

Abstract
The UL17 and UL25 proteins (pUL17 and pUL25, respectively) of herpes simplex virus 1 are located at the external surface of capsids and are essential for DNA packaging and DNA retention in the capsid, respectively. The current studies were undertaken to determine whether DNA packaging or capsid assembly affected the pUL17/pUL25 interaction. We found that pUL17 and pUL25 coimmunoprecipitated from cells infected with wild-type virus, whereas the major capsid protein VP5 (encoded by the UL19 gene) did not coimmunoprecipitate with these proteins under stringent conditions. In addition, pUL17 (i) coimmunoprecipitated with pUL25 in the absence of other viral proteins, (ii) coimmunoprecipitated with pUL25 from lysates of infected cells in the presence or absence of VP5, (iii) did not coimmunoprecipitate efficiently with pUL25 in the absence of the triplex protein VP23 (encoded by the UL18 gene), (iv) required pUL25 for proper solubilization and localization within the viral replication compartment, (v) was essential for the sole nuclear localization of pUL25, and (vi) required capsid proteins VP5 and VP23 for nuclear localization and normal levels of immunoreactivity in an indirect immunofluorescence assay. Proper localization of pUL25 in infected cell nuclei required pUL17, pUL32, and the major capsid proteins VP5 and VP23, but not the DNA packaging protein pUL15. The data suggest that VP23 or triplexes augment the pUL17/pUL25 interaction and that VP23 and VP5 induce conformational changes in pUL17 and pUL25, exposing epitopes that are otherwise partially masked in infected cells. These conformational changes can occur in the absence of DNA packaging. The data indicate that the pUL17/pUL25 complex requires multiple viral proteins and functions for proper localization and biochemical behavior in the infected cell.
Immature herpes simplex virus (HSV) capsids, like those of all herpesviruses, consist of two protein shells. The outer shell comprises 150 hexons, each composed of six copies of VP5, and 11 pentons, each containing five copies of VP5 (23, 29, 47). One vertex of fivefold symmetry is composed of 12 copies of the protein encoded by the UL6 gene and serves as the portal through which DNA is inserted (22, 39). The pentons and hexons are linked together by 320 triplexes composed of two copies of the UL18 gene product, VP23, and one copy of the UL38 gene product, VP19C (23). Each triplex arrangement has two arms contacting neighboring VP5 subunits (47). The internal shell of the capsid consists primarily of more than 1,200 copies of the scaffold protein ICP35 (VP22a) and a smaller number of protease molecules encoded by the UL26 open reading frame, which self-cleaves to form VP24 and VP21 derived from the amino and carboxyl termini, respectively (11, 12, 19, 25; reviewed in reference 31). The outer shell is virtually identical in the three capsid types found in HSV-infected cells, termed types A, B, and C (5, 6, 7, 29, 43, 48). It is believed that all three are derived from the immature procapsid (21, 38). Type C capsids contain DNA in place of the internal shell, type B capsids contain both shells, and type A capsids consist only of the outer shell (15, 16). Cleavage of viral DNA to produce type C capsids requires not only the portal protein, but all of the major capsid proteins and the products of the UL15, UL17, UL28, UL32, and UL33 genes (2, 4, 10, 18, 26, 28, 35, 46). Only C capsids go on to become infectious virions (27).
The outer capsid shell contains minor capsid proteins encoded by the UL25 and UL17 open reading frames (1, 17, 20). These proteins are located on the external surface of the viral capsid (24, 36, 44) and are believed to form a heterodimer arranged as a linear structure, termed the C capsid-specific complex (CCSC), located between pentons and hexons (41). This is consistent with the observation that levels of pUL25 are increased in C capsids as opposed to in B capsids (30). On the other hand, other studies have indicated that at least some UL17 and UL25 proteins (pUL17 and pUL25, respectively) associate with all capsid types, and pUL17 can associate with enveloped light particles, which lack capsid and capsid proteins but contain a number of viral tegument proteins (28, 36, 37). How the UL17 and UL25 proteins attach to capsids is not currently known, although the structure of the CCSC suggests extensive contact with triplexes (41). It is also unclear when pUL17 and pUL25 become incorporated into the capsid during the assembly pathway. Less pUL25 associates with pUL17(−) capsids, suggesting that the two proteins bind capsids either cooperatively or sequentially, although this could also be consequential to the fact that less pUL25 associates with capsids lacking DNA (30, 36).
Both pUL25 and pUL17 are necessary for proper nucleocapsid assembly, but their respective deletion generates different phenotypes. Deletion of pUL17 precludes DNA packaging and induces capsid aggregation in the nuclei of infected cells, suggesting a critical early function (28, 34), whereas deletion of pUL25 precludes correct cleavage or retention of full-length cleaved DNA within the capsid (8, 20, 32), thus suggesting a critical function later in the assembly pathway.
The current studies were undertaken to determine how pUL17 and pUL25 associate with capsids by studying their interaction and localization in the presence and absence of other capsid proteins.
MATERIALS AND METHODS
Cell lines and viruses.
Vero and Hep2 cells were obtained from the American Type Culture Association and were propagated in Dulbecco's modified Eagle's medium supplemented with 10% newborn calf serum (NBCS) and antibiotics as described previously (45).
The genotypes of viruses used in these studies are indicated in Table 1. A UL18 deletion virus (Δ18) virus was constructed to remove the entire open reading frame of UL18 and replace it with an FLP recombination target-flanked kanamycin resistance (Kanr) cassette as follows. A gene encoding Kanr was PCR amplified with the following primers containing sequences homologous to the flanking regions, the UL18 open reading frame and the Kanr cassette: del18F (no. 64), CCCCCGTGGGTCTAGCCGGGCCGTGTAGGCTGGAGCTGCTTC; del18R (no. 65), CCCTGCCGCGTGGATCGGCGCCATTCCGGGGATCCGTCGAC (Kanr homology underlined). Amplicons were transformed into EL250 cells containing a chloramphenicol-resistant (Cmr), wild-type HSV-1(F) bacterial artificial chromosome (BAC) and Cmr/Kanr recombinants were selected on agar plates (33). Extracted BAC DNA was transfected with SuperFect (Qiagen) into and propagated on G5 cells containing UL16 to UL21 of the HSV genome (13). Stocks were obtained from G5 cells infected at a multiplicity of infection of 0.01 and grown in 890-cm2 roller bottles.
TABLE 1.
Description of viruses used in the current study
| Virus | Gene modified | Protein affected | Function | Reference |
|---|---|---|---|---|
| Δ17 mutant | UL17 | pUL17 | CCSC | 44 |
| Δ25 mutant | UL25 | pUL25 | CCSC | 20 |
| Δ18 mutant | UL18 | VP23 | Triplex component | This study |
| Δ19 mutant | UL19 | VP5 | Major capsid protein | 13 |
| Δ15 mutant | UL15 | pUL15 | DNA cleavage and packaging; terminase subunit | 3 |
| Δ32 mutant | UL32 | pUL32 | DNA cleavage and packaging | 18 |
| 17R | UL17 restored | pUL17 | CCSC | This study |
A UL17 null (Δ17) virus and Δ25 (KUL25NS) viruses were described previously and contain a kanamycin cassette inserted into UL17 and a stop codon inserted into UL25, respectively (20, 44). Δ17 and Δ25 viruses were grown on CV1-17 and 8-1 cells complementing UL17 and UL25, respectively (20, 44). Stocks were obtained from cells infected at a multiplicity of infection of 0.01 and grown in 890-cm2 roller bottles.
To rescue the UL17 deletion, the Δ17 BAC and a UL17 expression vector (pRB457) containing DNA starting at a BamHI site in exon II of UL15 and ending at a BglII site downstream of pUL17 (base pairs 31097 to 34129) were cotransfected into noncomplementing (Vero) cells. The virus was harvested 10 days posttransfection after virus plaques had begun to expand. Several plaques from this virus preparation yielded plaques on Vero cells. For stock preparations after a second round of plaque picking, Vero cells were infected with the virus at 0.01 PFU/cell. The repaired virus, designated UL17R, was verified both by its competency to infect noncomplementing cells and by expression of pUL17 as assessed by immunoblotting.
Immunoprecipitation assay.
