

Abstract
The original annotation of the vaccinia virus (VACV) genome was limited to open reading frames (ORFs) of at least 65 amino acids. Here, we characterized a 35-amino-acid ORF (O3L) located between ORFs O2L and I1L. ORFs similar in length to O3L were found at the same genetic locus in all vertebrate poxviruses. Although amino acid identities were low, the presence of a characteristic N-terminal hydrophobic domain strongly suggested that the other poxvirus genes were orthologs. Further studies demonstrated that the O3 protein was expressed at late times after infection and incorporated into the membrane of the mature virion. An O3L deletion mutant was barely viable, producing tiny plaques and a 3-log reduction in infectious progeny. A mutant VACV with a regulated O3L gene had a similar phenotype in the absence of inducer. There was no apparent defect in virus morphogenesis, though O3-deficient virus had low infectivity. The impairment was shown to be at the stage of virus entry, as cores were not detected in the cytoplasm after virus adsorption. Furthermore, O3-deficient virus did not induce fusion of infected cells when triggered by low pH. These characteristics are hallmarks of a group of proteins that form the entry/fusion complex (EFC). Affinity purification experiments demonstrated an association of O3 with EFC proteins. In addition, the assembly or stability of the EFC was impaired when expression of O3 was repressed. Thus, O3 is the newest recognized component of the EFC and the smallest VACV protein shown to have a function.
Vaccinia virus (VACV), the best-studied member of the poxvirus family of cytoplasmic DNA viruses, encodes ∼200 genes, some of which are still uncharacterized (27). The focus of the present study is VACV O3L, a short 35-amino-acid open reading frame (ORF) that was recognized by homology to a 41-amino-acid ORF in molluscum contagiosum virus (37) but not previously investigated. Here, we show that O3L is conserved in all chordopoxviruses, expressed late in infection, and involved in cell entry.
Considerable information regarding VACV entry has been obtained during the past several years (28). There are two related infectious forms of VACV: the mature virion (MV) and the enveloped virions (EV). The MV is comprised of a lipoprotein membrane enclosing a nucleoprotein core, whereas the EV has an additional outer membrane that must be disrupted before fusion can occur (24). The MV can enter cells either by fusion at the plasma membrane (7) or by a low-pH-mediated endosomal route involving macropinocytosis (20, 26, 44). Regardless of which route is used, the ability of VACV to enter cells depends on a large number of proteins in the MV membrane that form or are associated with the entry/fusion complex (EFC) (39). Using genetic and biochemical methods, 11 entry/fusion proteins have been identified: A16 (33), A21 (43), A28 (40), F9 (4), G3 (21), G9 (32), H2 (38), I2 (31), J5 (39), L1 (3), and L5 (42). Eight of these proteins (A16, A21, A28, G3, G9, H2, J5, and L5) comprise the EFC, which depends on multiple interactions for assembly or stability. Although the structure of the EFC remains to be elucidated, there is evidence for direct interactions between A28 and H2 (30) and between A16 and G9 (50). An additional role for A16 and G9 involves an interaction with the A56/K2 heterodimer, which is present on the surface of infected cells, to prevent spontaneous cell-cell fusion and superinfection by progeny virus (45, 46, 48-50). Binding of L1 to an unidentified cell receptor has been suggested (16). Roles in membrane fusion have also been considered for A17 and A27 (23).
Here we provide physical and functional evidence that O3 (VACWR069.5) is an integral component of the EFC and participates in virus entry and membrane fusion. With just 35 amino acids, O3 is the smallest VACV protein with a defined function.
MATERIALS AND METHODS
Cells and virus.
Cells were maintained in minimum essential medium with Earle's salts supplemented with 10% fetal bovine serum, 100 U of penicillin, and 100 μg of streptomycin per ml (Quality Biologicals, Gaithersburg, MD). The Western Reserve (WR) strain of VACV (ATTC VR1354) and the recombinant virus vT7LacOI (1) were propagated as described previously (12). MVs were purified by sedimentation twice through 36% sucrose cushions and banding once in a 25 to 40% sucrose density gradient (13).
Plasmid and recombinant VACV construction.
Overlap PCR was used to assemble DNA segments for construction of plasmids and recombinant VACV. Transfections were carried out using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Recombinant viruses were clonally purified as described previously (14), and their structures were verified by PCR and DNA sequencing of relevant segments.
Plaque assay and virus yield determinations.
