

Abstract
The human herpesvirus 8 (HHV-8) viral G protein-coupled receptor (vGPCR) has been implicated in virus-associated disease pathogenesis due principally to its ability to induce the production of angiogenic cytokines involved in this process. However, the role of the vGPCR in normal virus biology is understudied and remains unknown. Here we provide evidence from vGPCR gene knockout and depletion experiments that vGPCR is a positive regulator of HHV-8 productive replication and, through experimental utilization of Gα-coupling variants of vGPCR, that signaling via Gαq activation and targeted mitogen-activated protein kinase pathways is of particular relevance to this activity.
Several herpesviruses encode one or more viral G protein-coupled receptors (vGPCRs), structurally related to chemokine receptors, some of which are directly orthologous and others of which are evolutionarily distinct (12, 17, 19). Even where orthologous, such as ORF74-type vGPCRs in the HHV-8-related gamma-2-herpesviruses, the vGPCRs are fairly highly diverged structurally and have unique functional properties (19, 21, 22). The role of vGPCRs in herpesvirus replication has been investigated in other viral systems, principally for those viruses that undergo efficient productive replication in culture and for which in vivo studies can be undertaken. For example, vGPCRs specified by murine gammaherpesvirus 68 ORF74 and rat cytomegalovirus (CMV) R78 have identifiable proreplication functions both in vitro and in vivo, as revealed by phenotypic analyses of vGPCR gene-disrupted mutant viruses (2, 11, 15). Other vGPCRs investigated, such as rat CMV R33 and mouse CMV M33, influence virus replication and viral pathogenesis in experimentally infected animals but are not required for optimal virus replication in culture (3, 7). For human herpesvirus 8 (HHV-8) vGPCR, one study using engineered HHV-8 latently infected primary effusion lymphoma (PEL) cells conditionally expressing the chemokine receptor found that induced vGPCR expression led to cell cycle arrest and to reduced viral lytic gene expression in tetradecanoyl phorbol acetate (TPA)/butyrate-reactivated cultures (4). While the former finding is fully compatible with productive replication (8, 25), the latter seems counterintuitive; however, it may have resulted from the particular timing and levels of vGPCR expression prior to chemical induction or other specific features of the system and conditions used.
For our investigations of vGPCR function in lytic replication, two main approaches were taken: vGPCR gene (ORF74) disruption in the context of the viral genome (“knockout”) and short hairpin RNA (shRNA)-mediated vGPCR depletion in the context of HHV-8-infected PEL and endothelial cells (“knockdown”). Gain-of-function (vGPCR overexpression) and complementation of endogenous vGPCR depletion also were employed. In the knockout approach, we first generated an ORF74-deleted HHV-8 genome, utilizing recombinase A-mediated techniques applied to bacmid HHV-8 genome BAC36 (1, 14, 18, 23, 26). The general strategy used is illustrated in Fig. 1A, showing the structure of the pST76KSR-based transfer vector employed for introduction of Flp recombinase-cleavable Frt-flanked Tetr gene in place of ORF74. Following selection of resolved cointegrants on sucrose and isolation of tetracycline-resistant recombinants, the Tetr expression cassette was removed by transfection of a temperature-sensitive Flp recombinase-expressing plasmid, pCP20 (5), which was subsequently selected against by growth at 43°C. Tetracycline-sensitive colonies were then selected and screened for appropriate recombination by restriction digestion. Examples of diagnostic restriction and Southern blot analyses for identification of ORF74-deleted recombinant and revertant (repaired genome, control) are shown in Fig. 1B. A similar strategy was used to generate HHV-8 bacmid genomes in which native ORF74 was replaced with point-mutated reading frames encoding Gα-coupling vGPCR variants vGPCR.8 (R322W) and vGPCR.15 (M325S) (13) or containing a single nucleotide (frameshifting) insertion after the first ATG codon. Here, the mutated ORFs 74 were cointroduced with the Tetr cassette into the ΔORF74 genome and Tetr was subsequently removed with Flp recombinase. Repaired counterparts of these mutated genomes were generated by essentially the same strategy, to replace mutated ORFs with wild-type ORF74 (introduced into genomes first deleted of mutated ORFs 74). General integrity of recombinant genomes (mutated and repaired) was verified by restriction digestion (Fig. 1B). In addition, transcription from the K14/ORF74 region (10, 16) was checked by quantitative reverse transcription-PCR (RT-qPCR) analysis of bacmid-transfected HEK293T cells, mock treated or treated with TPA to induce lytic cycle (Fig. 1C).
