LAK.
Plasmids and site-directed mutagenesis.
The full-length human STX11 cDNA was inserted into a pFLAG–CMV-4 vector (Sigma-Aldrich) with the BD In-Fusion PCR Cloning Kit (BD Biosciences; Clontech). The full-length human STXBP2 WT and the IVS14-1–splice mutant were amplified by PCR using control and F5P1 patient cDNA, respectively (see Supplemental Table 1 for primers). PCR products were cloned into a pEGFPC1 vector (Clontech) using EcoRI and BamHI restriction sites and the pFLAG–CMV-4 vector. A pECFP-C1 vector (Clontech) was used in the cotransfection assay as a marker of transfected cells.
Cells, cell culture, and transfection.
PBMCs were isolated from blood samples taken from FHL5 patients, STX11-deficient patients, and controls. The STX11-deficient patients carried either a homozygous deletion or a homozygous nonsense mutation in STX11 similar to those previously described (4), with a predicted early (P2-STX11: V124fsX60) or late (P1-STX11: Q268X) premature stop codon. Phytohemagglutinin-induced T lymphoblasts were obtained by incubating PBMCs for 24 hours with phytohemagglutinin (1:400 dilution; Sigma-Aldrich) and IL-2 (20 IU/ml; PeproTech) in Panserin medium (Biotech GmbH) supplemented with 10% human AB serum (Etablissement Français du Sang). We then added more IL-2 (40 IU/ml) and cultured the cells for 6–7 days in Panserin medium with 5% human AB serum. Alternatively, LAKs were produced by culturing PBMCs (Panserin, 5% human AB serum) with large amounts of IL-2 (2,000 IU/ml) for up to 25 days. Restoration of patient NK cell exocytic activity was evaluated with LAKs cultured for 15–20 days with IL-2 (2,000 IU/ml) and (co-)transfected with an insert-less pECFP vector with or without WT STXBP2-FLAG–tagged or an empty-FLAG–tagged vector, using the Amaxa nucleofection technique (Amaxa Biosystems) in accordance with manufacturer’s instructions. Electroporated cell populations were incubated in complete medium for 4 hours, and IL-2 (2,000 IU/ml) was then added. For each individual PBMC population electroporated, the “transfected” cell population designated the cell fraction that co-expressed the vectors, whereas the “nontransfected” cell fraction designated cells that did not express the vectors. Efficacy of transfection was around 30%. The degranulation assay was performed 24 hours after transfection. To adjust for the slight variation level of spontaneous CD107 expression among patients or following electroporation, in all experiments gates were individually set up on the basis of PBMCs incubated with P815 alone, a setting resulting in CD107 expression by less than 1% of cells degranulating. The murine mastocytoma cell line P815 (ATCC) was maintained in RPMI plus 10% FCS. The 293T cell line was maintained in DMEM plus 10% FCS and transfected using Lipofectamine 2000 Transfection Reagent (Invitrogen).
Degranulation assay.
To quantify cytotoxic granule exocytosis, 2 × 105 PBMCs were mixed with 2 × 105 P815 cells supplemented with 10 μg/ml mAbs for stimulation and in the presence of anti-CD107a–FITC and anti-CD107b–FITC mAbs, according to a standard technique (18). Cells were incubated for 3 hours at 37°C in 5% CO2. Thereafter, cells were harvested, washed once with PBS-Azide (Azide 0.02%), stained with anti-CD3–APC, anti-CD8–PE, and anti-CD56–PE mAbs (BD Biosciences), and analyzed by flow cytometry. For NK cell analysis, lymphocytes were gated on forward-scatter/side-scatter plots, followed by gating of the CD3–CD56+ cell subset corresponding to the NK population. Cytotoxic granule exocytosis was measured by the induction of CD107 surface expression on NK cells (ΔCD107), calculated as the percentage of CD107+ NK cells stimulated with anti-CD16 and P815 cells subtracted from the percentage of CD107+ NK cells incubated with P815 cells alone. CD16+ cells in the CD3–CD56+ population were similar in all samples (80%–90%). Data were analyzed with FlowJo 8.8.4 software (TreeStar). NK cell cytotoxic activity was assessed using a standard 51Cr-release assay.
