

Abstract
Regulatory T cells (Treg) are a subpopulation of CD4+ T cells characterized by the suppressive activity they exert on effector immune responses, including human immunodeficiency virus (HIV)-specific immune responses. Because Treg express CXCR4 and CCR5, they represent potential targets for HIV; however, Treg susceptibility to HIV infection is still unclear. We therefore performed an extensive study of Treg susceptibility to HIV, using lab strains and primary isolates with either CCR5 or CXCR4 tropism. Furthermore, we quantified HIV infection at early and late time points of the virus life cycle. We found that Treg were clearly susceptible to HIV infection. Circulating Treg were not preferentially infected with HIV compared to effector T cells (Teff) in vivo. Conversely, in vitro infection with either CCR5-using (R5) or CXCR4-using (X4) viruses occurred with different dynamics. For instance, HIV infection by R5 viruses (lab strains and primary isolates) resulted in lower levels of infection in Treg compared with Teff at both early and late time points. In contrast, X4 viruses induced higher levels of infection in Treg compared to Teff at early time points, but this difference disappeared at the late time points of the virus life cycle. Our results suggest that the relative susceptibility of Treg to HIV infection compared to Teff varies, depending on both viral and host factors. These variations may play an important role in HIV pathogenesis.
Regulatory T cells (Treg) constitute a subset of CD4+ T cells that play a major role in the homeostasis of the immune system (11, 25, 29, 31, 32). The frequency of natural Treg is low in the circulating blood of healthy adults, constituting ∼1 to 2% of CD4+ T cells (4). Expression of the forkhead family transcription factor FOXP3 is the most definitive marker of Treg activity described so far (11). However, because of its intracellular localization, functional Treg cannot be purified based on FOXP3 expression. Recently, it has been described that Treg express high levels of the interleukin-2 (IL-2) receptor α chain (CD25) and low levels of the IL-7 receptor (CD127), thus allowing for isolation and functional characterization of highly enriched Treg (13).
Treg exert suppressive activity on effector responses to pathogens, particularly in the setting of chronic infections (17, 19, 20, 24). Importantly, Treg suppress cell-mediated immunity against human immunodeficiency virus (HIV) (1, 7, 9, 17-19, 36). We have shown that Treg accumulate in the lymphoid tissues of HIV-infected individuals with ongoing viral replication (2, 22). Other reports showed a higher Treg frequency in mucosal tissues than in the peripheral blood of untreated HIV-infected individuals, suggesting that Treg could attenuate HIV-induced immune hyperactivation and reduce the availability of target cells for HIV replication (9, 18).
Treg susceptibility to HIV infection remains unclear. Data on Treg infection by HIV in vivo are scarce. During acute simian immunodeficiency virus (SIV) infection in rhesus macaques, productive infection was detected in some FOXP3+ cells, present in the gut-associated lymphoid organs (10). A recent study performed in HIV-infected humanized mice documented that FOXP3+ Treg supported high levels of HIV-1 infection and were preferentially depleted by HIV-1 isolates (15). In chronically HIV-infected patients, two recent studies reported that Treg were not preferentially infected compared to non-Treg (6, 8), although a third study reported discrepant data (33). Similarly, few studies have evaluated the susceptibility of natural Treg to infection with full-length HIV, and reported results are discrepant (3, 23).
We therefore performed an extensive study of Treg susceptibility to HIV, using lab strains with either CCR5 or CXCR4 tropism. Furthermore, we analyzed HIV infection at early and late time points of the virus life cycle. We showed that human Treg were clearly susceptible to HIV infection. Importantly, Treg were not preferentially infected with HIV compared to effector T cells (Teff) in vivo. Conversely, in vitro infection with either CCR5-tropic (R5) or CXCR4-tropic (X4) virus occurred with different dynamics. These results suggest that the level of Treg infection and their relative susceptibility to HIV infection compared to non-Treg vary depending on both viral and host factors. These variations may play an important role in HIV pathogenesis.
MATERIALS AND METHODS
Human subjects.
Peripheral blood was obtained from seven treatment-naïve patients with documented HIV infection from the Infectious Diseases Center at the University of Cincinnati (Cincinnati, OH). Patients had CD4 counts of < 758 cells/mm3 with viral loads of >1,148 copies/ml (Ultrasensitive RT-PCR 1.0, detection limit of 50 copies/ml; Roche Diagnostic Systems, Indianapolis, IN). The main clinical features of the enrolled patients are shown in Table 1. In addition, peripheral blood from nine healthy HIV-negative subjects was obtained from the Hoxworth Blood Center (Cincinnati, OH). All subjects provided written informed consent to protocols approved by the corresponding Institutional Review Boards.
TABLE 1.
Clinical data of the HIV-infected individuals included in the study
| Donor | Age (yr) | Racea | Gender | No. of CD4+ cells/mm3 | Viral load (copies/ml) |
|---|---|---|---|---|---|
| 1 | 36 | AA | Female | 546 | 7,558 |
| 2 | 27 | AA | Male | 331 | 9,380 |
| 3 | 46 | AA | Male | 354 | 86,391 |
| 4 | 35 | Caucasian | Male | 720 | 1,148 |
| 5 | 30 | AA | Male | 758 | 4,193 |
| 6 | 40 | AA | Male | 413 | 25,200 |
| 7 | 62 | AA | Male | 488 | 14,097 |
AA, African-American.
Cell isolation and culture.
Peripheral blood mononuclear cells from healthy HIV-seronegative subjects were separated from blood by centrifugation through Ficoll-Hypaque (GE, Fairfield, CT). Resting CD4+ T cells were purified by negative selection using the Miltenyi CD4 separation kit (Auburn, CA), according to the manufacturer's instructions. To isolate Treg, purified CD4+ T cells were stained with anti CD8-fluorescein isothiocyanate (FITC), anti-CD25-allophycocyanin (APC) (BD Pharmingen, San Diego, CA), and anti-CD127-phycoerythrin (PE) (Beckman Coulter Fullerton, CA) and sorted using a FACS Vantage fluorescence-activated cell sorter (FACS) (BD). The purity of Treg (CD8neg CD25hi CD127lo cells) and Teff (CD8neg CD25lo CD127hi cells) was characterized by intracellular detection of FOXP3, using the anti-FOXP3-FITC antibody clone PCH101 (e-Bioscience, San Diego, CA) and analyzed on a FACS LSRII (BD). Purity of the sorted populations was 90%, as shown in Fig. 2. Purified Treg and Teff were activated for 3 days using anti-CD3/CD28 beads (1 cell per 3 beads) (Invitrogen; Carlsbad, CA) in the presence of 100 IU/ml IL-2 (Hoffmann La Roche, Inc., NIH AIDS Research and Reference Reagent Program) in RPMI supplemented with 100 U/ml penicillin, 10 μg/ml streptomycin, 2 mM glutamine, 10 mM HEPES, and 10% fetal calf serum (Life Technologies, Carlsbad, CA). In some experiments, cells were stimulated with 10 μg/ml phytohemagglutinin (PHA) (Sigma, St. Louis, MO) in the presence of 100 IU/ml IL-2.