FIG. 1.

Shuttle vector and recA approach (14, 18) for generation of mutations in HHV-8 bacmid BAC36 (26). (A) A Tetr cassette was introduced individually or together with engineered vGPCR open reading frame (ORF) sequences into the ORF74 locus and subsequently excised for generation in Flp recombinase-expressing bacteria to generate ORF74-deleted or -mutated HHV-8 genomes. A single nucleotide was inserted after the initiator ATG codon of ORF74 (genome position 129374) (20) to generate frameshifted (vGPCR null) ORF74; ORFs mutated in codons 322 and 325 (genome positions 130337 to 130339 and 130346 to 130348, respectively) and specifying signaling-altered vGPCRs R322W (m8) and M325S (m15) were made previously (13). Generation of genomes containing these point-mutated ORFs was achieved by cointroduction of each engineered ORF74 together with Tetr into ORF74-deleted bacmid (HHV-8.ΔORF74) and subsequent Flp-mediated deletion of the Tetr cassette to arrive at the final recombinant genomes. Each was repaired by substitution with wild-type ORF74 using analogous procedures. The various steps used to generate and select for intermediate and final bacmid recombinants are illustrated; the reagents, strategies and experimental approaches are explained fully in published papers (1, 14, 18, 23). (B) Examples of restriction (KpnI) and Southern analyses of bacmid recombinants, outlining the diagnostic principles used for the identification of ORF74 deletion (HHV-8.ΔORF74), introduction of point-mutated ORFs 74, and reintroduced wild-type (WT) (repaired [rep]) ORF74. Panels i and ii show agarose gel and Southern blot analyses of the KpnI-restricted HHV-8.ΔORF74 bacmid recombinant and its repaired counterpart, and panel iii provides a comparison of the KpnI restriction patterns of the point-mutated genomes (and repaired versions) with wild-type, ΔORF74, and ΔORF74rep bacmids. (C) Verification of appropriate mRNA production from the K14/ORF74 transcription unit of recombinant genomes was checked by RT-PCR detection of K14- and ORF74-encoding transcripts. K14 transcripts from all recombinant genomes were detected in the presence of TPA, and all bacmids, except HHV-8.ΔORF74, produced vGPCR transcripts, induced by TPA. The primer pairs used for PCR were 5′-CAAGCAGGCCATGTGTTATG-3′ and 5′-AGCACCACAGCAACAATCAC-3′ for ORF74 and 5′-GATGGAGGGGCTGTTACCTGTGT-3′ and 5′-CGTCTTCTGTGCTATTTACCT-3′ for K14. RT, reverse transcriptase.