Vero cells in 10-cm or 15-cm dishes were infected with various viruses at 5 PFU per cell and incubated at 37°C. Sixteen hours following infection, cells were removed by scraping, transferred to 15-ml conical tubes, and pelleted at 4,000 rpm in an Eppendorf 5810R centrifuge with an A-4-62 rotor at 4°C. Supernatants were discarded, and pellets were resuspended in 5 ml phosphate-buffered saline (PBS) with Complete protease inhibitors (Roche). Cells were then pelleted at 4,000 rpm at 4°C for 10 min, and supernatants were decanted. Cell pellets were stored at −80°C overnight. After thawing, the infected cell pellets were lysed in RIPA buffer (1.0% NP-40, 0.25% sodium deoxycholate, 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA [pH 8.0], 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4) on ice for at least 20 min. Infected cell lysates were centrifuged to pellet insoluble material at 4°C for 10 min in a microcentrifuge, and supernatants were transferred to new tubes. One-twentieth of each soluble lysate volume was removed and combined with 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) buffer (100 mM Tris-HCl [pH 6.8], 4.0% SDS, 0.2% bromophenol blue, 20% glycerol, 200 mM fresh dithiothreitol) to assess protein expression in the lysates. The remaining lysate was precleared by reaction with normal mouse immunoglobulin G (IgG) with constant rotation at 4°C for 30 to 60 min, after which GammaBind G beads (GE Life Sciences) in a 50% PBS slurry were added. The mixture was then constantly rotated at 4°C for 1 h, after which samples were centrifuged for 5 min at 4°C to remove the beads bearing normal mouse IgG, as well as any proteins capable of nonspecific binding. The cleared lysates were then reacted with the target antibody (anti-pUL25 or anti-pUL17) either for 30 to 60 min at 4°C or overnight with constant rotation before GammaBind G beads were added. Immunoprecipitation with chicken anti-pUL17 IgY required an additional incubation with a rabbit anti-IgY bridging antibody to adhere to the Bind G beads. The beads were then allowed to react with antibody complexes for 2 h at 4°C with constant rotation, after which they were pelleted at 4°C for 5 min, resuspended in RIPA solution, and rotated for 10 min at 4°C. This washing step was repeated three more times before elution of proteins by boiling in 2× SDS-PAGE denaturation buffer for 5 min. Soluble lysate samples and immunoprecipitation samples were electrophoretically separated on a 10% polyacrylamide gel and transferred to nitrocellulose at 30 V for 5 h at 4°C.
Transfections.
Adherent Hep2 cells were transfected with the indicated expression plasmids using Lipofectamine 2000 (Invitrogen) or TurboFect (Fermentas) according to the protocol of the manufacturer.
Immunofluorescence assay.
Hep2 cells were seeded onto glass coverslips and cultured in Dulbecco's modified Eagle's medium with 10% supplemental NBCS. Cells were either transfected in Opti-MEM (Invitrogen) medium or infected with 5 PFU per cell using low-serum-containing medium (1% NBCS). At various times following transfection or infection, cells were briefly washed with PBS and fixed with 3% paraformaldehyde in PBS for 15 min at room temperature. Fixed cells were washed with PBS before quenching autofluorescence with a 15-min incubation in 50 mM ammonium chloride. Cells were then washed with PBS and permeabilized for 5 min in 1.0% Triton X-100, washed again with PBS, and then incubated for 30 min in PBS with 1.0% bovine serum albumin, 5% donkey serum, and 5% human serum to block nonspecific antibody binding. Primary antibody was diluted in blocking solution at the following concentrations: anti-pUL17, between 1:1,000 and 1:500; anti-pUL25 (25E10), 1:200; anti-capsid 8F5 monoclonal antibody, 1:100 (40). After reaction of the primary antibodies with the fixed, permeabilized cells for 45 min, unbound antibody was removed by washing in PBS. The coverslips were then reacted for 45 min with the indicated fluorescence-conjugated secondary antibodies diluted 1:1,000 in blocking solution. Subsequently, cells were washed extensively with PBS, dipped briefly in distilled H2O to remove any excess salt, and mounted on glass slides with Vectashield mounting medium. Mounted coverslips were sealed with nail polish and were examined with either an Olympus IX70 or Zeiss LSM510 laser scanning confocal microscope equipped with argon, krypton, and helium/neon lasers. Images were recorded digitally and imported into Adobe Photoshop for display.
Immunoblotting.
Immunoblots were blocked in PBS with Tween 20 (PBST) with 5% milk for 20 to 30 min and rinsed with PBST. Primary antibodies were diluted in PBST with 1% bovine serum albumin to the following concentrations: anti-pUL17, 1:10,000; anti-pUL25, 1:1,000; anti-VP5 (Virusys), 1:500; anti-VP23 (NC-5), 1:2,000; anti-VP19C (NC-2), 1:2,000 (NC-2 and NC-5 antibodies were gifts from Gary Cohen and Roselyn Eisenberg) (9). Excess antibody was removed with three 10-min washes in PBST. Horseradish peroxidase-conjugated secondary antibodies were diluted 1:5,000 in PBST with 5% milk and allowed to react with immunoblots for at least 1 h. Excess antibody was again removed with PBST washes, and conjugate binding was detected with the Pierce chemiluminescent ECL substrate and subsequent exposure to X-ray film. Bound antibody was stripped from membranes with 2% SDS and 0.71% β-mercaptoethanol in 62.5 mM Tris (pH 6.8) at 50°C for 15 to 20 min, and membranes were washed extensively with PBST before reprobing.
Expression and purification of pUL17-His.
Insect Sf21 cells were infected with 5 PFU/cell of a recombinant baculovirus expressing pUL17 fused in frame at the C terminus with a six-His tag. Infected cells were collected at 24 h postinfection, pelleted by centrifugation, and washed once with PBS. The cells were then lysed in 50 mM NaH2PO4-300 mM NaCl-10 mM imidazole-1% NP-40 (pH 8.0) by sonication for 15 s on ice. Insoluble material was removed by centrifugation for 10 min at 4,000 rpm in an Eppendorf 5810R centrifuge with an A-4-62 rotor at 4°C. A 50% slurry of nickel-nitrilotriacetic acid resin in PBS was added to the solubilized proteins and mixed by rotation at 4°C for 90 min. pUL17-His-bound nickel-nitrilotriacetic acid resin was pelleted and washed twice in 50 mM NaH2PO4-300 mM NaCl-20 mM imidazole (pH 8.0). Protein was eluted with 50 mM NaH2PO4-300 mM NaCl-250 mM imidazole (pH 8.0). Eluted protein was dialyzed stepwise at 4°C down to 50 mM NaH2PO4-50 mM NaCl-10% glycerol to remove imidazole and stored at −80°C.
RESULTS
pUL17 and pUL25 interact in the absence of assembled capsids.
Experiments were undertaken to determine whether pUL17 interacted with pUL25 in the absence of DNA packaging or major capsid components. To this end, cells were infected with HSV-1(F), and deletion mutants lacking functional UL17 (Δ17), UL25 (Δ25), UL18 encoding VP23 (Δ18), UL19 encoding VP5 (Δ19), or UL32 (Δ32). The infected cells were lysed at 16 h postinfection, and lysates were reacted with anti-pUL17 antibody and a rabbit anti-IgY bridging antibody for 2 hours. Immunoprecipitated material and aliquots of soluble cell lysates were electrophoretically separated on a denaturing polyacrylamide gel, transferred to nitrocellulose, and probed with antibodies to pUL17, the major capsid protein VP5, and a monoclonal antibody to pUL25.
We noted variable expression of pUL17 and pUL25 in clarified infected cell lysates. Specifically, the ratios of levels of pUL25 to pUL17 were higher in lysates of cells infected with the UL19 null (Δ19) virus and UL32 null (Δ32) virus compared to those in HSV-1(F)-infected cells (Fig. 1C versus Fig. 1A). We cannot know whether the discrepancies are specific effects related to each deleted gene or whether they reflect differences in expression of these proteins from different viral strains.
FIG. 1.