For plaque assays, BS-C-1 monolayers in six-well tissue culture plates were infected with 10-fold serial dilutions of virus. After 1 h of adsorption, the medium containing unbound virus was removed and replaced with medium containing 0.5% methylcellulose with or without 50 μM isopropyl-β-d-1-thiogalactopyranoside (IPTG). After 48 h, the infected cells were stained with crystal violet and the plaques were counted.
For comparative determination of virus yield, BS-C-1 or RK13 cells in 24-well dishes were incubated for 1 h with equal numbers of viral particles based on the light scattering at A260. After adsorption, the medium was removed and the cells were washed and incubated in medium with or without 50 μM IPTG. Cells were harvested and lysed by freeze-thawing. Virus titers were determined by plaque assay in BS-C-1 cells in the presence of 50 μM IPTG.
Western blotting.
Whole-cell lysates or purified proteins were resolved by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) on 4 to 12% Novex NuPAGE acrylamide gels with 2-(N-morpholino)ethanesulfonic acid-SDS running buffer and transferred to nitrocellulose membranes using mini iBlot gel transfer stacks (Invitrogen). The membrane was blocked with 5% nonfat milk in phosphate-buffered saline (PBS) containing 0.05% Tween 20 and then incubated with a primary antibody for 1 h at room temperature and washed with PBS containing Tween 20 followed by PBS without detergent. For chemiluminescence detection, the appropriate secondary antibody conjugated with horseradish peroxidase (Pierce, Rockford, IL) was added and the blot was washed and developed using Dura or Femto chemiluminescent substrate (Pierce). For fluorescent detection, donkey anti-mouse IRDye 680/800 and donkey anti-rabbit IRDye 680/800 were used and developed using a LI-COR Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE). To reprobe the blots with additional antibodies, the nitrocellulose membrane was stripped at 55°C for 20 min using Restore buffer (Pierce) or Newblot Nitro stripping buffer (LI-COR). Mouse monoclonal antibody against human influenza virus hemagglutinin (HA) epitope tag was obtained from Covance (Princeton, NJ). Rabbit polyclonal antibodies against A3 (R. Doms and B. Moss, unpublished data), A4 (10), A16 (33), A17 (51), A21 (43), A26 (19), A27 (15), A28 (29), F9 (4), L1 (25), and L5 (42) and mouse monoclonal antibody to D8 (34) were used.
Protein IP.
For immunopurification (IP), BS-C-1 or HeLa cells were infected with recombinant viruses expressing affinity-tagged proteins. After 24 h, cells were harvested and then lysed in 50 mM Tris (pH 8.0), 200 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail (Roche, Indianapolis, IN) for 1 h at 4°C. Soluble extract obtained by centrifugation of the lysate at 15,000 × g was incubated with appropriate affinity beads for 2 h at 4°C. The beads were washed thoroughly with the same lysis buffer, and the remaining bound proteins were eluted with either lithium dodecyl sulfate sample loading buffer or buffer containing 1 mg/ml d-biotin when the affinity tag was streptavidin-binding peptide. Input, unbound, and bound proteins were separated by SDS-PAGE, transferred to a membrane, and probed with relevant antibodies.
Detergent extraction of purified virions.
Proteins associated with the MV membrane were extracted by incubating the purified virions in 50 mM Tris (pH 8.0) containing 150 mM NaCl and 1% Nonidet P-40 detergent in the absence and presence of 50 mM dithiothreitol (DTT) for 1 h at 37°C. Soluble and insoluble materials were separated by centrifugation at 15,000 × g for 30 min at 4°C, resolved by SDS-PAGE in the absence of reducing agent, and analyzed by Western blotting.
Confocal microscopy.
HeLa cells grown on coverslips were infected for 18 h or the times specified in the figure legends, fixed with 4% paraformaldehyde, and permeabilized with PBS containing 0.1% Triton X-100. The sample was blocked with 10% fetal bovine serum, followed by incubation with primary antibodies in serum for 1 h. Cells were washed with PBS and incubated with appropriate secondary antibodies conjugated to dyes (Molecular Probes, Eugene, OR) for an additional 1 h. Coverslips were washed and mounted on a glass slide using Prolong Gold (Invitrogen). For the viral core entry assay, HeLa cells grown on coverslips were infected with purified virions at 4°C for 1 h. The cells were either fixed after absorption or incubated for 2 h at 37°C in the presence of 300 μg/ml cycloheximide and fixed. Virion membrane or cores were labeled with a mouse monoclonal antibody 7D11 to L1 (52) and rabbit polyclonal antibody to A4, followed by fluorescent secondary antibodies. The number of cores was quantified with Imaris software (Bitplane Scientific Software, St. Paul, MN).