The various mutated bacmid genomes and their repaired counterparts were transfected into HEK293T cells, and their abilities to replicate were compared following lytic cycle induction with TPA treatment or replication and transcription activator (RTA) expression vector transfection. Replication competence was assessed by qPCR determination of viral genome copy numbers in cultures 3 days postinduction. Initial experiments compared the replication competence of the ΔORF74 recombinant with its repaired/wild-type counterparts; the data showed significant decreases in both TPA- and RTA-induced genome copy numbers of the ORF74-null genomes relative to native and repaired (R) genomes, indicating a positive role of vGPCR in HHV-8 productive replication (Fig. 2A). The reversion to the wild-type phenotype for the repaired genome confirmed lack of functionally significant coincident alteration in the ORF74-deleted genome during its generation. Further experiments were undertaken utilizing the point-mutated HHV-8 genomes in analogous experiments; ORF74X is vGPCR null due to a single-nucleotide frameshift insertion, whereas the other ORFs 74 specified vGPCR coupling specifically to Gαi (R322W, vGPCR.8) or predominantly to Gαq (M325S, vGPCR.15). HHV-8.ORF74X, like HHV-8.ΔORF74, had reduced replication capacity (shown for RTA induction), demonstrating that vGPCR deficiency, rather than ORF74 locus disruption, leads to this phenotype (Fig. 2B, bottom panel). The HHV-8 genome encoding vGPCR.8 (Gαi specific) was impaired in virus production, whereas an HHV-8 genome specifying vGPCR.15 (coupling preferentially with Gαq) was essentially unaffected (Fig. 2B). As vGPCR.8 and vGPCR.15 signal predominantly via phosphatidylinositol 3-kinase/NF-κB and protein kinase C/mitogen-activated protein kinase (MAPK) pathways, respectively, in HEK293T cells (13), our data indicate that MAPK signaling may be of particular relevance to proreplication activities of vGPCR and that NF-κB signaling is not of primary importance.
FIG. 2.
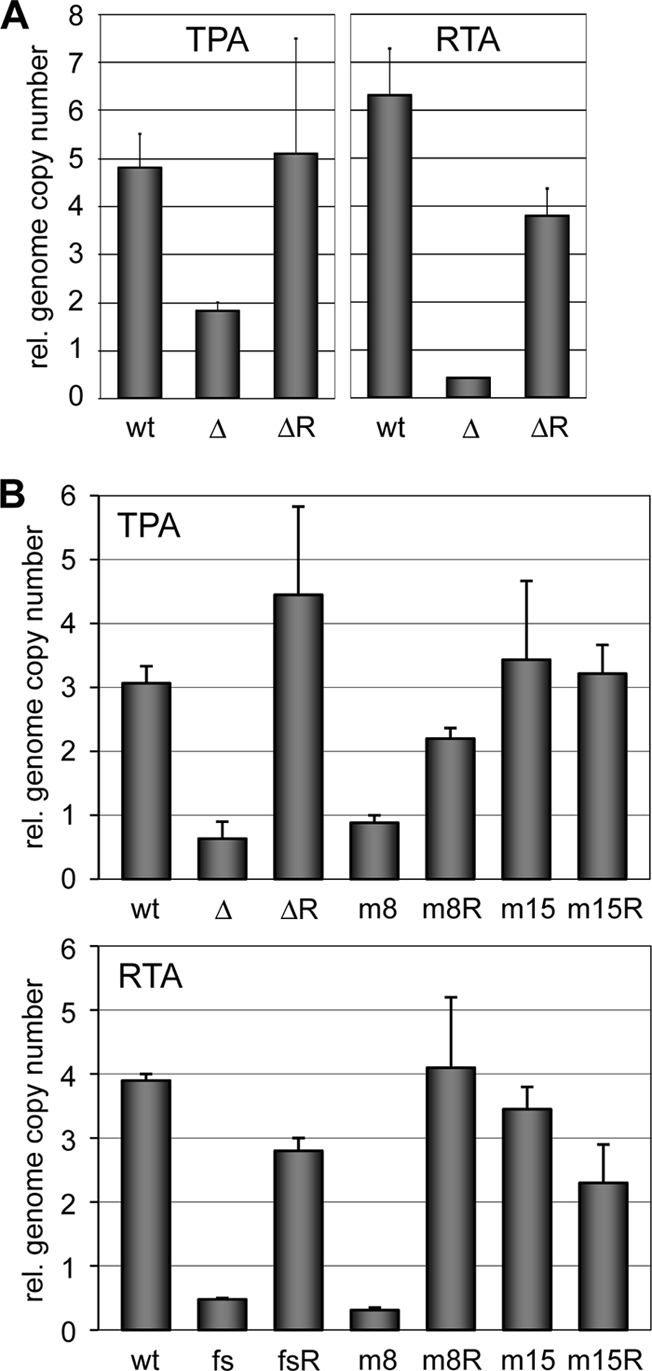
Effects of ORF74 knockout and substitution in reactivated lytic replication of bacmid genomes. (A) Initial analysis of ORF74-deleted (Δ) and -repaired (ΔR; reverted to wild type [wt]) genomes for replication competence relative (rel.) to wild-type BAC36. HEK293T cells were transfected with the bacmids, transformants were selected in hygromycin, and virus lytic replication was induced either by TPA treatment or RTA expression vector transfection. Cells were harvested 3 days postinduction, DNA was extracted, and virus DNA was quantified using a primer pair directed to ORF73. (B) Analogous experiments were carried out using ΔORF74, point-mutated genomes containing a frameshift (fs) insertion into ORF74, or altered codons R322W (m8) and M325S (m15) and repaired (fsR, m8R, and m15R) counterparts. Representative results from multiple experiments are shown. Values were normalized to uninduced controls; error bars represent divergence from the mean of duplicate values.