FIG. 2.

Phenotypic characterization of isolated Teff and Treg. Purified CD4+ T cells were stained with anti-CD127-PE and anti-CD25-APC antibodies. The CD25hi CD127lo Treg and CD25lo CD127hi Teff subsets were identified, as shown in the left panel. Freshly isolated Treg and Teff were stained with anti-FOXP3-FITC to confirm their phenotype. Data from a representative experiment of nine independent experiments are shown. Percentages indicate the levels of expression of each marker defined in relation to the unstained cells.
Virus production and infection using lab strains and primary isolates.
HIV viruses were prepared from 293T cells transfected with plasmids encoding R5-tropic (YK-JRCSF and YU2) or X4-tropic (NL4-3) HIV lab strains, using FuGENE transfection technology (Roche) according to the manufacturer's protocol. After 2 days, supernatants were harvested, and the viruses were precipitated using polyethylene glycol. HIV primary isolates (clade A, KNH1088, 92USSN20, and 92UG029; clade B, 92HT599; clade BF, 93BR019; and clade C, HIV301965) were obtained from the NIH AIDS Research and Reference Reagent Program and expanded in PHA blasts. Virus titers were determined using TZM-bl indicator cells (16). Treg and Teff were infected at a multiplicity of infection (MOI) of 1. Briefly, activated Treg and Teff were incubated with HIV for 2 h at 37°C, washed twice with complete RPMI, and cultured for 24 h to measure HIV DNA integration or for 3 and 7 days for p24Gag quantification. As a negative control, cells were treated with zidovudine (AZT; 1 μM) at the time of HIV infection. For the HIV DNA integration experiments, the infection was carried out with viruses pretreated with DNase I at 20 U/ml in 10 mM MgCl2 for 1 h at room temperature to eliminate any cellular DNA carryover from virus production.
Determination of p24Gag levels by ELISA and flow cytometry.
Supernatants were collected from infected cultures at 3 and 7 days postinfection, and p24Gag levels were quantified by enzyme-linked immunosorbent assay (ELISA) (sensitivity limit of 150 pg/ml; SAIC Frederick, Inc., Frederick, MD). To evaluate intracellular HIV p24Gag, HIV receptors, and cell proliferation, Treg and Teff were treated with 20 μg/ml of human immunoglobulin G to block Fc receptors and stained with anti-CD3-peridinin chlorophyll protein-Cy5.5, anti-CD4-PB, anti-CCR5-APC-Cy7 (BD), and anti-CXCR4-APC (R&D Systems, Minneapolis, MN) for 30 min at 4°C in phosphate-buffered saline containing 2% fetal calf serum and 0.1% sodium azide. Cells were washed twice, fixed with 2% formaldehyde for 30 min at 4°C, and stained using anti-p24Gag HIV-PE (KC-57; Beckman Coulter) in 0.3% saponin buffer for 30 min at 4°C. To determine cell proliferation, cells were labeled before culture with 0.625 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) (Molecular Probes, Eugene, OR). Proliferation was measured 3 and 5 days postactivation. Cells were analyzed on FACS LSRII using FACS DIVA software. At least 10,000 events were recorded for each sample. Live cells were gated based on forward- and side-scatter properties.
Quantification of HIV DNA by real-time PCR.
Treg and Teff from 7 healthy donors were suspended in lysis buffer containing 10 mM Tris HCl (pH 9), 0.1% Tween 20-NP-40, and 400 μg/ml proteinase K (Invitrogen) after 24 h of infection. Cellular lysates were subsequently used to quantify cell-associated HIV-1 DNA by nested real-time PCR (5). Briefly, the first round of amplification used Alu sequence-specific primers and the HIV-1 long terminal repeat (LTR). In the same reaction, the CD3 gene was quantified to precisely determine the number of input cells. This reaction was followed by a second round of amplification with specific primers and a specific labeled probe against the LTR performed in a Light Cycler (Roche). In parallel, CD3 was reamplified and detected using SYBR green. The ACH-2 cell line, a line of human T-lymphocytic leukemia that contains a single copy of HIV-1 proviral DNA (LAV strain) was used to determine the efficiency of the primers (NIH AIDS Research and Reference Reagent Program) (detection limit of 3 copies of HIV DNA).
Chemokine quantification in culture supernatants.
Supernatants were collected after 3 days of stimulation, and chemokine levels were quantified using a commercially available Luminex assay for CCL3 (MIP1-α), CCL4 (MIP1-β), and CCL5 (RANTES) (sensitivity limit of 3.2 pg/ml; Millipore, Billerica, MA). CXCL12 (SDF-1) levels were measured by ELISA (sensitivity limit of 156 pg/ml; R&D Systems, Minneapolis, MN).
Suppression assay.
Purified Treg and Teff from two healthy donors were activated for 3 days using anti-CD3/CD28 beads (1 cell per 3 beads) (Invitrogen) in the presence of 100 IU/ml IL-2 in complete RPMI. After 3 days, Treg were infected or not with YK-JRCSF-JRCSF virus at an MOI of 1 and incubated for 2 h at 37°C. Teff were labeled with CFSE. After 2 h of incubation, Treg were washed twice and cocultured with Teff at a 1:1 ratio. After 3 days, the suppressive capacity of Treg was determined by comparing the CFSE mean fluorescent intensity (MFI) in Teff cultured alone to that of the Teff cultured with Treg.
Statistical analysis.
Statistical analysis was performed using Prism (GraphPad Software 5). HIV p24Gag levels in supernatants and HIV DNA levels were compared using the paired t tests, after log10 transformation. Levels of CD4, CCR5, and CXCR4 expression, as well as chemokine levels in the culture supernatants, were compared using the paired t test. P values of <0.05 were considered to be significant.
RESULTS
Percentages of infected Treg and Teff are similar in vivo.
Few studies have evaluated the level of HIV infection in Treg in vivo. We therefore compared the percentages of cells expressing HIV-p24Gag in circulating CD4+ CD25+ CD127− and CD4+ CD25− CD127+ T cells from seven HIV-infected individuals. All subjects were chronically infected and had detectable viral loads at the time of analysis. None were treated with highly active antiretroviral therapy (Table 1). Analysis of FOXP3 expression in purified cells confirmed that >80% of the CD4+ CD25+ CD127− cells were FOXP3+, whereas only ∼30% of the CD4+ CD25− CD127+ cells were FOXP3+. Within these populations, we found an equivalent proportion of cells that expressed p24Gag (Fig. 1), in agreement with a recent study (8). Moreover, Siliciano et al. reported similar results when sorted FOXP3+ and FOXP3− cells were evaluated (6). Because two of the donors had well-controlled HIV infection—both exhibited viremia of <5,000 copies/ml and CD4 T-cell counts of >700 cells/mm3—we repeated the analysis after excluding these two donors. Similar results were found (i.e., no differences in the proportion of p24Gag+ cells in Treg and Teff; P = 0.08).