Electron microscopy.
For conventional transmission electron microscopy, infected BS-C-1 cells in 60-mm-diameter wells were fixed with 2% glutaraldehyde and embedded in EmBed-182 resin (Electron Microscopy Sciences, Hatfield, PA). Specimens were viewed with FEI-CM100 and FEI Tecnai Spirit transmission electron microscopes (FEI, Hillsboro, OR).
RESULTS
Homologs of the O3L ORF are present in all chordopoxviruses.
Due to small size, the O3L ORF was not originally annotated in VACV (17). However, it was subsequently identified by low homology to an ORF in molluscum contagiosum virus (37). Since gene order is highly conserved in chordopoxviruses, we examined representatives of each genus for a short ORF at the same genetic locus as O3L (Fig. 1A). ORFs from 29 to 48 amino acids were found in each case. Although the amino acid sequences were poorly conserved (Fig. 1B), each contained a helical hydrophobic domain near the N terminus, as shown for O3L (Fig. 1C), suggesting that they are orthologs. The strategy used to identify chordopoxvirus orthologs of O3 could not be used for entomopoxviruses because of the different arrangement of their genes.
FIG. 1.

Computational analysis of the O3L ORF. (A) Diagram of the VACV WR genome indicating ORFs from O1L to I2L. (B) Clustal alignment of VACV O3L ORF with orthologs from representative members of each genus of the Chordopoxvirinae. VACV, vaccinia virus strain WR; VARV, variola virus strain Bangladesh 1975 v75-55-Banu; SWPV, swinepox virus strain Nebraska 17077-99; SPPV, sheeppox virus strain NISKHI; MYXV, myxoma virus strain Lausanne; YMTV, Yaba monkey tumor virus strain Amano; FWPV, fowlpox virus strain Iowa; MOCV, molluscum contagiosum virus strain subtype 1; ORV, Orf virus strain OV-SA00. Amino acid residues conserved in all viruses are denoted by asterisks, those conserved in all but one virus are denoted by colons, and similar amino acids are denoted by periods. (B) Kyte/Doolittle hydrophilicity plot of O3 amino acid sequence analyzed using MacVector 10.6.0 software.
The characteristic TAAATG sequence (9) overlapping the start of the O3L ORF suggested that it was expressed at late times after infection. As we wished to experimentally confirm this prediction but had no antibody, we constructed vO3-HA, a recombinant VACV with DNA encoding the influenza virus HA epitope of 9 amino acids at the C terminus of the O3L ORF. To maintain the normal transcriptional regulation of O3L, neither the genetic locus nor the sequences immediately upstream of the ORF were altered. To aid in isolating the recombinant virus, DNA encoding green fluorescent protein (GFP) with the VACV P11 promoter was inserted in the rightwards direction within the intergenic region between end of O3L and O2L. BS-C-1 cells were infected with vO3-HA, harvested at several times, and analyzed by SDS-PAGE and Western blotting. Expression of the well-characterized A3 late protein served as a reference. A small protein that reacted with antibody to the HA tag and migrated just above the dye front was detected at 6 h after infection and increased at later times, similar to that of A3 (Fig. 2A). Moreover, neither protein was synthesized when cells were infected in the presence of the DNA synthesis inhibitor AraC (Fig. 2A). Thus, the O3L ORF was expressed as a late postreplicative protein.
FIG. 2.

Temporal expression and localization of O3. (A) Kinetics of expression of O3. BS-C-1 cells were infected with vO3-HA, harvested at the indicated times, and analyzed by SDS-PAGE and Western blotting with anti-HA antibody. Lysates of cells infected in the presence of AraC and harvested at 24 h were analyzed on the same gel. The blot was stripped and probed with antibody to the A3 protein and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control. Proteins were detected by chemiluminescence. (B) Confocal microscopy. HeLa cells grown on coverslips were infected with either vO3-HA or control vH7-FS (36). After 18 h, the cells were stained with antibodies against L1 and the HA epitope tag on O3 and corresponding fluorescent dye-conjugated secondary antibodies. DAPI, 4′,6-diamidino-2-phenylindole. (C) Association of O3 with purified virions. MVs were purified from cells infected with vO3-HA, incubated at 37°C with buffers containing the indicated components, and centrifuged to obtain soluble (S) pellet (P) fractions. The pellets were resuspended in the same volume as the soluble fraction, and equivalent amounts were analyzed by SDS-PAGE and Western blotting as in panel A. DTT, dithiothreitol.