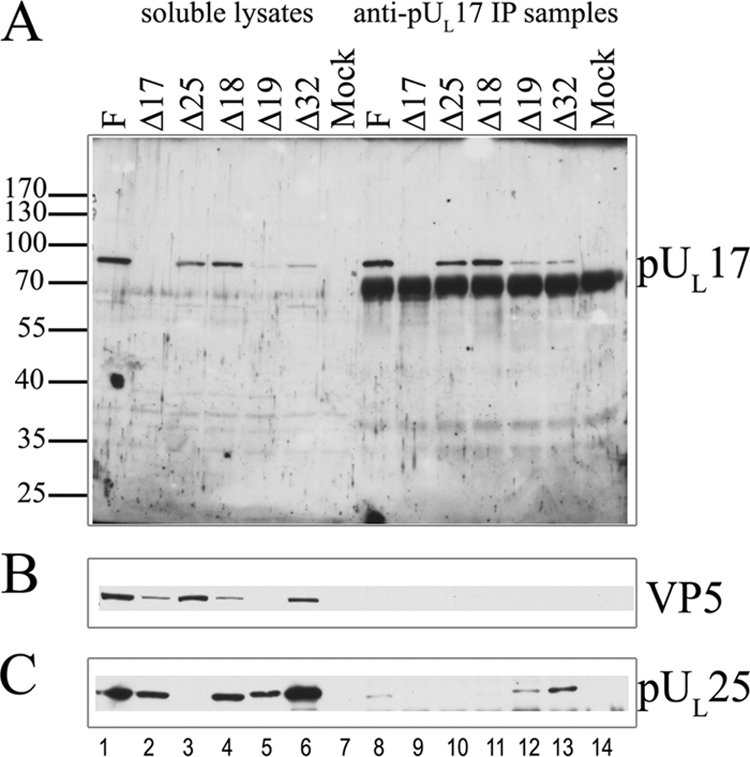
Coimmunoprecipitation of pUL25 with pUL17-specific antibody from lysates of cells infected with HSV-1(F) and capsid null viruses. Cells were mock infected or were infected with the viruses indicated above each lane. Lysates of these cells were reacted with pUL17-specific IgY antibody and a rabbit anti-IgY bridging antibody for 2 h, and immune complexes were purified, denatured in SDS, electrophoretically separated, transferred to nitrocellulose and probed with antibodies to pUL17 (A), VP5 (B), or pUL25 (C). Bound IgY was revealed by reaction with peroxidase-conjugated anti-IgY antibody, and signals were revealed by chemiluminescence. Lanes 1 to 7 contain samples of soluble lysates that were reacted with pUL17 antibody. Lanes 8 to 14 contain immunoprecipitation reactions from the corresponding lysates. The position of molecular weight markers in the gel and their sizes (in thousands) are indicated to the left. F, HSV-1(F); IP, immunoprecipitation.
Despite these caveats, and as shown in Fig. 1A, pUL17 was immunoprecipitated specifically from lysates of cells infected with HSV-1(F), Δ25, Δ18, Δ19, and Δ32 viruses at levels that reflected their relative amounts in the respective cell lysates. The absence of a signal from the lysates of cells infected with the UL17 deletion virus indicated that the signal represented the UL17 protein.
Immunoblotting pUL17 coimmunoprecipitated material with mouse monoclonal anti-pUL25 antibody indicated that pUL25 coimmunoprecipitated with pUL17 from lysates of cells infected with HSV-1(F), UL19 null (Δ19), and UL32 null (Δ32) viruses (Fig. 1C). Barely detectable levels of pUL25 were coimmunoprecipitated from lysates of cells infected with the UL18 null (Δ18) virus, suggesting that the UL18 gene-encoded VP23 augmented pUL17/pUL25 coimmunoprecipitation. In a control reaction, no detectable pUL25 was immunoprecipitated from lysates of Δ17 virus-infected cells despite high levels of pUL25 in soluble lysates, indicating that pUL25 was specifically coimmunoprecipitated through interactions with pUL17 (Fig. 1C).
We also wanted to determine whether or not major capsid proteins were associated with the pUL17/pUL25 complex. To address this question, we reprobed the pUL17 immunoprecipitated material with antibodies to the capsid protein VP5 (Fig. 1B). In no case were detectable levels of VP5 coimmunoprecipitated with pUL17 antibody.
Taken together, these data suggest that pUL17 and pUL25 interact independently of assembled capsids and the major capsid protein VP5, whereas VP23 either directly or indirectly facilitates the pUL17/pUL25 interaction.
Reciprocal coimmunoprecipitation of pUL17 with anti-pUL25 antibody.
To confirm the capsid-independent interaction of pUL17/pUL25, cells were infected with HSV-1(F), Δ17, Δ25, Δ18, Δ19, and Δ32 viruses, and lysates prepared at 16 h postinfection were reacted with a pUL25-specific monoclonal antibody, 25E10. The presence of various proteins in immunoprecipitated material was then determined by immunoblotting with monospecific antibodies. The results are shown in Fig. 2.
FIG. 2.
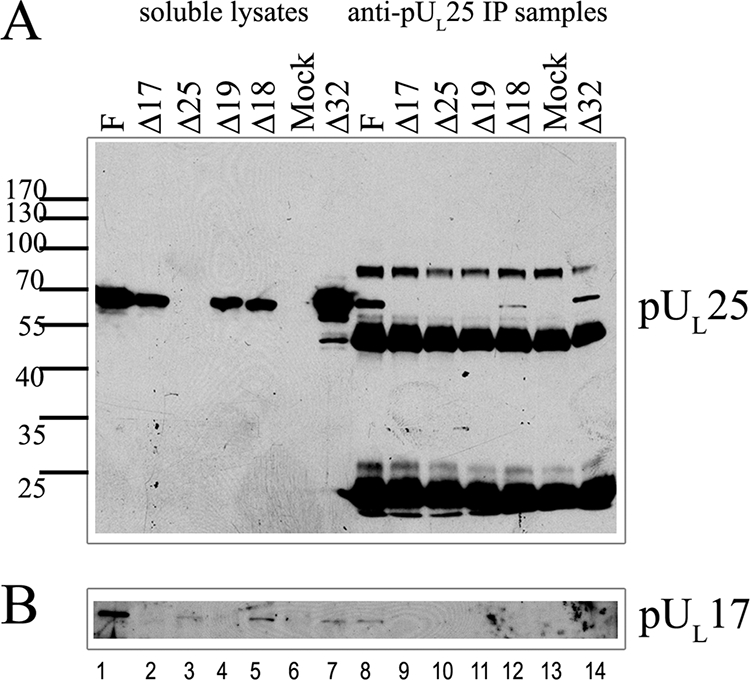
Coimmunoprecipitation of pUL17 with anti-pUL25 antibody. Cells were infected with the indicated viruses, and lysates were precleared and reacted with a monoclonal antibody directed against pUL25. Immunocomplexes were purified, electrophoretically separated, transferred to nitrocellulose, and probed with antibodies to pUL25 (A) or pUL17 (B). Bound immunoglobulins were revealed as indicated in the legend to Fig. 1. Lanes 1 to 7 contain samples of lysates used in the immunoprecipitation reactions. Lanes 8 to 14 contain immunoprecipitation reactions. F, HSV-1(F); IP, immunoprecipitation.
The levels of pUL25 were highest in lysates of cells infected with wild-type HSV-1(F) and the UL32 deletion virus, but pUL25 was also readily detected in lysates of cells infected with the UL17, UL18, and UL19 deletion viruses. Moreover, pUL25 was immunoprecipitated with its cognate antibody from HSV-1(F)-, Δ18 virus-, and Δ32 virus-infected cell lysates, although significantly less pUL25 was immunoprecipitated from the Δ18 virus-infected samples despite the fact that ample pUL25 was present in the corresponding infected cell lysate (Fig. 2A). Surprisingly, pUL25 was not immunoprecipitated to detectable levels with its cognate monoclonal antibody from Δ19 (VP5 null)-infected cell lysates. Thus, UL17, UL18, UL19, and UL32 genes encode functions that augment the immunoprecipitation of pUL25 with its cognate monoclonal antibody. One possibility to explain this result is that pUL25 adopts at least two conformations, one of which is reactive with monoclonal 25E10 antibody and one that is not reactive. It follows that UL17, UL18, UL19, and UL32 augment production of the immunoreactive pUL25 species.
When the 25E10-immunoprecipitated proteins on the nitrocellulose sheet were reacted with anti-pUL17 antibody, coimmunoprecipitation of pUL17 was noted only from HSV-1(F)-infected cell lysates (Fig. 2B). These data confirmed the interaction between pUL17 and pUL25 in lysates of cells infected with wild-type virus. Conclusions concerning the pUL17/pUL25 interaction in the Δ18 virus- and Δ19 virus-infected cell lysates were hampered by the poor immunoprecipitation of pUL25 with the monoclonal antibody 25E10 from those lysates and the relatively small amounts of pUL17-specific immunoreactivity detected in the clarified lysates (Fig. 2B).
To overcome the problems with the monoclonal 25E10 immunoprecipitation, we reacted infected cell lysates with this antibody overnight, and the presence of various proteins in the immunoprecipitated material was determined by immunoblotting. The results are shown in Fig. 3. Under these conditions, the monoclonal antibody 25E10 immunoprecipitated substantial amounts of pUL25 from lysates of cells infected with the UL18 deletion and UL17 deletion viruses and the wild-type viruses, HSV-1(F) and 17R. Thus, prolonged exposure to the monoclonal antibody rescued the ability of pUL25 to be immunoprecipitated with its cognate antibody from lysates of cells infected with the UL17 and UL18 deletion mutants.
FIG. 3.