FIG. 1.
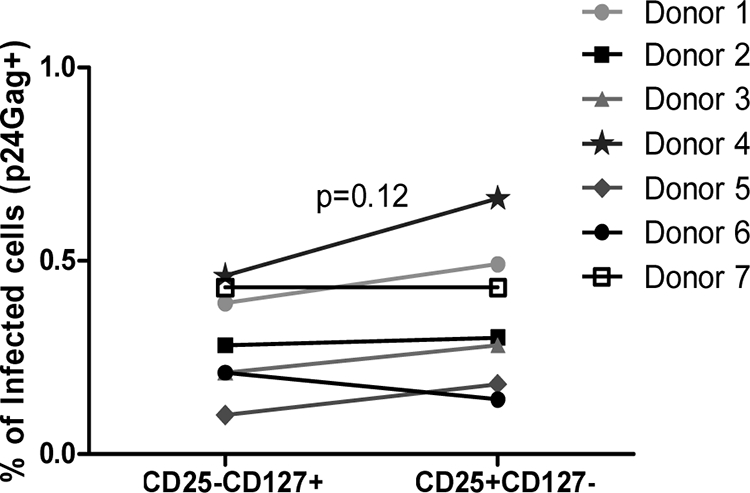
Percentages of infected Treg and Teff are similar in vivo. CD25− and CD25+ cells were isolated from seven HIV-infected donors using magnetic beads and stained with anti-CD25, anti-CD127, and anti-p24Gag antibodies and analyzed by flow cytometry. Percentages of HIV p24Gag+ cells in each cell subset are shown.
Treg can be infected by HIV, but replication levels vary depending on the HIV strain.
In vivo levels of infection are the result of many viral and host factors, which make the determination of the contribution of each factor difficult. We therefore used an in vitro infection model to explore the mechanisms involved in regulating Treg infection by HIV. We isolated Treg (defined as CD25hi CD127lo) and Teff (CD25lo CD127hi) from nine healthy donors. As shown in Fig. 2, isolated Treg expressed high levels of FOXP3 (>85%), whereas few Teff expressed FOXP3 (<5%). Purified Treg and Teff activated with anti-CD3/CD28 beads and IL-2 were then infected with either the R5 YK-JRCSF(four individuals) or X4 NL4-3 (five individuals) HIV strains.
Infection by R5 virus resulted in higher levels of p24Gag in supernatants from cultured Teff than Treg at both days 3 and 7 postinfection, as shown for a representative donor in Fig. 3A. Because of the high variability of p24Gag production between individuals, we calculated the ratio of p24Gag detected in Teff versus Treg for each individual. R5 virus infection resulted in 10.6-fold-lower p24Gag levels in Treg compared with Teff at day 3 (P = 0.01). At day 7, HIV infection of Treg was 4.3-fold lower than that in Teff (P = 0.08) (Fig. 3B). As expected, R5 infection was slow, with less p24Gag produced by both Treg and Teff at day 3 than at day 7 postinfection. As expected, treatment with the nucleoside reverse transcriptase inhibitor AZT reduced the amount of p24Gag detected in the supernatants of all infected cells by ∼90% (data not shown). The percentage of infected cells was also quantified by intracellular detection of p24Gag by flow cytometry. HIV infection could not be detected at day 3 postinfection with the R5 virus by flow cytometry. At day 7 postinfection, the percentage of p24Gag+ cells was lower in Treg and Teff (1.6% ± 0.3% and 2.9% ± 0.7% in Treg and Teff, respectively; P = 0.06) (Fig. 3C).
FIG. 3.

Treg are less susceptible than Teff to the HIV R5 strain. (A) HIV p24Gag levels in culture supernatants were measured by ELISA. p24Gag levels from one representative experiment of infection by R5 YK-JRCSF of four different experiments are shown. HIV p24Gag levels from AZT-treated cells were measured at day 7. ND, not detectable. (B) p24Gag levels in Treg were expressed as a percentage of the p24Gag levels produced by the Teff from the same donor. P values indicate the differences between Treg and Teff. (C) A representative experiment of intracellular p24Gag staining at day 7 postinfection is shown. (D) Treg and Teff from seven healthy donors were infected, and HIV proviral DNA levels were measured by nested real-time HIV-LTR-Alu PCR 24 h postinfection; HIV DNA levels were normalized based on CD3 quantification and log10 transformed.
Since production of new viral particles, as represented by p24Gag levels, is the last step in the HIV life cycle, we next quantified HIV proviral DNA integration after 24 h of infection. Higher levels of integrated HIV-1 DNA were found in Teff than in Treg (P = 0.01, paired t test, n = 7) (Fig. 3D).
In contrast to cells infected with R5 virus, more p24Gag was detected in cultures of Treg infected with X4 virus than in infected Teff at day 3 postinfection. Similar amounts of p24Gag were found at day 7, as shown for a representative individual in Fig. 4A. On average, X4 virus induced higher levels of p24Gag in Treg cultures compared to Teff at day 3 (Teff/Treg ratio of 0.5; P = 0.04). At day 7, p24Gag levels induced by X4 virus were similar in Treg and Teff (Teff/Treg ratio of 1.4; P = 0.21) (Fig. 4B). Of note, X4 virus induced a more rapid infection than R5 virus, with higher levels of p24Gag measured in day 3 supernatants. We also found a higher percentage of p24Gag+ Treg than p24Gag+ Teff at day 3 (4.8% ± 6.4% versus 8.6% ± 6.04%; P = 0.006), whereas a similar percentage of p24Gag+ cells was found in both populations at day 7 (14% ± 5.5% versus 11.3% ± 9.1% for Treg and Teff, respectively; P = 0.52) (Fig. 4C), corroborating the results obtained by ELISA. Higher levels of integrated HIV DNA were also found in X4-infected Treg compared with infected Teff (P = 0.0006, paired t test, n = 7) (Fig. 4D). Collectively, these results support the hypothesis that the relative level of HIV infection in Treg is strongly dependent on the HIV strain used.
FIG. 4.