O3 localizes to cytoplasmic viral factories and is associated with the virion membrane.
The N-terminal hydrophobic domain suggested that O3 would be associated with cellular or viral membranes. The location of O3 in cells infected with vO3-HA was analyzed by confocal microscopy. At 18 h after infection, anti-HA antibody detected O3 predominantly in viral factories, similar to VACV L1 MV membrane protein (Fig. 2B). As a control for the specificity of the anti-HA antibody staining, cells were also infected with vH7-FS, a recombinant VACV chosen because it lacks the HA epitope (36); only a faint background was noted (Fig. 2B).
The late expression, viral factory localization, and predicted transmembrane domain suggested that O3 might be a component of the MV membrane. To investigate such an association, virions from BS-C-1 cells infected with vO3-HA were purified by sedimentation through two successive sucrose cushions followed by banding in a 20 to 40% sucrose density gradient. The purified virions were treated with buffer alone, with NP-40 detergent, or with detergent and dithiothreitol. Soluble and insoluble fractions were separated by centrifugation and analyzed by Western blotting. O3 remained associated with purified virions after incubation with Tris and NaCl but was partially extracted with NP-40 detergent and completely extracted with detergent and reducing agent (Fig. 2C). This procedure, for distinction of membrane and core proteins, was validated by analyzing the distribution of the L1 membrane and A3 core proteins in the same samples (Fig. 2C). The similar extraction of O3 and L1 suggested that O3 is an MV membrane protein. In addition, the mobility of O3 was unaltered by reducing agent, indicating the absence of intermolecular disulfide bonds.
O3 is required for efficient VACV replication.
The conservation of the O3L ORF in all chordopoxviruses and its presence in the MV membrane suggested that the protein has a role in replication. To investigate such a role, homologous recombination was used to delete the O3L ORF by transfection of a GFP expression cassette flanked by O2L and O1L sequences into cells infected with VACV WR. Tiny green plaques were detected, and the vO3Δ deletion mutant was clonally purified. PCR and DNA sequencing confirmed the absence of the O3L ORF. The small-plaque phenotype of vO3Δ in BS-C-1 cells is shown in Fig. 3A. In addition, there was a 3-log reduction in 24-h virus yield relative to the parental virus (Fig. 3B).
FIG. 3.
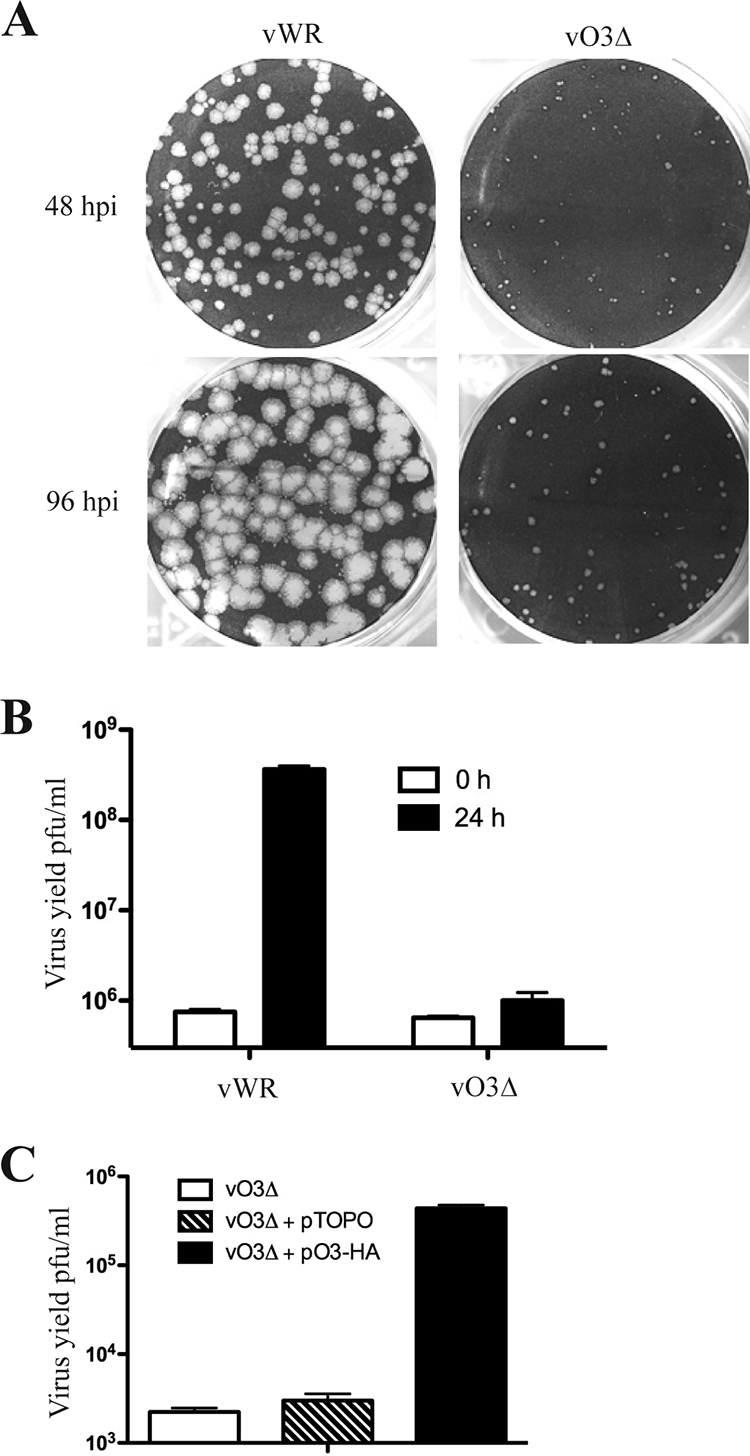