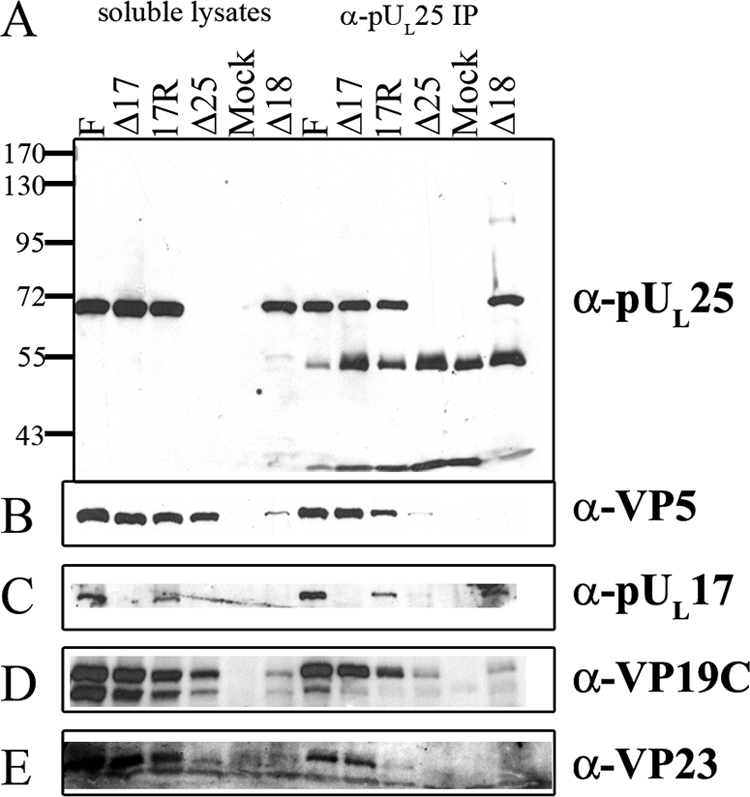
Coimmunoprecipitation of major capsid proteins and pUL17 with pUL25-specific antibody. Cells were infected with the indicated viruses, and lysates were prepared 18 h after infection. Immunoprecipitations were performed as described in the legend to Fig. 2 except that the precleared lysates were reacted with pUL25-specific antibody overnight. Immune complexes were purified, denatured, electrophoretically separated, and subjected to immunoblotting with antibodies to pUL25 (A), VP5 (B), pUL17 (C), VP19C (D), or VP23 (E). Lanes 1 to 6 contain samples of lysates, whereas lanes 7 to 12 contain immunoprecipitation reactions. The positions of molecular weight markers and their sizes (in thousands) are indicated to the left in panel A. F, HSV-1(F); IP, immunoprecipitation; α, anti.
Probing the 25E10-immunoprecipitated material with antibody to capsid proteins revealed that capsid proteins were not coimmunoprecipitated from lysates of cells infected with the UL25 null virus, although background levels of VP5 and VP19C were detectable. In contrast, the major capsid proteins VP5, VP23, and VP19C, as well as the minor capsid protein pUL17, were readily coimmunoprecipitated with the monoclonal antibody 25E10 from lysates of cells infected with wild-type viruses, HSV-1(F) and 17R. Moreover, the coimmunoprecipitation of VP5, VP23, and VP19C did not require pUL17 inasmuch as these proteins were immunoprecipitated with the pUL25-specific antibody from lysates of cells infected with the UL17 deletion virus. On the other hand, it is unclear whether the coimmunoprecipitation of major capsid proteins with pUL25 antibody reflects direct interactions, because the anti-pUL25 antibody could conceivably immunoprecipitate intact capsids. Potentially more revealing were the results obtained from viruses that could not form intact capsids. Specifically, the pUL25 antibody could coimmunoprecipitate pUL17 from lysates of cells infected with the UL18 null virus, whereas levels of VP19c and VP5 remained below or at background levels. We therefore conclude that pUL17 and pUL25 can interact in infected cells in the absence of intact capsids and the major capsid proteins.
To confirm these results, the same experiment was repeated, except that infected cell lysates were reacted overnight with the UL17-specific antibody. As shown in Fig. 4, and like the results obtained when the anti-pUL25 antibody was used, the UL17-specific antibody readily immunoprecipitated pUL17 and coimmunoprecipitated VP5 and VP23 from lysates of cells infected with wild-type viruses, HSV-1(F) and 17R. High background levels conferred by the IgY heavy chain precluded assessment of whether VP19C coimmunoprecipitated with these proteins. pUL17 was not detected in the lanes bearing 1/20 of the amount of cell lysates present in the immunoprecipitation reactions, indicating that the immunoprecipitation enriched pUL17 to detectable levels. Only background levels of VP5, VP23, and pUL25 were coimmunoprecipitated from lysates of cells infected with the UL17 null virus reacted with the UL17-specific antibody. Importantly, pUL25 was coimmunoprecipitated from lysates of cells infected with the UL18 deletion virus, whereas VP5 was not coimmunoprecipitated above background levels. We conclude that a complex of pUL17 and pUL25 can form in infected cells in the absence of intact capsids and that VP5 does not readily associate with this complex in the absence of VP23.
FIG. 4.
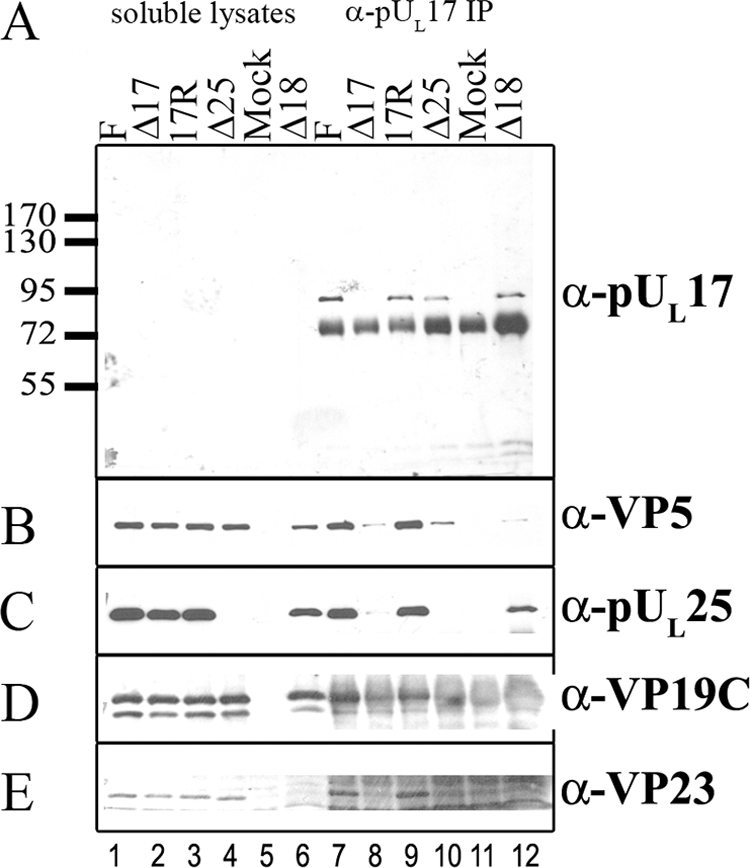
Coimmunoprecipitation of major capsid proteins and pUL25 with pUL17-specific antibody. Cells were infected with the indicated viruses, and lysates were prepared 18 h after infection. Immunoprecipitations were performed as described in the legend to Fig. 1 except that the precleared lysates were reacted with pUL17-specific antibody overnight. Immune complexes were purified, denatured, electrophoretically separated, and subjected to immunoblotting with antibodies to pUL17 (A), VP5 (B), pUL25 (C), VP19C (D), or VP23 (E). Lanes 1 to 6 contain samples of lysates, whereas lanes 7 to 12 contain immunoprecipitation reactions. The positions of molecular weight markers and their sizes (in thousands) are indicated to the left of panel A. F, HSV-1(F); IP, immunoprecipitation; α, anti.
Solubility of pUL17 in the absence of pUL25 and capsids.
The results shown in Fig. 2A suggest that less pUL17 was present in clarified lysates of cells infected with viruses lacking UL19 or UL18. To determine whether this reflected lower levels of pUL17 in cells infected with these mutant viruses, or a difference in solubility, cells infected with HSV-1(F) or the UL25, UL18, or UL19 null virus-infected cell lysates were denatured and subjected to immunoblotting with pUL17-specific antibodies. As a control for infection, the immunoblot was probed with antibody to ICP8, and antibody to lamin A/C served as a loading control. As shown in Fig. 5, low levels of pUL17 and pUL25 were detected in lysates of cells infected with the UL17 and UL25 null viruses, respectively. We speculate that these low levels reflect input protein that is associated with complemented virions during the initial infection because genetic revertants were not detected in the viral stocks used in the experiment (data not shown). In any event, despite the lower levels of pUL17 in soluble lysates, steady-state levels of pUL17 did not vary significantly among the total lysates of cells infected with the different viruses. We conclude that at least UL18, UL19, and UL25 contribute to the solubility of pUL17 seen in cells infected with wild-type viruses.