Fluorescence microscopy.
P815 target cells were prelabeled with the Cell Tracker Blue CMAC (Invitrogen). LAKs and P815 target cells were mixed in equal quantities in the presence of anti-CD16 (10 μg/ml), incubated for 10 minutes at 37°C, plated on poly-L-lysine–coated glass coverslips (Sigma-Aldrich), and incubated for 20 minutes at 37°C. Coverslips were then placed on ice and conjugates were fixed by incubation in 3.7% paraformaldehyde on ice for 15 minutes. The cells were incubated first with specific antibodies and then with phalloidin (for 45 minutes in each case) in a permeabilizing buffer (PBS, BSA 1 mg/ml, 0.05% [wt/vol] saponin; Sigma-Aldrich) and then washed. Cells were mounted on slides in a medium containing Mowiol anti-fading agent (Calbiochem). Samples were analyzed with an LSM 5 Pascal confocal microscopy (Zeiss). For quantification, conjugates consisting of 1 NK cell and 1 blue CMAC-stained P815 target were identified by differential interference contrast and by increased phalloidin labeling (actin) at the cell-cell interface. Full z stacks of ×30 fields for each sample were acquired. Three-dimensional reconstructions of the image were used with confocal z series to determine whether the perforin-containing granules had polarized toward target cells. Conjugates were scored positive for polarization if the granules (based on perforin staining) were against the cell interface. The percentage of NK cells in conjugates showing perforin polarized to the targets was normalized for each individual to the percentage polarization of NK cells in the absence of target.
Protein blots.
Control, FHL4, and FHL5 LAKs or transiently transfected 293T cells were lysed in RIPA/glycerol buffer (50 mM Hepes, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, 1% sodium deoxycholate) supplemented with protease (Roche Applied Science) and phosphatase (Sigma-Aldrich) inhibitors. At the time of lysis, LAK populations from the various individuals comprised about 20%–25% CD3–CD56+ NK cells and 70%–75% CD3+ T lymphocytes. Cell extracts were separated by SDS-PAGE, blotted, and then stained with the specific antibodies. Polyclonal antibodies against STXBP2 (15) and STX11 were prepared by immunizing rabbits with specific peptides (STXBP2161–229, STX111–15) and then using affinity purification. Antibodies against STX3 and PI3K are described in Supplemental Table 2. An anti-FLAG antibody or a control IgG isotype was used to immunoprecipitate STX11 from transfected 293T cell lysates. After staining with an HPRT-conjugated secondary antibody, the immunoblot was developed with an ECL kit (Amersham). The intensity of the immunoblot bands was quantified using ImageJ 1.4.3.67 Launcher Symmetry Software.
URLs.
PSI-BLAST searches (BLASTP 2.2.18+) were performed in the nonredundant (6,933,853 sequences) database at NCBI (http://www.ncbi.nlm.nih.gov/blast) (23) using the default parameters. Protein sequence alignments were performed using ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html).
Manipulation of 3-dimensional structures was done with Swiss-PdbViewer (24). The STXBP2 model was obtained from the SWISS-MODEL Repository Database (http://swissmodel.expasy.org/repository/) with the search keyword STXB2_HUMAN.
Accession codes.
The GenBank reference sequence for STXBP2 mRNA was NM_006949. The Protein Data bank reference sequence for peptide STXBP2 was NP_008880.
Statistics.
Results are expressed as mean ± SD. Statistical analysis was performed using the 2-tailed unpaired Student’s t test. Data were considered statistically significant at P < 0.05.
Supplementary Material
Acknowledgments
We acknowledge M. Garfa-Traore for help with the imaging study and B.A. Algarbi, M. Kurowska, N. Lambert, M. Salmon, V. Grandin, C. Jacques, C. Harré, and S. Ngaba for technical assistance. We are grateful to S. Rigaud, N. Nehme, J. Pachlopnik, and F. Legrand for discussion and assistance and to all the staff nurses who cared for the patients. We also thank the patients and their families for their participation. We thank I. Callebaut for critical reading of the manuscript. The study was funded by INSERM, the French National Research Agency (ANR), and the Fondation pour la Recherche Médicale (FRM). M. Côte, A. Burgess, and M.M. Ménager were supported by doctoral fellowships from the Ministère de l’Education Nationale de la Recherche et de la Technologie and l’Association de Recherche contre le cancer (ARC). Core facilities were partly financed by the Imagine Foundation.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J. Clin. Invest. 119:3765–3773 (2009). doi:10.1172/JCI40732.