We next investigated the effects of vGPCR depletion on virus replication in PEL cells, naturally infected (latently) with HHV-8. First, two vGPCR mRNA-specific shRNAs were generated, cloned into a green fluorescent protein-positive (GFP+) lentiviral vector, and tested for their abilities to deplete vGPCR protein levels in appropriately transfected HEK293T cells (Fig. 3A). Both shRNAs were effective in vGPCR-red fluorescent protein (RFP) depletion (as identified by decreased RFP fluorescence in the transfected [GFP+] cells) and were used in functional assays. Parallel cultures of BCBL-1 cells were transduced with lentiviral vectors expressing each of the vGPCR-directed shRNAs or a nonsilencing (NS) shRNA (control), and these cultures were then treated with TPA to induce lytic reactivation. Quantitation of encapsidated viral genomes released into culture media during a 3-day period (before excessive PEL cell death) was done by application of ORF73-directed qPCR to ultracentrifuge-pelleted and DNase I-resistant viral DNA. Significant reductions in virus titers were detected in PEL cultures expressing each of the vGPCR-targeted shRNAs relative to virus production from NS shRNA-transduced cultures (Fig. 3B). Analogous testing of the effects of vGPCR depletion on virus replication was undertaken in NS or vGPCR shRNA+ endothelial (TIME) cells infected with BCBL-1-derived virus. Infection and establishment of latency were confirmed by immunofluorescence staining for latency-associated nuclear antigen (LANA) in parallel cultures (data not shown). The results of these experiments demonstrated depletion of cumulative HHV-8 titers derived over a 6-day period (shown previously to allow for maximum virus production [6]) following TPA treatment in vGPCR sh1-transduced relative to NS-shRNA-transduced cultures (Fig. 3C). Thus, consistent with our bacmid-based experiments, the data from vGPCR depletion in both PEL and TIME cells indicated a positive role of vGPCR in virus productive replication.
FIG. 3.