FIG. 5.

Immunoblot of total infected cell lysates probed with pUL17- and pUL25-specific antibodies. Cells were infected with the indicated viruses, and 18 h after infection, the cells were denatured in SDS-containing buffer. The proteins were electrophoretically separated, transferred to nitrocellulose, and probed with pUL17- and pUL25-specific antibodies (A) or ICP8- and lamin A/C-specific antibodies (B) as infection and loading controls, respectively. The exposure of the lamin A/C immunoblot was generated separately. For illustrative purposes, the lamin A/C immunoblot was superimposed on the ICP8 immunoblot at the approximate position of the lamin A/C signal. The positions of molecular weight markers are indicated to the left of each panel and correspond to molecular weights of (top to bottom) 170,000, 130,000, 95,000, 72,000, 55,000, and 43,000, respectively. 17, pUL17; 25, pUL25; F, HSV-1(F); LMN A/C, lamin A/C.
Distribution of pUL17 and pUL25 in the infected cell nucleus is codependent.
Having established pUL17 and pUL25 association, we next asked whether either protein affected the location of the other in infected cells. To assess this possibility, we infected Hep2 cells with 5.0 PFU of HSV-1(F), Δ17, or Δ25 virus per cell, fixed and permeabilized the cells at various times after infection, and reacted them with chicken polyclonal antibody to pUL17 and the 25E10 monoclonal antibody directed against pUL25. After extensive washing, bound immunoglobulins were revealed by reaction with the appropriately conjugated antibodies, and the cells were examined by confocal microscopy.
As shown in Fig. 6, upon infection with wild-type HSV-1(F) virus, pUL17- and pUL25-specific immunostaining colocalized extensively in nuclei at 12 and 16 h postinfection (Fig. 6C and F). Despite extensive colocalization, individual foci containing only pUL17 or pUL25 immunostaining were also observed. Cytoplasmic staining of both pUL17 and pUL25 increased slightly by 16 h after infection, although the great majority of immunoreactivity remained in the nucleus. Of the cytoplasmic signals, very little colocalized. The intranuclear compartment containing pUL25 and pUL17 also immunostained with ICP8-specific antibody (not shown, but see Fig. 8A to D). The distribution of pUL17 differed from that reported by others, in which pUL17 localized in larger foci, similar to previously termed assemblons that accumulate peripheral to the replication compartment (17, 42). Although we noted an association of pUL17 in assemblon-like foci in some cells, this was much less common than the localization of pUL17 in the replication compartment.
FIG. 6.

Intranuclear localization of pUL17/pUL25 requires expression of both proteins. Hep2 cells were infected with HSV-1(F) (A to I), the UL17 deletion virus (G to I), or the UL25 deletion virus (J to O). The cells were fixed and permeabilized at the indicated times and were reacted with antibodies to pUL17 and pUL25 and then with Texas Red-conjugated anti-chicken IgY and fluorescein-conjugated anti-mouse IgG. Images in the red and green channels were collected with an Olympus confocal microscope. The red channel indicating the position of pUL17 is shown in the leftmost column, while the channel corresponding to pUL25 is shown in the middle column. A merge of these two images is shown in the rightmost column, and convergence of the two signals is indicated by a yellow color. Relative to the images in other panels, fluorescence in panels G to I was increased by increasing the excitation laser power to clearly illustrate pUL25 distribution.
FIG. 8.

Localization of pUL17 and pUL25 in cells infected with viral mutants defective in nucleocapsid assembly. Hep2 cells were infected with the viruses indicated to the left of each row and were fixed and permeabilized at 14 h after infection. The cells were then immunostained with antibodies to pUL25, pUL17, and ICP8 as a marker of the viral DNA replication compartment. Bound immunoglobulins were recognized by the appropriate conjugates, and induced fluorescence was recorded by a confocal microscope by the use of identical settings for each image. The conjugates recognized antibodies for pUL25 (fluorescein isothiocyanate [FITC]; leftmost column), pUL17 (Texas Red; middle left column), or ICP8 (cyan5 pseudocolored white; right middle column). The rightmost column (merge) contains superimposed unaltered images from the three panels to the left. Single optical sections obtained near the middle of the cells are shown. F, HSV-1(F).
Immunostaining for either pUL17 or pUL25 in the absence of the other was different from that seen in cells infected with wild-type HSV-1(F) virus. Specifically, in Δ17 virus-infected cells, pUL25-specific immunostaining was generally at a much lower intensity (the fluorescence intensity in Fig. 6G to I was increased to illustrate the distribution pattern); the distributions of pUL25-specific immunoreactivity in Δ17 virus-infected cells were nearly equal between the nucleus and cytoplasm; and in cells infected with the UL25 null virus, much of the pUL17-specific immunoreactivity was mislocalized to intensely staining foci near the periphery of the nucleus at both 12 and 16 h postinfection. However, this distribution was more pronounced at the earlier (12 h) time point, presumably due to increased levels of immunoreactivity at 16 h that obscured the presence of the foci (Fig. 6J and M).
We conclude from these data that both pUL17 and pUL25 colocalize primarily in the nuclei of infected cells and that the localization of both proteins to nuclei is partially dependent on the interacting partner.
pUL17 colocalizes with capsids in Δ25 virus infection.
The pUL17-containing foci at the nuclear periphery of Δ25 virus-infected cells were reminiscent of capsid aggregates observed upon infection with a UL17 null virus (28). To determine whether the foci containing pUL17 were also associated with capsids, we immunostained Δ25 virus-infected cells with pUL17-specific antibody and a monoclonal antibody (8F5), which recognizes VP5 in capsid pentons (40). As shown in Fig. 7, HSV-1(F)-infected cells displayed both pUL17-specific and capsid-specific staining in the nuclear interior (Fig. 7A to C). However, in Δ25 virus-infected cells, both capsid- and pUL17-specific immunoreactivities were observed to colocalize at the nuclear periphery (Fig. 7D to F). Examination at higher magnification revealed that pUL17-specific and 8F5 immunostaining colocalized not only in the intensely staining peripheral regions, but also in smaller puncta distributed throughout the central intranuclear replication compartment. The smaller puncta were of a size and distribution similar to those of capsids (14). This result was consistent with the observation that pUL17 and VP5 coimmunoprecipitated (albeit weakly) when lysates of cells infected with the Δ25 virus were reacted with the pUL17 antibody overnight (Fig. 4). The observation was not consistent with the absence of pUL17/VP5 coimmunoprecipitation from lysates of UL25 null virus-infected cells when reacted with the pUL17 antibody for 2 h (Fig. 1). We favor the hypothesis that pUL17 interacts with capsids in the presence and absence of pUL25, but the interaction is of low affinity in the absence of pUL25 such that capsid proteins do not coimmunoprecipitate with pUL17 antibody efficiently. This assessment is consistent with the conclusions of others that each protein augments capsid association of the other (36).
FIG. 7.

Peripheral nuclear aggregates in cells infected with the UL25 null virus contain pUL17 and completely assembled capsids. Hep2 cells were infected with HSV-1(F) (A to C) or the UL25 deletion virus (D to I) and were fixed and permeabilized at 14 h after infection. The cells were then reacted with a mouse monoclonal antibody, 8F5, that recognizes pentons in intact capsids or pUL17-specific IgY. The cells were then reacted with Texas Red-conjugated anti-IgY and fluorescein-conjugated anti-mouse antibodies, and images were collected with an Olympus confocal microscope. The red channel indicating the position of pUL17 is shown in the left column. The middle column displays the fluorescent signal emitted by excitation of fluorescein. A merge of these images is shown in the rightmost column. Arrows indicate nuclear aggregates that stain with both antibodies. The white bars at the bottom of panels A, D, and G correspond to 5 μm in length.
Capsid formation, but not DNA encapsidation, aids nuclear localization of pUL17 and pUL25.
Since pUL17 and pUL25 are both capsid associated and necessary for DNA cleavage and packaging, we questioned whether the mislocalization of these proteins in Δ25 and Δ17 virus infections was a specific effect of each protein or a general effect of either capsid malformation or lack of DNA encapsidation. To distinguish between these possibilities, we determined the distributions of pUL17 and pUL25 upon infection with capsid null and DNA encapsidation-deficient viruses by indirect immunofluorescence.