Treg are more susceptible than Teff to the HIV X4 strain. (A) HIV p24Gag levels in culture supernatants were measured by ELISA. p24Gag levels from one representative experiment of infection by X4 NL4-3 are shown. HIV p24Gag levels from AZT-treated cells were measured at day 7. (B) p24Gag levels in Treg were expressed as a percentage of the p24Gag levels produced by the Teff from the same donor. P values indicate the differences between Treg and Teff. (C) A representative experiment of intracellular p24Gag staining at days 3 and 7 postinfection is shown. (D) Treg and Teff from seven healthy donors were infected, and HIV proviral DNA levels were measured by nested real-time HIV-LTR-Alu PCR 24 h postinfection; HIV DNA levels were normalized based on CD3 quantification and log10 transformed.
To determine whether the results seen with the R5 and X4 lab strains were representative of R5-tropic and X4-tropic isolates in general, we next infected Treg and Teff from three separate individuals with a panel of primary isolates and analyzed the levels of integrated HIV DNA. Because of the variability in levels of HIV DNA integration between individuals, we calculated the ratio of HIV DNA integrated in Teff versus Treg for each individual. In all individuals, R5 primary isolates showed similar results to lab strains: i.e., all R5 viruses were less efficient at infecting Treg than Teff (all mean levels below the 100% bar), although the levels of integration greatly varied, depending on the virus used. In contrast all X4 viruses were more efficient at infecting Treg than Teff (all means above the 100% bar). However, the intraindividual heterogeneity was higher for X4 viruses than for R5 viruses, as reflected by the large standard error. Of interest, the dualtropic strain 92USSN20 behaved similarly to R5-tropic viruses in all individuals (Fig. 5).
FIG. 5.
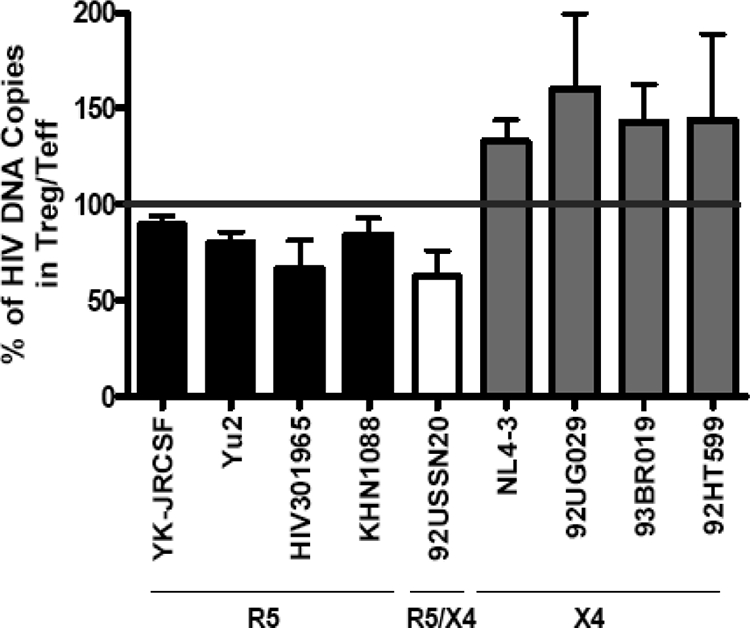
Treg susceptibility to HIV primary isolates resembles their susceptibility to HIV lab strains. Treg and Teff were infected by R5 viruses (clade C, YK-JRCSF, YU2, and HIV301965; and clade A, KNH1088), the dualtropic isolate 92USSN20 (clade A), or X4 viruses (clade A, 92UG029; clade BF, 93BR019; clade B, 92HT599; and NL4-3). HIV infection was analyzed at 24 h postinfection by nested real-time HIV-LTR-Alu PCR. Similar levels of CD3 DNA were detected under all conditions. Integrated HIV levels in Treg were expressed as percentages of the levels detected in the Teff from the same donor and represent each virus. Results show the mean (± standard error) percentages for the three donors studied.
Treg express similar levels of CD4, CCR5, and CXCR4 to Teff.
To better understand the differences in HIV susceptibility between Treg and Teff, we evaluated the surface expression of HIV receptors. CD4 expression was high in both Treg and Teff at the time of infection. The percentage of CD4+ cells decreased during the course of infection, particularly during X4 infection, although it similarly decreased in both populations (31.8% ± 3.6% versus a 27.3% ± 8.1% decrease at day 3 postinfection in Treg and Teff, respectively). During R5 infection, the percentage of CD4+ cells in Teff did not change dramatically (13.5% ± 0.8% decrease at day 3 postinfection), whereas it decreased more substantially in Treg (55.7% ± 7.2% decrease at the same time point). Levels of CCR5 were not significantly different between Treg and Teff, neither at the time of infection (P = 0.25) nor at day 3 postinfection (P = 0.16), although there was a consistent trend toward higher levels in Treg (Fig. 6A). Similar results were observed when levels of expression of CCR5 per cell were quantified (data not shown). The same pattern was observed for CXCR4 expression, with Treg expressing consistently more CXCR4 than Teff, although the differences were not significantly different (P = 0.09 and P = 0.28) (Fig. 6B). As shown in Fig. 6C, Treg and Teff secreted similar levels of the CCR5 ligands MIP1-α, MIP1-β, and RANTES. We also measured the levels of the CXCR4 ligand SDF-1; as expected, neither Treg or Teff produced this chemokine (M. Bermejo, J. Martin-Serrano, J. L. de Pablos, J. M. Alonso, C. Gamallo, F. Arenzana-Seisdedos, and J. Alcami, presented at the 9th Conference on Retroviruses and Opportunistic Infections, Seattle, WA, 24 to 28 February 2002).
FIG. 6.