Depletion of vGPCR expression in PEL and HHV-8 infected TIME cells. (A) Two shRNAs directed to vGPCR mRNA sequences (5′-AGCGGATATGACTACTCTGGA-3′, sh1; 5′-AACGTTGGAATACTCTCTCTGAT-3′, sh2) were tested for their efficacies in HEK293T cells transfected with lentiviral-based expression vectors (GFP+) encoding each shRNA together with vGPCR-RFP expression plasmid. Fluorescence micrography revealed substantially decreased vGPCR-RFP expression in vGPCR shRNA-transduced cells relative to levels in NS shRNA-transfected cells. The panels containing sh1/2 plus vGPCR have been deliberately overexposed to demonstrate shRNA efficacy. (B) Both shRNAs, cloned into and expressed from puromycin-selectable lentiviral vectors, were utilized to investigate the functional significance of vGPCR for HHV-8 productive replication in TPA-treated PEL (BCBL-1) cultures. Lentivirus-infected cultures were grown in the presence of puromycin to ensure 100% shRNA transduction prior to TPA treatment. Media were harvested 3 days postinduction for qPCR analysis of ultracentrifuge-pelleted and DNase I-resistant encapsidated viral DNA. Virus replication was diminished in each case, relative to NS shRNA-transduced control cultures. Data were obtained from triplicate qPCR reactions; error bars show standard deviations from mean values. (C) Analogous experiments were undertaken in latently infected endothelial (TIME) cells, using one of the vGPCR-directed shRNAs (sh1). Similar results were obtained, demonstrating vGPCR shRNA-specific negative effects on virus replication. The data presented are from duplicate samples and qPCRs. (D) Testing of resistance of shRNA target site-altered vGPCR coding sequences to effects of shRNA-mediated depletion in HEK293T-based cotransfection experiments using wild-type (wt) and engineered ORFs 74 fused to RFP coding sequences. The ORF74 sequence (AGCGGATATGACTACTCTGGA) targeted by shRNA 1 was changed to AGT GGT TAC GAT TAT TCC GGA (changes in codons [triplets] are underlined). (E) Rescue of the vGPCR mRNA depletion phenotype in TIME cells by (shRNA resistant) vGPCR complementation but not by vOX2/CD200, encoded in HHV-8 by a bicistronic message also containing ORF74 (10, 16). Data were derived from duplicate samples and qPCRs; error bars represent deviations from mean values.
As vGPCR is produced from a bicistronic transcript, additionally encoding ORF K14 (vOX2/vCD200), we wanted to confirm that observed phenotypic effects were due to vGPCR specifically, although expression of the vOX2 receptor, CD200R, through which vOX2 functions, is known to be restricted mainly to myeloid and T-cell subpopulations and to be absent from endothelial cells (9, 10, 16, 24). Lentiviral-based expression vectors for vGPCR and vOX2 were used in complementation assays. Coding sequences for vGPCR were engineered to be resistant to the shRNA (vGPCR sh1) by introduction of codon-synonymous mutations in the shRNA target sequence; the resulting mRNA was indeed resistant to depletion by the shRNA (Fig. 3D). Empty lentivirus vector or lentiviruses encoding vGPCR or vOX2 were used to infect vGPCR-shRNA-transduced HHV-8+ TIME cultures. Virus titers following TPA induction of lytic replication were undertaken by qPCR analysis of medium-derived encapsidated genomes, as before. The vGPCR depletion phenotype was rescued by vGPCR transduction, specifically, confirming the functional relevance of vGPCR signaling in HHV-8 productive replication (Fig. 3E).
Finally, we investigated the significance of vGPCR and of particular vGPCR-activated signaling pathways to HHV-8 productive replication through a “gain-of-function” experimental approach. Thus, we tested whether vGPCR overexpression could boost normal levels of virus production in TPA-treated HHV-8 (latently)-infected TIME cultures. Consistent with the results obtained in the depletion and rescue experiment (Fig. 3E), overexpression of vGPCR, but not vOX2, was able to increase the efficiency of virus production, confirming the functional significance of vGPCR and inactivity of vOX2 in our assay (Fig. 4A). Experiments utilizing vGPCR.8 (R322W, Gαi specific) and vGPCR.15 (M325S, predominantly Gαq coupled) revealed that Gαi coupling was insufficient to support vGPCR-promoted HHV-8 replication in TIME cells, whereas Gαq-coupled vGPCR.15 was able to enhance TPA-induced virus replication equivalently to wild-type vGPCR (Fig. 4B). As Gαq-mediated signal transduction is known to activate MAPK signaling strongly via phospholipase C activation, our data indicate that this pathway is of primary importance in promotion of HHV-8 replication by vGPCR.
FIG. 4.