To address the effect of capsid formation upon pUL17/pUL25 localization, we infected cells with the Δ18 or Δ19 viruses and fixed and immunostained them to reveal the distribution of pUL17 and pUL25 proteins within cells. Cells infected with wild-type HSV-1(F), Δ17, and Δ25 viruses were processed in parallel as controls, and all samples were stained with antibody against ICP8 to indicate that the cells were infected and to mark the DNA replication compartment. The results are shown in Fig. 8.
Both pUL17 and pUL25 immunostaining colocalized in the intranuclear replication compartment marked by ICP8 in cells infected with HSV-1(F) (Fig. 8A to D). In cellular nuclei infected with wild-type virus (not shown) or the UL18 or UL19 null viruses that immunostained to less intense levels, numerous small foci of pUL17 or pUL25 were discernible within the ICP8-containing compartment (Fig. 8). Many of these individual foci localized adjacent to but not coincident with similarly sized foci of intranuclear ICP8 (e.g., Fig. 8H and L).
Several differences in distribution and staining intensity of pUL17 and pUL25 were noted in cells infected with wild-type virus compared to those infected with the UL18 and UL19 null viruses. Specifically, we noted the following. (i) The relative amounts of cytoplasmic staining of both pUL17 and pUL25 were increased relative to that seen in cells infected with wild-type virus (Fig. 8E, I, F, and J). (ii) Overall, the immunostaining of pUL25 in cells infected with the UL17, UL18, and UL19 null viruses was diminished compared to that in cells infected with HSV-1(F) (Fig. 8E and I). (iii) pUL17 immunostaining was greatly diminished in cells infected with the UL18 and UL19 null viruses (Fig. 8F and J). The pUL17-specific immunostaining was somewhat more intense in cells infected with the UL25 null virus (Fig. 8V), although this may reflect high protein concentrations in the foci at the nuclear periphery rather than overall levels. (iv) Although the aberrant localizations of pUL17 were similar in cells infected with the UL18 and UL19 null viruses, the mislocalization of pUL17 into peripheral nuclear aggregates was seen only in Δ25 virus-infected cells (Fig. 8V).
Taken together, these data indicate that VP5 and VP23 (encoded by UL19 and UL18 genes, respectively) affect the immunoreactivity and nuclear localization of pUL17 and pUL25. These data suggest that major capsid proteins or capsid formation greatly affects the distribution of pUL17 and pUL25 in infected cells.
To test the hypothesis that encapsidation of viral DNA was involved in proper localization of pUL25 and pUL17, a similar analysis was performed using cells infected with a virus lacking pUL32, which is required for DNA cleavage and packaging but dispensable for formation of double-shelled capsids (18). In cells infected with the Δ32 virus (Fig. 8M to P), the pUL17-specific signal localized in the intranuclear replication compartment in a pattern similar to that seen in infections with the wild-type virus. Although pUL25-specific signals localized primarily in the nucleus, significant levels of pUL25-specific signal were also present in the cytoplasm. We also noted that pUL25 and pUL17 specific immunoreactivity was greater in Δ32 virus-infected cells compared to that seen in cells infected with the capsid null viruses. These levels of immunostaining were similar to that of cells infected with wild-type HSV-1(F). We conclude that UL32 augments nuclear localization of pUL25 during viral infection but does not detectably affect pUL17 localization.
To further test the role of DNA packaging in pUL25 and pUL17 localization, these proteins were assayed by indirect immunofluorescence in cells infected with a virus lacking the terminase subunit encoded by the UL15 gene, which is also required for DNA packaging. As shown in Fig. 9, pUL25 and pUL17 colocalized within the nuclei of cells infected with the UL15 null virus in a pattern similar to that of ICP8 and similar to that seen in cells infected with HSV-1(F) (Fig. 9D to F). The intensity of the immunostaining of both proteins was similar to that in cells infected with wild-type virus. We conclude from these studies that proper localization of pUL17 and pUL25 does not require the UL15 protein or the process of viral DNA cleavage and packaging.
FIG. 9.

Localization of pUL25 in the DNA replication compartment does not require successful DNA packaging. Hep2 cells were infected with HSV-1(F) (A to C) or a UL15 null mutant that does not package DNA (D to F). Cells were permeabilized and fixed at 16 h after infection and were immunostained with mouse anti-pUL25, chicken anti-pUL17, and, in panels D to F, a rabbit antibody to ICP8. The respective antibodies were recognized by reaction with FITC-conjugated anti-mouse IgG, Texas Red-conjugated anti-chicken IgY, and cyan5-conjugated anti-rabbit IgG. Fluorescence in separate channels was recorded using a confocal microscope. (A and D) pUL25-specific immunostaining. (B and E) pUL17-specific staining. (C) A merge of panels A and B. (F) ICP8-specific staining.
pUL17 and pUL25 partially colocalize in the absence of other viral proteins.
The immunoprecipitation experiments described above indicate that pUL17 immunoprecipitates with pUL25 independently of capsid assembly. Next, we asked whether expression of any other viral protein was necessary for pUL17/pUL25 colocalization. We therefore transfected Hep2 cells with expression constructs pcDNA3-UL17 and pcDNA3-UL25(pJB71), either alone or together. At 24 to 36 h posttransfection, the cells were fixed and immunostained with anti-pUL17 and anti-pUL25 antibodies. In a control experiment, cells were transfected with Lipofectamine without plasmid DNA. Representative data showing the most commonly observed distribution of pUL17 and pUL25 are shown in Fig. 10.
FIG. 10.

Partial colocalization of transiently expressed pUL17 and pUL25 in the cytoplasm of Hep2 cells in the absence of other viral proteins. Hep2 cells were transfected with pUL17 (FITC) and pUL25 (Texas Red) expression plasmids. Twenty-four hours later, the cells were permeabilized, fixed in paraformaldehyde, and reacted with chicken anti-pUL17 and mouse monoclonal antibodies directed against pUL25. Following extensive washing, the cells were reacted with fluorescein-conjugated anti-chicken IgY and anti-mouse IgG conjugated with Texas Red. The green and red channels were collected separately and represent pUL17 (A) and pUL25 (B). The images are merged in panel C.
The UL17 protein localized primarily in the nucleus (Fig. 10A), but a substantial amount of pUL17-specific immunofluorescence also localized in the cytoplasm in brightly staining foci. While much of the pUL25-specific immunostaining localized in the cytoplasm, many of the regions containing pUL25 also contained pUL17-specific immunostaining. These data indicate that pUL17 and pUL25 can colocalize in the cytoplasm of infected cells in the absence of other viral proteins. Inasmuch as pUL25 is normally located in the nucleus in infected cells (Fig. 6), these data lend further support to the hypothesis that functions in the infected cell other than those mediated by pUL17 are involved in recruiting or retaining pUL25 to the nucleus.
pUL17 and pUL25 coimmunoprecipitate in the absence of other viral proteins.
To prove that the colocalization of pUL17/pUL25 reflected an interaction between these proteins, we tested whether pUL25 could coimmunoprecipitate with pUL17. Preliminary experiments suggested that pUL17 was somewhat cytotoxic and mostly insoluble when expressed transiently in Hep2 cells (not shown). To overcome this difficulty, pUL17 with a C-terminal six-His tag was purified from insect cells as indicated in Materials and Methods. We then transfected Hep2 cells with either a plasmid encoding full-length UL25 (pJB71), an N-terminal UL25 truncation (amino acids [aa] 51 to 580), a C-terminal UL25 truncation (aa 1 to 444), or the expression vector pcDNA3 and collected the transfected cells at 24 h postinfection. Cells were lysed in RIPA buffer, and 1/20 of the sample was mixed with 2× SDS-PAGE sample buffer to indicate the amount of pUL25 expressed in the total cell lysate. Purified pUL17-His was added to the remainder of the clarified lysates, followed by reaction with the anti-pUL25 monoclonal antibody 25E10 for 2 h. Total cell lysates and immunoprecipitated material samples were then subjected to immunoblotting with anti-pUL25 and anti-pUL17 antibodies. The results are shown in Fig. 11.
FIG. 11.