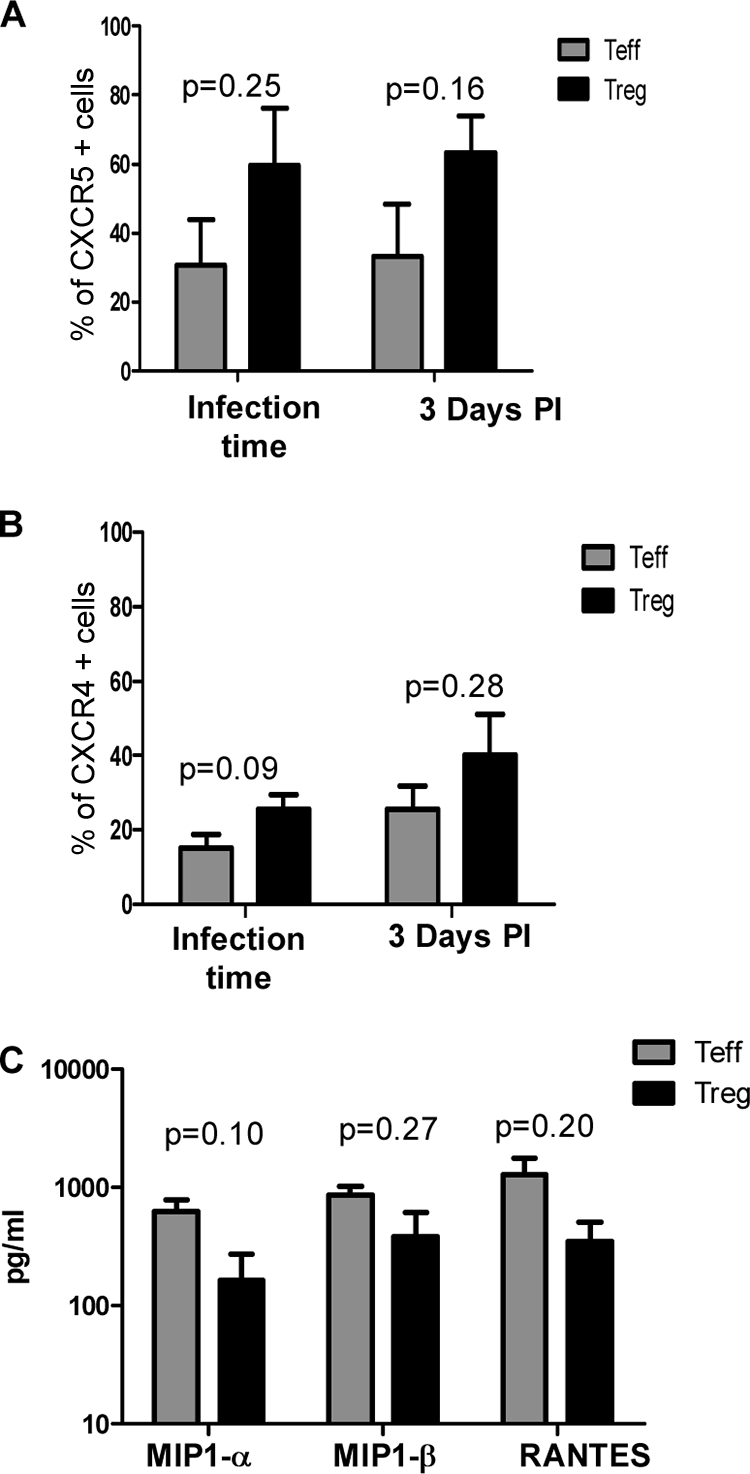
Treg express similar levels of CCR5 and CXCR4 and secrete similar levels of MIP1-α, MIP1-β, and RANTES compared to Teff. (A) CCR5 expression; (B) CXCR4 expression. Sorted Treg and Teff were activated with anti-CD3/CD28 beads for 3 days and then infected with HIV-1. Teff and Treg were stained with anti-CD4, anti-CCR5, and anti-CXCR4 antibody at the time of infection or 3 days postinfection and analyzed by flow cytometry. The percentage of positivity for each marker was defined in relation to the isotype control. Results are shown as means ± standard deviations (n = 4 or 5). (C) Supernatants were collected 3 days postactivation, and chemokine levels were analyzed using a Luminex assay.
Decreased Treg proliferation affects their susceptibility to infection by R5 viruses but not by X4 viruses.
Other cellular processes are important in determining the levels of HIV replication once HIV has penetrated the cell membrane. One of these processes is cell proliferation, which influences HIV replication by providing new targets for infection. We therefore determined whether Treg and Teff exhibit different capacities to proliferate. As shown in Fig. 7A, Treg proliferated more slowly than Teff, as shown by dilution of CFSE. Indeed, most of the Teff had divided three to four times by day 3 poststimulation, whereas the Treg had divided only twice. Nevertheless, Treg underwent proliferation and only a few cells remained undivided after 5 days of infection.
FIG. 7.

Decreased Treg proliferation affects their susceptibility to infection by R5 viruses but not by X4 viruses. Teff and Treg proliferation was measured by labeling the cells with CFSE before stimulation with anti-CD3/CD28 beads in the presence of 100 U/ml IL-2. (A) Levels of CFSE expression were analyzed by flow cytometry 3 days postactivation as well as 3 and 5 days after infection. The results shown are from one experiment, representative of nine separate experiments. (B and C) The percentage of p24Gag+ cells was measured in the highly proliferating (CFSE low) Treg and Teff at day 5 postinfection with the R5 strain YK-JRCSF (B) and at day 3 postinfection with the X4 strain NL4-3 (C).
Therefore, we determined whether decreased Treg proliferation is playing a role in the differential susceptibility of Treg and Teff to HIV infection. To do that, we analyzed the percentage of p24Gag+ cells in the highly proliferating Treg and Teff at day 5 postinfection with the R5 strain YK-JRCSF and at day 3 postinfection with the X4 strain NL4-3. These time points were chosen because they were previously determined to allow optimal detection of p24Gag+ cells (data not shown). As shown in Fig. 7B, the percentages of infected cells were similar in these two populations following YK-JRCSF infection (P = 0.21), suggesting the absence of intrinsic difference in the susceptibility to this virus between proliferating Treg and Teff. In contrast, when NL4-3-infected cells were analyzed, the percentage of infected cells in the proliferating Treg was higher than that in the proliferating Teff (P = 0.01) (Fig. 7C). Taken together, these results suggest that proliferation plays an important, but not unique, role in determining the respective susceptibility of Treg and Teff to HIV infection.
HIV-infected Treg remain suppressive.
Treg play an important role in suppressing immune activation; therefore, we evaluated whether HIV infection affected Treg suppressive activity. Activated Treg were infected or not by the R5 strain YK-JRCSF and cocultured with activated Teff, and Teff proliferation was determined by measuring CFSE MFI. As shown in Fig. 8, Treg infected by YK-JRCSF decreased Teff proliferation with a similar efficiency to uninfected Treg, suggesting that exposure of Treg to HIV does not affect their function.
FIG. 8.