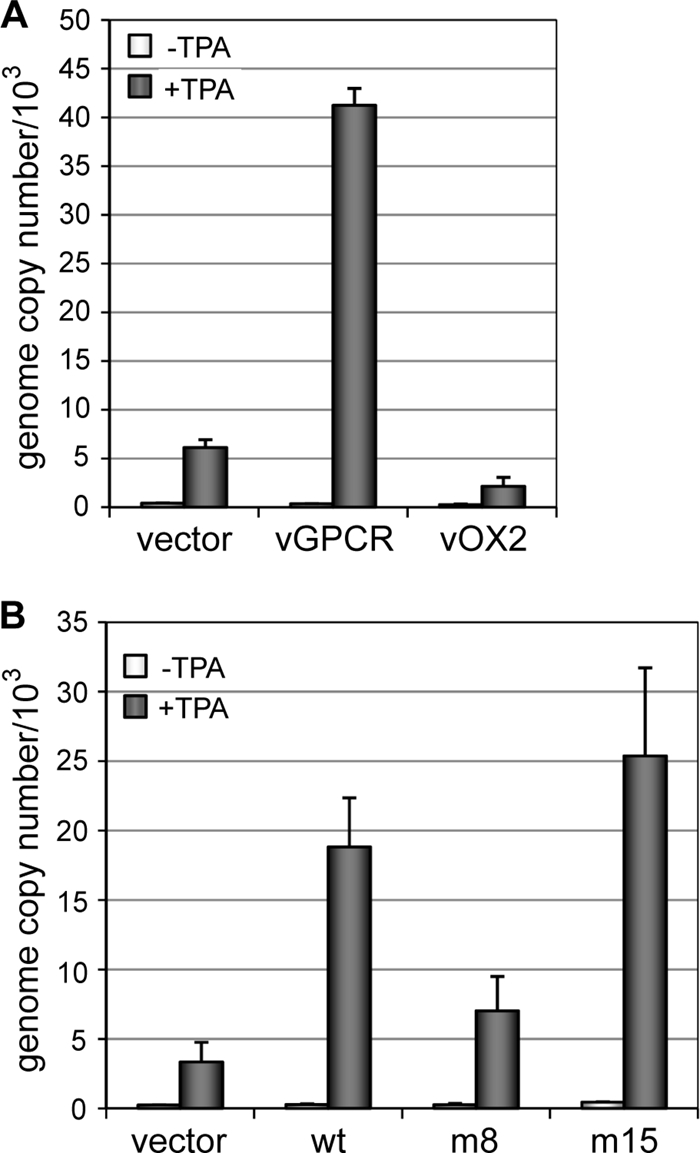
Gain-of-function analysis of wild-type and Gα coupling-altered vGPCRs. (A) Effects of lentiviral vector-mediated vGPCR and vOX2 transduction on TPA-induced virus reactivation in HHV-8-infected TIME cells. Cumulative titers of encapsidated viral genomes present in culture media were determined by qPCR applied to ultracentrifuge sedimentation-derived and DNase I-treated virion preparations from cultures transduced with empty lentiviral expression vector, lenti-vGPCR, and lenti-vOX2 and then mock or TPA treated for 6 days. Average values from triplicate qPCR reactions from duplicate cultures are expressed relative to encapsidated viral genome titers in parallel uninduced cultures. Overexpression of vGPCR, but not vOX2, was able to enhance virus production from TPA-treated TIME cells. (B) An analogous experiment was performed to investigate the abilities of vGPCR.8 (m8, R322W, exclusively Gαi coupled) and vGPCR.15 (m15, M325S, predominantly Gαq coupled), relative to wild-type vGPCR (wt), to support enhanced virus replication following TPA-induced lytic reactivation.