References
- 1.Henter J.I., Elinder G., Ost A. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. The FHL Study Group of the Histiocyte Society. Semin. Oncol. 1991;18:29–33. [PubMed] [Google Scholar]
- 2.Stepp S., et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 3.Feldmann J., et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115:461–473. doi: 10.1016/S0092-8674(03)00855-9. [DOI] [PubMed] [Google Scholar]
- 4.zur Stadt U., et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum. Mol. Genet. 2005;14:827–834. doi: 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]
- 5.Menasche G., Feldmann J., Fischer A., de Saint Basile G. Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol. Rev. 2005;203:165–179. doi: 10.1111/j.0105-2896.2005.00224.x. [DOI] [PubMed] [Google Scholar]
- 6.Voskoboinik I., Smyth M.J., Trapani J.A. Perforin-mediated target-cell death and immune homeostasis. Nat. Rev. Immunol. 2006;6:940–952. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 7.Stinchcombe J.C., Griffiths G.M. Secretory mechanisms in cell-mediated cytotoxicity. Annu. Rev. Cell Dev. Biol. 2007;23:495–517. doi: 10.1146/annurev.cellbio.23.090506.123521. [DOI] [PubMed] [Google Scholar]
- 8.Hata Y., Sudhof T.C. A novel ubiquitous form of Munc-18 interacts with multiple syntaxins. Use of the yeast two-hybrid system to study interactions between proteins involved in membrane traffic. J. Biol. Chem. 1995;270:13022–13028. doi: 10.1074/jbc.270.22.13022. [DOI] [PubMed] [Google Scholar]
- 9.Riento K., Kauppi M., Keranen S., Olkkonen V.M. Munc18-2, a functional partner of syntaxin 3, controls apical membrane trafficking in epithelial cells. J. Biol. Chem. 2000;275:13476–13483. doi: 10.1074/jbc.275.18.13476. [DOI] [PubMed] [Google Scholar]
- 10.Toonen R.F., Verhage M. Vesicle trafficking: pleasure and pain from SM genes. Trends Cell Biol. 2003;13:177–186. doi: 10.1016/S0962-8924(03)00031-X. [DOI] [PubMed] [Google Scholar]
- 11.Misura K.M., Scheller R.H., Weis W.I. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 2000;404:355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- 12.Kauppi M., Wohlfahrt G., Olkkonen V.M. Analysis of the Munc18b-syntaxin binding interface. Use of a mutant Munc18b to dissect the functions of syntaxins 2 and 3. J. Biol. Chem. 2002;277:43973–43979. doi: 10.1074/jbc.M208315200. [DOI] [PubMed] [Google Scholar]
- 13.Verhage M., Sorensen J.B. Vesicle docking in regulated exocytosis. Traffic. 2008;9:1414–1424. doi: 10.1111/j.1600-0854.2008.00759.x. [DOI] [PubMed] [Google Scholar]
- 14.Verhage M., et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Verdeaux S., et al. Evidence of a role for Munc18-2 and microtubules in mast cell granule exocytosis. J. Cell Sci. 2003;116:325–334. doi: 10.1242/jcs.00216. [DOI] [PubMed] [Google Scholar]
- 16.de Saint Basile G., Fischer A. The role of cytotoxicity in lymphocyte homeostasis. Curr. Opin. Immunol. 2001;13:549–554. doi: 10.1016/S0952-7915(00)00257-0. [DOI] [PubMed] [Google Scholar]
- 17.Bryceson Y.T., et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110:1906–1915. doi: 10.1182/blood-2007-02-074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Betts M.R., et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods. 2003;281:65–78. doi: 10.1016/S0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 19.Jordan M.B., Hildeman D., Kappler J., Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 20.Sudhof T.C., Rothman J.E. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toonen R.F., et al. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. 2006;25:3725–3737. doi: 10.1038/sj.emboj.7601256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 23.Altschul S.F., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.