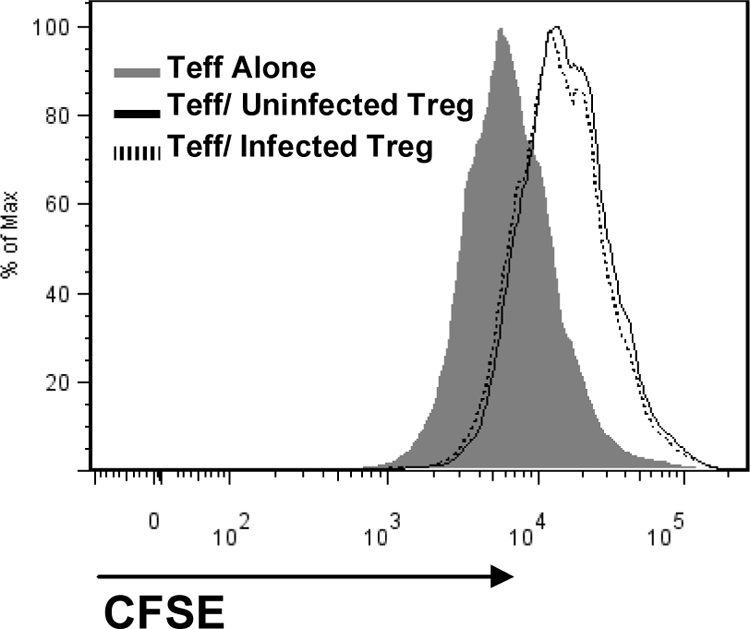
HIV-infected Treg remain suppressive. Purified Treg and Teff were separately activated for 3 days using anti-CD3/CD28 beads (1 cell per 3 beads) in the presence of 100 IU/ml IL-2. Treg were infected or not with YK-JRCSF virus at an MOI of 1 and incubated for 2 h at 37°C. Teff were labeled with CFSE. Treg and Teff were cocultured at a 1:1 ratio for 3 days. The suppressive capacity of Treg was determined by comparing the CFSE MFI between Teff cultured alone to those cultured with Treg. Results shown are from one experiment, representative of two separate experiments.
DISCUSSION
Although several previous studies have indicated that Treg can serve as a potential target cell population for HIV-1 infection in vivo, Treg and HIV interactions remain poorly understood. We therefore used a more systematic approach to study Treg susceptibility to infection, comparing lab strains with R5 or X4 tropism, as well as confirming the data with primary isolates. Collectively, our data convincingly demonstrate that Treg are infected in vitro by infectious HIV viruses, but their susceptibility depends on the tropism of the virus. Treg were less susceptible to R5 viruses, using both lab strains and primary isolates, compared to Teff, whereas Treg were more susceptible to X4 viruses.
Importantly, we have shown that Treg were less susceptible to several primary R5 isolates from different clades as well as to R5 lab strains, although the levels of infection vary greatly depending on the virus used. These data suggest that Treg could be relatively protected from HIV infection during the acute phase of mucosal transmission, which is thought to predominantly involve R5 viruses (26). Of note, Estes et al. showed that 13% of lymph node FOXP3+ cells were infected 2 weeks after vaginal infection by the R5 SIVmac251. Although a direct comparison of the infection levels to non-Treg was not made, such reported frequency of Treg infection is lower than that reported by the same group for lamina propria CD4+ cells (30% cells harboring SIV RNA) in a similar infection model (21). These results suggest that the decreased susceptibility to R5 HIV infection that we found in vitro reflects what happens in vivo. This important hypothesis will have to be confirmed in future studies of acute SIV infection. Our results are also in agreement with a previous study that used an R5 lab strain to infect memory and naïve Treg, as well as memory non-Treg (3). However, conflicting data have also been reported: i.e., that Treg are more susceptible to HIV infection and are more prone to HIV-induced death than memory non-Treg when high virus concentrations (MOI of 5) are used (23). Such discrepant results could be explained by the fact that many critical experimental parameters differed in these studies. Indeed, activation methods, cell purification techniques, and virus concentrations were different between the latter study and ours. However, it should be noted that the type of cell activation may not represent the most critical parameter, because we did not find differences in HIV susceptibility when cells were stimulated with PHA and IL-2 instead of anti-CD3/CD28 beads. Discrepant results were also found when interactions between FOXP3 and HIV LTR were studied in FOXP3-transfected CD4+ T cells. For instance, one study showed that FOXP3 limits HIV-1 LTR and human T-cell leukemia virus type 1 transcription by interfering with activation of NFAT and CREB pathways. The inhibitory effect was not absolute, and low-level LTR activation persisted (12). In contrast, Cron et al. (28) reported increased LTR activity in FOXP3-expressing cells, using a different LTR from the one used in the aforementioned study. Because the LTR sequence can vary significantly between HIV strains (30), it is possible that FOXP3 interactions with distinct HIV LTRs may have different functional consequences, which would explain this apparent discrepancy.
In contrast to infection with R5 viruses, Treg were more susceptible to infection by X4 viruses than Teff. To our knowledge, our study is the first one to assess primary Treg susceptibility to infection by full-length X4 viruses. Importantly, similar results were obtained with primary isolates or the lab strain NL4-3. Our results are also supported by data provided by other experimental systems. Indeed, in vivo infection of humanized DKO mice by the highly pathogenic dualtropic R3A HIV isolate led to an increased level of infection in Treg compared to non-Treg (15). Furthermore, overexpression of FOXP3 in CD4+ T cells enhanced NL4-3 LTR activity, by modulating chromatin structure as well as enhancing NF-κB activity (14).
Variations in levels of p24Gag production by Treg and Teff could arise from differences in multiple steps of the virus life cycle, as p24Gag release is at the end of a complex developmental program that takes place in a cycling cell. To identify the potential mechanisms underlying the differences between Treg and Teff, we first analyzed the expression of HIV receptors by these two cell subsets. Similar expression levels of CD4 and a trend toward higher levels of CCR5 per cell were found in activated Treg compared with activated Teff, a result in agreement with previous studies (8, 23, 27). Treg produced similar levels of CCR5 ligands to Teff. Our data thus suggest that an altered balance of HIV receptors and CCR5 ligands is not the main mechanism explaining decreased Treg susceptibility to R5 HIV. Similar differences between Treg and Teff were found when infection levels were analyzed at an early time point postinfection. One potential explanation of our data could be the decreased activation state of Treg compared to Teff, as the activation state of the target cell markedly affects the efficiency of the early steps of HIV replication (34). The majority of the Treg underwent proliferation during the 7-day culture (Fig. 7), but they clearly proliferated less than Teff. Therefore, to better understand the contribution of decreased proliferation to decreased infection of Treg by R5 viruses, we analyzed the percentage of p24Gag+ cells in the highly proliferating Treg and Teff. Similar levels of infection were found, a result that suggests the absence of intrinsic differences in the susceptibility of proliferating Treg and Teff to this virus. Considering all our data together, decreased proliferative capacity of Treg likely constitutes a major mechanism explaining their decreased susceptibility to infection by R5 viruses.
Despite the fact that Treg proliferated less than Teff, they were more infected than Teff, showing that proliferation is not the only mechanism regulating Treg infection by X4 viruses. Interestingly, a higher percentage of infection was observed in the highly proliferating Treg compared to that in the proliferating Teff, suggesting that some cellular factors may potentiate X4 replication in Treg. Although not statistically significant, there was a trend toward Treg expressing higher levels of CXCR4 than Teff at the time of infection, a result in agreement with previous studies (23, 27). This characteristic could have mediated their higher susceptibility to HIV X4, which was particularly striking at the early times after infection. Alternatively, differences in the expression of some cellular factors critical for HIV infection could also be involved. Of interest, Th1 and Th2 cells have been shown to express different levels of APOBEC3G, and these differences determined their susceptibility to infection by HIV, including by Vif-competent viruses (35). How these cellular factors are expressed in activated Treg is not yet known, and differences in their expression could contribute to the regulation of Treg susceptibility to HIV. An additional level of regulation may involve modulation of HIV LTR activity by FOXP3. Indeed, it was previously shown that FOXP3 could enhance the LTR activity of an X4 virus, by modulating chromatin structure as well as enhancing NF-κB activity (14). Future experiments will be needed to confirm whether differences in receptor-mediated entry/fusion explain the difference in susceptibility of Treg to different HIV strains, or whether other early steps of the life cycle are also implicated.