The data presented here identify proreplication activity of HHV-8 vGPCR. As vGPCR is expressed during lytic replication, a positive role of the chemokine receptor in virus production would have been expected, perhaps, at least in the context of natural infection in vivo. The culture-based proreplication activity we have observed demonstrates direct autocrine effects of vGPCR signaling in the support of lytic replication, and incorporation of Gα-coupling variants of vGPCR into our experiments has implicated MAPK pathway activation, mediated predominantly by Gαq coupling, as of particular significance in respect of this function. Interestingly, MAPK/extracellular signal-regulated kinase signaling was identified as important for MHV-68 vGPCR-mediated proreplication activity also (11), suggesting that this may be a general means by which viral chemokine receptors can promote replication. While further studies are necessary to elucidate the precise mechanisms of HHV-8 vGPCR proreplication activity via MAPK signaling, the data presented here provide the foundation for these more detailed investigations. Of particular importance, perhaps, is to understand whether the viral chemokine receptor activates HHV-8 lytic gene expression, whether it contributes to cell cycle state and/or prosurvival signaling via direct effects on cellular genes and proteins, and whether vGPCR may contribute via indirect mechanisms, perhaps involving the induction of other signal transducers, such as cytokines. Clearly, precise mechanisms of action may, to some extent at least, be dependent on cell type; thus, future studies with cell types relevant to HHV-8 replication, such as endothelial and B cells, are warranted. These should provide useful information about the processes required for virus production and therefore indicate potential therapeutic avenues directed at inhibiting these mechanisms. Our data provide a first-step contribution to this goal, demonstrating that HHV-8 vGPCR, like some other herpesvirus-encoded chemokine receptors (2, 11, 15), plays an important, autocrine role in support of productive replication.
Acknowledgments
The work presented in this article was supported by grant R21-CA119887 from the National Cancer Institute.
Footnotes
Published ahead of print on 30 September 2009.
REFERENCES
- 1.Adler, H., M. Messerle, M. Wagner, and U. H. Koszinowski. 2000. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J. Virol. 74:6964-6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beisser, P. S., G. Grauls, C. A. Bruggeman, and C. Vink. 1999. Deletion of the R78 G protein-coupled receptor gene from rat cytomegalovirus results in an attenuated, syncytium-inducing mutant strain. J. Virol. 73:7218-7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beisser, P. S., C. Vink, J. G. Van Dam, G. Grauls, S. J. V. Vanherle, and C. A. Bruggeman. 1998. The R33 G protein-coupled receptor gene of rat cytomegalovirus plays an essential role in the pathogenesis of viral infection. J. Virol. 72:2352-2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cannon, M., E. Cesarman, and C. Boshoff. 2006. KSHV G protein-coupled receptor inhibits lytic gene transcription in primary-effusion lymphoma cells via p21-mediated inhibition of Cdk2. Blood 107:277-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherepanov, P. P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9-14. [DOI] [PubMed] [Google Scholar]
- 6.Choi, Y. B., and J. Nicholas. 2008. Autocrine and paracrine promotion of cell survival and virus replication by human herpesvirus 8 chemokines. J. Virol. 82:6501-6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis-Poynter, N. J., D. M. Lynch, H. Vally, G. R. Shellam, W. D. Rawlinson, B. G. Barrell, and H. E. Farrell. 1997. Identification and characterization of a G protein-coupled receptor homolog encoded by murine cytomegalovirus. J. Virol. 71:1521-1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flemington, E. K. 2001. Herpesvirus lytic replication and the cell cycle: arresting new developments. J. Virol. 75:4475-4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foster-Cuevas, M., G. J. Wright, M. J. Puklavec, M. H. Brown, and A. N. Barclay. 2004. Human herpesvirus 8 K14 protein mimics CD200 in down-regulating macrophage activation through CD200 receptor. J. Virol. 78:7667-7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirshner, J. R., K. Staskus, A. Haase, M. Lagunoff, and D. Ganem. 1999. Expression of the open reading frame 74 (G-protein-coupled receptor) gene of Kaposi's sarcoma (KS)-associated herpesvirus: implications for KS pathogenesis. J. Virol. 73:6006-6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee, B. J., U. H. Koszinowski, S. R. Sarawar, and H. Adler. 2003. A gammaherpesvirus G protein-coupled receptor homologue is required for increased viral replication in response to chemokines and efficient reactivation from latency. J. Immunol. 170:243-251. [DOI] [PubMed] [Google Scholar]