Coimmunoprecipitation of pUL17 and pUL25 in the absence of other viral proteins. Hep2 cells were transfected with the indicated pUL25 expression plasmids or pcDNA3. Lysates of the transfected cells were prepared 24 h later and reacted with purified pUL17 fused at the C terminus with a histidine tag. After 2 h, the lysates were prepared and precleared. They were then reacted with pUL25-specific antibody, and immune complexes were purified, denatured, and electrophoretically separated on a denaturing polyacrylamide gel. The proteins were transferred to nitrocellulose and probed with pUL25- and pUL17-specific antibodies. (A) Immunoblot probed with pUL25-specific antibody. Lanes 1 to 4, total cellular lysates of Hep2 cells transfected with the indicated plasmids; lanes 5 to 9, proteins reacted with purified pUL17-His and immunoprecipitated with pUL25-specific antibody. The nature of the relevant protein expressed in each sample is indicated above each lane. (B) Immunoblot probed with pUL17-specific antibody. The nitrocellulose sheet used in panel A was stripped of bound immunoglobulin and probed with pUL17-specific antibody.
Large amounts of full-length, N-terminally truncated, and C-terminally truncated pUL25 from total transfected cell lysates were detected with anti-pUL25 antibody (Fig. 11A, lanes 1 to 3). The mouse IgG heavy chain was readily recognized in the lanes subjected to immunoprecipitation (Fig. 11A, lanes 5 to 8). Anti-pUL25 antibody immunoprecipitated each of these forms of pUL25, as revealed by a novel band migrating slower than the mouse heavy chain in reactions containing full-length pUL25 and containing truncated pUL25 bearing aa 51 to 580 (Fig. 11A, lanes 5 and 7). The truncation at aa 1 to 444 of pUL25 was found to migrate slightly faster than the heavy chain of mouse IgG, thus broadening the corresponding protein band (Fig. 11, lane 6).
The same immunoblot was stripped of antibody and reprobed with anti-pUL17 IgY antibody. Approximately 0.1 μg of purified, dialyzed pUL17-His was loaded as an immunoblotting control (Fig. 11B, lane 9). In the control experiment, very little pUL17 was noted in the control sample in which pcDNA3 was transfected into Hep2 cells, and the lysate was mixed with purified pUL17, and then immunoprecipitated with pUL25-specific antibody. Most importantly for the purposes of the experiment, coimmunoprecipitation of pUL17 was detected with all pUL25 forms (Fig. 11B, lanes 5 to 7), confirming an interaction between pUL17 and pUL25 in the absence of other viral proteins. This experiment also suggests that the first 50 and last 136 amino acids of pUL25 are dispensable for interaction with pUL17.
DISCUSSION
As revealed by coimmunoprecipitation and colocalization in the nuclei of infected cells, the presented data indicate the conditions under which pUL17 and pUL25 interact. Their interaction can occur in infected cells independently of capsid formation and in the absence of the major capsid protein VP5 or successful viral DNA cleavage and packaging. Moreover, VP5 was not detectably coimmunoprecipitated with pUL17 upon reaction with antibodies for 2 h. Because it is known that pUL17 and pUL25 interact with VP5 indirectly through their association with capsids, the lack of coimmunoprecipitated VP5 suggests that reactions at 2 h were highly stringent. In contrast, reaction with pUL17 or pUL25 antibodies overnight caused coimmunoprecipitation of all capsid proteins, suggesting that in these reactions, capsids or capsid assembly intermediates were immunoprecipitated.
In contrast to the results with the VP5 null virus, experiments with the VP23 null virus indicated that the triplex protein VP23 greatly augmented pUL17/pUL25 coimmunoprecipitation when reacted with pUL17 and pUL25 antibodies for 2 h. This observation indicates that VP23 augments the pUL17/pUL25 interaction and is consistent with the localization of pUL17/pUL25 on triplexes at the capsid surface (41). On the other hand, it is unclear whether this is a direct function of VP23 or whether the effect is mediated through VP19C, VP23's interaction partner. Further studies will be necessary to assess direct interactions between the pUL25/pUL17 complex and specific triplex components.
We also showed that the major capsid proteins, VP5 and VP23, were necessary for normal localization of pUL17 and pUL25 in infected cell nuclei, proficient immunoreactivity with pUL17 and pUL25-specific antibodies in indirect immunofluorescence assays, immunoprecipitation with a monoclonal antibody to pUL25 reacted for 2 h, and solubility of pUL17. Thus, the absence of capsid binding of the pUL17/pUL25 complex may alter the conformation of the complex and the biochemical behavior of pUL17 and pUL25. The observation that the pUL17/pUL25 complex requires the presence of capsids for their proper intranuclear localization is consistent with the possibility that pUL25 nuclear import is partly dependent on VP5, VP23, or other capsid components. Alternatively, it is possible that both proteins, when free from capsids, are exported to the cytoplasm as opposed to their nuclear retention when capsids are present. Further studies will be needed to distinguish between these possibilities.
Our observation that the first 50 amino acids of pUL25 are dispensable for interaction with pUL17, at least in transient expression assays, is of interest because these amino acids have been shown to be critical for the association of pUL25 with capsids (8). Thus, pUL25 domains that mediate interactions with capsids and pUL17 are experimentally separable. Based on the localization of the pUL25/pUL17 complex on the capsid surface, we would predict that the first 50 amino acids of pUL25 likely interact with capsid triplexes (41).
The data presented here also document the effects of pUL32 on the pUL17/pUL25 complex. This study is one of the first to implicate pUL32 in a specific aspect of the DNA packaging reaction—specifically, that it ensures proper nuclear localization of pUL25. Striking to us was the observation that in lysates of the UL32 null virus, the ratio of soluble pUL25 to pUL17 was greatly increased. Although pUL17 and pUL25 interacted in the absence of pUL32, pUL25 was found abundantly and aberrantly in the cytoplasm of cells infected with a UL32 deletion mutant. The precise mechanism by which pUL32 mediates its effects on pUL25 is unknown. The possibilities include effects on pUL25 expression or chaperone-like functions. Further studies will be necessary to distinguish between these and other possibilities.
Finally, the proper nuclear localization of both pUL17 and pUL25 each requires the expression of the corresponding interacting partner. Taken together, these studies reveal that multiple functions and proteins in the infected cell are required to mediate the proper behavior, interactions, and localization of the pUL17/pUL25 complex.
Acknowledgments
We thank Fred Homa for the UL25 expression plasmids, the UL25 deletion virus, and the cells for its propagation and Fred Homa and Jay Brown for the antibody to pUL25. We thank Gary Cohen and Roselyn Eisenberg for the antibodies to HSV-1 capsid proteins. We also thank Sandra Weller for the UL32 deletion virus and the cells required for its propagation, as well as Preshant Desai for the UL19 null virus and the complementing G5 cells.
This study was supported by NIH R01 grant GM50740 to J.D.B. and NIH training grant 5 T32 AI07618 to L.S.
Footnotes
Published ahead of print on 7 October 2009.