Of importance, infected Treg were as suppressive as noninfected Treg, as evidenced by their capacity to inhibit Teff proliferation. Our data are in agreement with our previous studies showing that exposure of Treg to noninfectious HIV did not affect their suppressive capacity (22). Other studies have also shown that circulating Treg purified from most HIV-infected patients exhibited suppressive activity (1, 9). However, it should be noted that our assay did not assess the functionality of infected Treg on a per-cell basis as viable infected Treg could not be separated and tested for their function. Nevertheless, these data suggest that Treg remain functional in an HIV- infected host and they likely regulate the homeostasis of the immune system as well as control HIV-specific immune responses.
Our in vitro data, as well as those from other laboratories, suggest that FOXP3 and HIV interactions are tightly regulated by both virus and host factors and that minor changes in either side will greatly impact the outcome of infection. These results may explain why no major differences in the level of infection of Treg and non-Treg were seen in vivo in chronically infected patients, both in our study and in two other recent studies (6, 8). Indeed, both the level of T-cell activation and the level of virus heterogeneity are known to vary among patients, and changes in these critical parameters are expected to impact infection of Treg by HIV.
Acknowledgments
This study was supported by NIH grant RO1 AI068524 (to C.C.) and the Dean's Scholar Award from the University of Cincinnati, College of Medicine (to J.T.B.).
We thank the NIH AIDS Research and Reference Reagent Program for the IL-2, cell lines, and HIV primary isolates as well as Nicolas Chomont (Laboratoire d'Immunologie, Département de Microbiologie et d'Immunologie, Université de Montréal, Québec, Canada) for assistance with the HIV-Alu PCR. We also thank Jun Ying (Department of Public Health Sciences, University of Cincinnati College of Medicine) for assistance with the statistics, Carl Fichtenbaum and Eva Moore (Division of Infectious Diseases, University of Cincinnati College of Medicine) for kindly supplying HIV patients' samples, and the Cincinnati Children's Hospital Research Foundation Sorting Core as well as Kevin Ma and Laura Rusie for help with virus production. We also thank Kris Orsborn, Pietro Presicce, and Celine Silva-Lages for constructive comments.
Footnotes
Published ahead of print on 14 October 2009.
REFERENCES
- 1.Aandahl, E. M., J. Michaëlsson, W. J. Moretto, F. M. Hecht, and D. F. Nixon. 2004. Human CD4+ CD25+ regulatory T cells control T-cell responses to human immunodeficiency virus and cytomegalovirus antigens. J. Virol. 78:2454-2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersson, J., A. Boasso, J. Nilsson, R. Zhang, N. J. Shire, S. Lindback, G. M. Shearer, and C. A. Chougnet. 2005. The prevalence of regulatory T cells in lymphoid tissue is correlated with viral load in HIV-infected patients. J. Immunol. 174:3143-3147. [DOI] [PubMed] [Google Scholar]
- 3.Antons, A. K., R. Wang, K. Oswald-Richter, M. Tseng, C. W. Arendt, S. A. Kalams, and D. Unutmaz. 2008. Naive precursors of human regulatory T cells require FoxP3 for suppression and are susceptible to HIV infection. J. Immunol. 180:764-773. [DOI] [PubMed] [Google Scholar]
- 4.Baecher-Allan, C., J. A. Brown, G. J. Freeman, and D. A. Hafler. 2001. CD4+CD25high regulatory cells in human peripheral blood. J. Immunol. 167:1245-1253. [DOI] [PubMed] [Google Scholar]
- 5.Brussel, A., and P. Sonigo. 2003. Analysis of early human immunodeficiency virus type 1 DNA synthesis by use of a new sensitive assay for quantifying integrated provirus. J. Virol. 77:10119-10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chase, A. J., H.-C. Yang, H. Zhang, J. N. Blankson, and R. F. Siliciano. 2008. Preservation of FoxP3+ regulatory T cells in the peripheral blood of human immunodeficiency virus type 1-infected elite suppressors correlates with low CD4+ T-cell activation. J. Virol. 82:8307-8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dittmer, U., H. He, R. J. Messer, S. Schimmer, A. R. Olbrich, C. Ohlen, P. D. Greenberg, I. M. Stromnes, M. Iwashiro, S. Sakaguchi, L. H. Evans, K. E. Peterson, G. Yang, and K. J. Hasenkrug. 2004. Functional impairment of CD8+ T cells by regulatory T cells during persistent retroviral infection. Immunity 20:293-303. [DOI] [PubMed] [Google Scholar]
- 8.Dunham, R. M., B. Cervasi, J. M. Brenchley, H. Albrecht, A. Weintrob, B. Sumpter, J. Engram, S. Gordon, N. R. Klatt, I. Frank, D. L. Sodora, D. C. Douek, M. Paiardini, and G. Silvestri. 2008. CD127 and CD25 expression defines CD4+ T cell subsets that are differentially depleted during HIV infection. J. Immunol. 180:5582-5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eggena, M. P., B. Barugahare, N. Jones, M. Okello, S. Mutalya, C. Kityo, P. Mugyenyi, and H. Cao. 2005. Depletion of regulatory T cells in HIV infection is associated with immune activation. J. Immunol. 174:4407-4414. [DOI] [PubMed] [Google Scholar]
- 10.Estes, J. D., Q. Li, M. R. Reynolds, S. Wietgrefe, L. Duan, T. Schacker, L. J. Picker, D. I. Watkins, J. D. Lifson, C. Reilly, J. Carlis, and A. T. Haase. 2006. Premature induction of an immunosuppressive regulatory T cell response during acute simian immunodeficiency virus infection. J. Infect. Dis. 193:703-712. [DOI] [PubMed] [Google Scholar]
- 11.Fontenot, J. D., M. A. Gavin, and A. Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4:330-336. [DOI] [PubMed] [Google Scholar]
- 12.Grant, C., U. Oh, K. Fugo, N. Takenouchi, C. Griffith, K. Yao, T. E. Newhook, L. Ratner, and S. Jacobson. 2006. Foxp3 represses retroviral transcription by targeting both NF-kappaB and CREB pathways. PLoS Pathog. 2:e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartigan-O'Connor, D. J., C. Poon, E. Sinclair, and J. M. McCune. 2007. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J. Immunol. Methods 319:41-52. [DOI] [PubMed] [Google Scholar]