- 12.Lesniewski, M., S. Das, Y. Skomorovska-Prokvolit, F. Z. Wang, and P. E. Pellett. 2006. Primate cytomegalovirus US12 gene family: a distinct and diverse clade of seven-transmembrane proteins. Virology 354:286-298. [DOI] [PubMed] [Google Scholar]
- 13.Liu, C., G. Sandford, G. Fei, and J. Nicholas. 2004. Gα protein selectivity determinant specified by a viral chemokine receptor-conserved region in the C tail of the human herpesvirus 8 G protein-coupled receptor. J. Virol. 78:2460-2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Messerle, M., I. Crnkovic, W. Hammerschmidt, H. Ziegler, and U. H. Koszinowski. 1997. Cloning and mutagenesis of a herpesvirus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 94:14759-14763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moorman, N. J., H. W. Virgin IV, and S. H. Speck. 2003. Disruption of the gene encoding the γHV68 v-GPCR leads to decreased efficiency of reactivation from latency. Virology 307:179-190. [DOI] [PubMed] [Google Scholar]
- 16.Nador, R. G., L. L. Milligan, O. Flore, X. Wang, L. Arvanitakis, D. M. Knowles, and E. Cesarman. 2001. Expression of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor monocistronic and bicistronic transcripts in primary effusion lymphomas. Virology 287:62-70. [DOI] [PubMed] [Google Scholar]
- 17.Nicholas, J. 2005. Human gammaherpesvirus cytokines and chemokine receptors. J. Interferon Cytokine Res. 25:373-383. [DOI] [PubMed] [Google Scholar]
- 18.Pósfai, G., M. D. Koob, H. A. Kirkpatrick, and F. R. Blattner. 1997. Versatile insertion plasmids for targeted genome manipulations in bacteria: isolation, deletion, and rescue of the pathogenicity island LEE of the Escherichia coli O157:H7 genome. J. Bacteriol. 179:4426-4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenkilde, M. M., M. Waldhoer, H. R. Luttichau, and T. W. Schwartz. 2001. Virally encoded 7TM receptors. Oncogene 20:1582-1593. [DOI] [PubMed] [Google Scholar]
- 20.Russo, J. J., R. A. Bohenzky, M. C. Chien, J. Chen, M. Yan, D. Maddalena, J. P. Parry, D. Peruzzi, I. S. Edelman, Y. Chang, and P. S. Moore. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 93:14862-14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verzijl, D., C. P. Fitzsimons, M. van Dijk, J. P. Stewart, H. Timmerman, M. J. Smit, and R. Leurs. 2004. Differential activation of murine herpesvirus 68- and Kaposi's sarcoma-associated herpesvirus-encoded ORF74 G protein-coupled receptors by human and murine chemokines. J. Virol. 78:3343-3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vischer, H. F., C. Vink, and M. J. Smit. 2006. A viral conspiracy: hijacking the chemokine system through virally encoded pirated chemokine receptors. Curr. Top. Microbiol. Immunol. 303:121-154. [DOI] [PubMed] [Google Scholar]
- 23.Wagner, M., Z. Ruzsics, and U. H. Koszinowski. 2002. Herpesvirus genetics has come of age. Trends Microbiol. 10:318-324. [DOI] [PubMed] [Google Scholar]
- 24.Wright, G. J., H. Cherwinski, M. Foster-Cuevas, G. Brooke, M. J. Puklavec, M. Bigler, Y. Song, M. Jenmalm, D. Gorman, T. McClanahan, M. R. Liu, M. H. Brown, J. D. Sedgwick, J. H. Phillips, and A. N. Barclay. 2003. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J. Immunol. 171:3034-3046. [DOI] [PubMed] [Google Scholar]
- 25.Wu, F. Y., S. E. Wang, Q.-Q. Tang, M. Fujimuro, C.-J. Chiou, Q. Zheng, H. Chen, S. D. Hayward, M. D. Lane, and G. S. Hayward. 2003. Cell cycle arrest by Kaposi's sarcoma-associated herpesvirus replication-associated protein is mediated at both the transcriptional and posttranslational levels by binding to CCAAT/enhancer-binding protein α and p21CIP-1. J. Virol. 77:8893-8914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou, F.-C., Y.-J. Zhang, J.-H. Deng, X.-P. Wang, H.-Y. Pan, E. Hettler, and S.-J. Gao. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]