REFERENCES
- 1.Ali, M. A., B. Forghani, and E. M. Cantin. 1996. Characterization of an essential HSV-1 protein encoded by the UL25 gene reported to be involved in virus penetration and capsid assembly. Virology 216:278-283. [DOI] [PubMed] [Google Scholar]
- 2.al-Kobaisi, M. F., F. J. Rixon, I. McDougall, and V. G. Preston. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180:380-388. [DOI] [PubMed] [Google Scholar]
- 3.Baines, J. D., C. Cunningham, D. Nalwanga, and A. J. Davison. 1997. The UL15 gene of herpes simplex virus type 1 contains within its second exon a novel open reading frame that is translated in frame with the UL15 gene product. J. Virol. 71:2666-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baines, J. D., A. P. W. Poon, J. Rovnak, and B. Roizman. 1994. The UL15 gene of herpes simplex virus encodes two proteins and is required for cleavage of viral DNA. J. Virol. 68:8118-8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker, T. S., W. W. Newcomb, F. P. Booy, J. C. Brown, and A. C. Steven. 1990. Three-dimensional structures of maturable and abortive capsids of equine herpesvirus 1 from cryoelectron microscopy. J. Virol. 64:563-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Booy, F. P., W. W. Newcomb, B. L. Trus, J. C. Brown, T. S. Baker, and A. C. Steven. 1991. Liquid crystalline, phage-like packaging of encapsidated DNA in herpes simplex virus. Cell 64:1007-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Booy, F. P., B. L. Trus, W. W. Newcomb, J. C. Brown, J. F. Conway, and A. C. Steven. 1994. Finding a needle in a haystack: detection of a small protein (the 12-kDa VP26) in a large complex (the 200-MDa capsid of herpes simplex virus). Proc. Natl. Acad. Sci. USA 91:5652-5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cockrell, S. K., M. E. Sanchez, A. Erazo, and F. L. Homa. 2009. Role of the UL25 protein in herpes simplex virus DNA encapsidation. J. Virol. 83:47-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen, G. H., M. Ponce de Leon, H. Diggelmann, W. C. Lawrence, S. K. Vernon, and R. Eisenberg. 1980. Structural analysis of the capsid polypeptides of herpes simplex virus types 1 and 2. J. Virol. 34:521-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cunningham, C., and A. J. Davison. 1993. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 197:116-124. [DOI] [PubMed] [Google Scholar]
- 11.Davison, M. D., F. J. Rixon, and A. J. Davison. 1992. Identification of genes encoding two capsid proteins (VP24 and VP26) of herpes simplex type 1. J. Gen. Virol. 73:2709-2713. [DOI] [PubMed] [Google Scholar]
- 12.Deckman, I. C., M. Hagen, and P. J. McCann III. 1992. Herpes simplex virus type 1 protease expressed in Eschericia coli exhibits autoprocessing and specific cleavage of the ICP35 assembly protein. J. Virol. 66:7362-7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desai, P., N. A. DeLuca, J. C. Glorioso, and S. Person. 1993. Mutations in herpes simplex virus type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J. Virol. 67:1357-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forest, T., S. Barnard, and J. D. Baines. 2005. Active intranuclear movement of herpesvirus capsids. Nat. Cell Biol. 7:429-431. [DOI] [PubMed] [Google Scholar]
- 15.Gibson, W., and B. Roizman. 1972. Proteins specified by herpes simplex virus VIII. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 10:1044-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibson, W., and B. Roizman. 1974. Proteins specified by herpes simplex virus. Staining and radiolabeling properties of B capsids and virion proteins in polyacrylamide gels. J. Virol. 13:155-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goshima, F., D. Watanabe, H. Takakuwa, K. Wada, T. Daikoku, H. Yamada, and Y. Nishiyama. 2000. Herpes simplex virus UL17 protein is associated with B capsids and colocalizes with ICP35 and VP5 in infected cells. Arch. Virol. 145:417-426. [DOI] [PubMed] [Google Scholar]
- 18.Lamberti, C., and S. K. Weller. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 72:2463-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu, F., and B. Roizman. 1991. The herpes simplex virus 1 gene encoding a protease also contains within its coding domain the gene encoding the more abundant substrate. J. Virol. 65:5149-5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNab, A. R., P. Desai, S. Person, L. L. Roof, D. R. Thomsen, W. W. Newcomb, J. C. Brown, and F. L. Homa. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated DNA. J. Virol. 72:1060-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newcomb, W. W., F. L. Homa, D. R. Thomsen, F. P. Booy, B. L. Trus, A. C. Steven, J. V. Spencer, and J. C. Brown. 1996. Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free capsid formation. J. Mol. Biol. 263:432-446. [DOI] [PubMed] [Google Scholar]
- 22.Newcomb, W. W., R. M. Juhas, D. R. Thomsen, F. L. Homa, A. D. Burch, S. K. Weller, and J. C. Brown. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 75:10923-10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Newcomb, W. W., B. L. Trus, F. P. Booy, A. C. Steven, J. S. Wall, and S. C. Brown. 1993. Structure of the herpes simplex virus capsid molecular composition of the pentons and the triplexes. J. Mol. Biol. 232:499-511. [DOI] [PubMed] [Google Scholar]
- 24.Newcomb, W. W., F. L. Homa, and J. C. Brown. 2006. Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J. Virol. 80:6286-6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Person, S., S. Laquerre, P. Desai, and J. Hempel. 1993. Herpes simplex virus type 1 capsid protein, VP21, originates within the UL26 open reading frame. J. Gen. Virol. 74:2269-2273. [DOI] [PubMed] [Google Scholar]
- 26.Poon, A. P. W., and B. Roizman. 1993. Characterization of a temperature-sensitive mutant of the UL15 open reading frame of herpes simplex virus 1. J. Virol. 67:4497-4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roizman, B., and D. Furlong. 1974. The replication of herpesviruses, p. 229-403. In H. Fraenkel-Conrat and R. R. Wagner (ed.), Comprehensive virology. Plenum Press, New York, NY.
- 28.Salmon, B., C. Cunningham, A. J. Davison, W. J. Harris, and J. D. Baines. 1998. The herpes simplex virus 1 UL17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J. Virol. 72:3779-3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schrag, J. D., B. V. Prasad, F. J. Rixon, and W. Chiu. 1989. Three-dimensional structure of the HSV1 nucleocapsid. Cell 56:651-660. [DOI] [PubMed] [Google Scholar]
- 30.Sheaffer, A. K., W. W. Newcomb, M. Gao, D. Yu, S. K. Weller, J. C. Brown, and D. J. Tenney. 2001. Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J. Virol. 75:687-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steven, A. C., and P. G. Spear. 1998. Herpesvirus capsid assembly and envelopment, p. 312-351. In W. Chiu, R. M. Burnett, and R. L. Garcea (ed.), Structural biology of viruses. Oxford Press, New York, NY.
- 32.Stow, N. D. 2001. Packaging of genomic and amplicon DNA by the herpes simplex virus type 1 UL25-null mutant KUL25NS. J. Virol. 75:10755-10765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka, M., H. Kagawa, Y. Yamanashi, T. Sata, and Y. Kawaguchi. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taus, N. S., B. Salmon, and J. D. Baines. 1998. The herpes simplex virus 1 UL17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology 252:115-125. [DOI] [PubMed] [Google Scholar]
- 35.Tengelsen, L. A., N. E. Pedersen, P. R. Shaver, M. W. Wathen, and F. L. Homa. 1993. Herpes simplex virus type 1 DNA cleavage and capsidation require the product of the UL28 gene: isolation and characterization of two UL28 deletion mutants. J. Virol. 67:3470-3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thurlow, J. K., M. Murphy, N. D. Stow, and V. G. Preston. 2006. Herpes simplex virus type 1 DNA-packaging protein UL17 is required for efficient binding of UL25 to capsids. J. Virol. 80:2118-2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thurlow, J. K., F. J. Rixon, M. Murphy, P. Targett-Adams, M. Hughes, and V. G. Preston. 2005. The herpes simplex virus type 1 DNA packaging protein UL17 is a virion protein that is present in both the capsid and the tegument compartments. J. Virol. 79:150-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trus, B. L., F. P. Booy, W. W. Newcomb, J. C. Brown, F. L. Homa, D. R. Thomsen, and A. C. Steven. 1996. The herpes simplex virus procapsid: structure, conformational changes upon maturation, and roles of the triplex proteins VP19C and VP23 in assembly. J. Mol. Biol. 263:447-462. [DOI] [PubMed] [Google Scholar]
- 39.Trus, B. L., N. Cheng, W. W. Newcomb, F. L. Homa, J. C. Brown, and A. C. Steven. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 78:12668-12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trus, B. L., W. W. Newcomb, F. P. Booy, J. C. Brown, and A. C. Steven. 1992. Distinct monoclonal antibodies separately label the hexons or the pentons of herpes simplex virus capsid. Proc. Natl. Acad. Sci. USA 89:11508-11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trus, B. L., W. W. Newcomb, N. Cheng, G. Cardone, L. Marekov, F. L. Homa, J. C. Brown, and A. C. Steven. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol. Cell 26:479-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ward, P. L., W. O. Ogle, and B. Roizman. 1996. Assemblons: nuclear structures defined by aggregation of immature capsids and some tegument proteins of herpes simplex virus 1. J. Virol. 70:4623-4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wildy, P., W. C. Russell, and R. W. Horne. 1960. The morphology of herpes virus. Virology 12:204-222. [DOI] [PubMed] [Google Scholar]
- 44.Wills, E., L. Scholtes, and J. D. Baines. 2006. Herpes simplex virus 1 DNA packaging proteins encoded by UL6, UL15, UL17, UL28, and UL33 are located on the external surface of the viral capsid. J. Virol. 80:10894-10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang, K., F. Homa, and J. D. Baines. 2007. Putative terminase subunits of herpes simplex virus 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J. Virol. 81:6419-6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu, D., A. K. Shaeffer, D. J. Tenny, and S. K. Weller. 1997. Characterization of ICP6::lacZ insertion mutants of the UL15 gene of herpes simplex virus type 1 reveals the translation of two proteins. J. Virol. 71:2656-2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou, Z. H., M. Dougherty, J. Jakana, J. He, F. J. Rixon, and W. Chiu. 2000. Seeing the herpesvirus capsid at 8.5 A. Science 288:877-880. [DOI] [PubMed] [Google Scholar]
- 48.Zhou, Z. H., B. V. Prasad, J. Jakana, F. J. Rixon, and W. Chiu. 1994. Protein subunit structures in herpes simplex virus A-capsid determined from 400 kV spot-scan electron cryomicroscopy. J. Mol. Biol. 242:456-469. [DOI] [PubMed] [Google Scholar]