- 14.Holmes, D., G. Knudsen, S. Mackey-Cushman, and L. Su. 2007. FoxP3 enhances HIV-1 gene expression by modulating NFkappaB occupancy at the long terminal repeat in human T cells. J. Biol. Chem. 282:15973-15980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang, Q., L. Zhang, R. Wang, J. Jeffrey, M. L. Washburn, D. Brouwer, S. Barbour, G. I. Kovalev, D. Unutmaz, and L. Su. 2008. FoxP3+CD4+ regulatory T cells play an important role in acute HIV-1 infection in humanized Rag2−/−γC−/− mice in vivo. Blood 112:2858-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimpton, J., and M. Emerman. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated β-galactosidase gene. J. Virol. 66:2232-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinter, A., J. McNally, L. Riggin, R. Jackson, G. Roby, and A. S. Fauci. 2007. Suppression of HIV-specific T cell activity by lymph node CD25+ regulatory T cells from HIV-infected individuals. Proc. Natl. Acad. Sci. USA 104:3390-3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinter, A. L., M. Hennessey, A. Bell, S. Kern, Y. Lin, M. Daucher, M. Planta, M. McGlaughlin, R. Jackson, S. F. Ziegler, and A. S. Fauci. 2004. CD25+CD4+ regulatory T cells from the peripheral blood of asymptomatic HIV-infected individuals regulate CD4+ and CD8+ HIV-specific T cell immune responses in vitro and are associated with favorable clinical markers of disease status. J. Exp. Med. 200:331-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinter, A. L., R. Horak, M. Sion, L. Riggin, J. McNally, Y. Lin, R. Jackson, A. O'Shea, G. Roby, C. Kovacs, M. Connors, S. A. Migueles, and A. S. Fauci. 2007. CD25+ regulatory T cells isolated from HIV-infected individuals suppress the cytolytic and nonlytic antiviral activity of HIV-specific CD8+ T cells in vitro. AIDS Res. Hum. Retrovir. 23:438-450. [DOI] [PubMed] [Google Scholar]
- 20.Li, L., and C. Y. Wu. 2008. CD4+ CD25+ Treg cells inhibit human memory gammadelta T cells to produce IFN-gamma in response to M tuberculosis antigen ESAT-6. Blood 111:5629-5636. [DOI] [PubMed] [Google Scholar]
- 21.Li, Q., L. Duan, J. D. Estes, Z. M. Ma, T. Rourke, Y. Wang, C. Reilly, J. Carlis, C. J. Miller, and A. T. Haase. 2005. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 434:1148-1152. [DOI] [PubMed] [Google Scholar]
- 22.Nilsson, J., A. Boasso, P. A. Velilla, R. Zhang, M. Vaccari, G. Franchini, G. M. Shearer, J. Andersson, and C. Chougnet. 2006. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood 108:3808-3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oswald-Richter, K., S. M. Grill, N. Shariat, M. Leelawong, M. S. Sundrud, D. W. Haas, and D. Unutmaz. 2004. HIV infection of naturally occurring and genetically reprogrammed human regulatory T-cells. PLoS Biol. 2:E198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rouse, B. T., and S. Suvas. 2007. Regulatory T cells and immunity to pathogens. Expert Opin. Biol. Ther. 7:1301-1309. [DOI] [PubMed] [Google Scholar]
- 25.Sakaguchi, S., N. Sakaguchi, J. Shimizu, S. Yamazaki, T. Sakihama, M. Itoh, Y. Kuniyasu, T. Nomura, M. Toda, and T. Takahashi. 2001. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol. Rev. 182:18-32. [DOI] [PubMed] [Google Scholar]
- 26.Schuitemaker, H., M. Koot, N. A. Kootstra, M. W. Dercksen, R. E. Y. de Goede, R. P. van Steenwijk, J. M. A. Lange, J. K. M. E. Schattenkerk, F. Miedema, and M. Tersmette. 1992. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 66:1354-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sebastiani, S., P. Allavena, C. Albanesi, F. Nasorri, G. Bianchi, C. Traidl, S. Sozzani, G. Girolomoni, and A. Cavani. 2001. Chemokine receptor expression and function in CD4+ T lymphocytes with regulatory activity. J. Immunol. 166:996-1002. [DOI] [PubMed] [Google Scholar]
- 28.Selliah, N., M. Zhang, S. White, P. Zoltick, B. E. Sawaya, T. H. Finkel, and R. Q. Cron. 2008. FOXP3 inhibits HIV-1 infection of CD4 T-cells via inhibition of LTR transcriptional activity. Virology 381:161-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shevach, E. M. 2001. Certified professionals: CD4+CD25+ suppressor T cells. J. Exp. Med. 193:F41-F46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simm, M., W. Chao, O. Pekarskaya, P. Sova, P. Gupta, R. Balachandran, and D. J. Volsky. 1996. Genetic variability and function of the long terminal repeat from syncytium-inducing and non-syncytium-inducing human immunodeficiency virus type 1. AIDS Res. Hum Retrovir. 12:801-809. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi, T., Y. Kuniyasu, M. Toda, N. Sakaguchi, M. Itoh, M. Iwata, J. Shimizu, and S. Sakaguchi. 1998. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10:1969-1980. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi, T., T. Tagami, S. Yamazaki, T. Uede, J. Shimizu, N. Sakaguchi, T. W. Mak, and S. Sakaguchi. 2000. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 192:303-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran, T. A., M. G. de Goer de Herve, H. Hendel-Chavez, B. Dembele, E. Le Nevot, K. Abbed, C. Pallier, C. Goujard, J. Gasnault, J. F. Delfraissy, A. M. Balazuc, and Y. Taoufik. 2008. Resting regulatory CD4 T cells: a site of HIV persistence in patients on long-term effective antiretroviral therapy. PLoS ONE 3:e3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Unutmaz, D., V. N. KewalRamani, S. Marmon, and D. R. Littman. 1999. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J. Exp. Med. 189:1735-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vetter, M. L., and R. T. D'Aquila. 2009. Cytoplasmic APOBEC3G restricts incoming Vif-positive human immunodeficiency virus type 1 and increases two-long terminal repeat circle formation in activated T-helper-subtype cells. J. Virol. 83:8646-8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weiss, L., V. Donkova-Petrini, L. Caccavelli, M. Balbo, C. Carbonneil, and Y. Levy. 2004. Human immunodeficiency virus-driven expansion of CD4+CD25+ regulatory T cells, which suppress HIV-specific CD4 T-cell responses in HIV-infected patients. Blood 104:3249-3256. [DOI] [PubMed] [Google Scholar]