Characterization of the O3L deletion mutant. (A) Plaque phenotypes of VACV WR and vO3Δ. BS-C-1 monolayers were infected with either VACV WR (vWR) or vO3Δ and incubated for 48 or 96 h (postinfection [hpi]) with a methylcellulose overlay and stained with crystal violet to visualize the plaques. (B) Yields of VACV WR and vO3Δ. RK13 cells were infected with VACV WR and vO3Δ at 3 PFU per cell. After 1 h of adsorption, cells were washed to remove the inoculum and harvested immediately or 24 h postinfection. Virus titers were determined by plaque assay in BS-C-1 cells. (C) Transcomplementation of vO3Δ. BS-C-1 cells were infected with vO3Δ and transfected with either the empty vector or plasmid expressing O3-HA under the control of the natural O3L promoter. Cells were harvested after 24 h, and the virus titers were determined by plaque assay.
A complementation experiment was carried out to prove that the deficiency in virus replication was due to the deletion of O3L and not to unrelated mutations. RK13 cells were infected with vO3Δ and transfected with a plasmid expressing O3 from its natural promoter or with the empty vector plasmid. Expression of O3 increased the virus yield by more than 2 logs, whereas the vector plasmid had no effect (Fig. 3C).
Phenotype of an O3 inducible virus is similar to that of vO3Δ.
While making vO3Δ, we were also constructing a mutant with an inducible O3L ORF. vT7LacOi, containing the T7 RNA polymerase gene regulated by the Escherichia coli lac operator and the lac repressor gene with a constitutive promoter (1), was used as the parent virus. The endogenous O3L ORF was replaced with an HA-tagged O3L ORF under the control of T7 promoter and lac operator with an adjacent GFP gene to enable selection of recombinant virus. In this construct, we reversed the original orientation of the O3L ORF to prevent read-through of neighboring genes and enhance the stringency of repression. In the absence of inducer, the O3 inducible mutant virus, vO3-HAi, made small plaques (Fig. 4A) similar to those made by the deletion mutant. The dependency of IPTG on vO3-HAi virus replication was determined by a virus yield experiment. An optimal yield was obtained with 20 μM IPTG (Fig. 4B).
FIG. 4.

Characterization of the O3 inducible mutant virus. (A) Plaque phenotypes of vO3-HAi virus. BS-C-1 monolayers were infected with vO3-HAi and incubated in the absence or presence of 50 μM IPTG with a methylcellulose overlay for 48 h. Plaques were visualized by staining with crystal violet. (B) IPTG dependence of vO3-HAi replication. BS-C-1 cells were infected with 3 PFU per cell of either VACV WR (vWR) or vO3-HAi and incubated with the indicated concentrations of IPTG. After 24 h, the virus yields were determined by plaque assay. (C) Plaque size of vO3-HAi in different cell lines. Confluent cells were infected with vO3-HAi in the absence or presence of inducer for 24 h. Plates were viewed with a fluorescence microscope to show GFP expression, and plaques of a typical size are shown. Cell lines are indicated on the left.
To determine whether the requirement for O3 is host cell dependent, eight different cell lines were infected with vO3-HAi in the absence and presence of 50 μM IPTG. Advantage was taken of the presence of the GFP reporter gene to ascertain virus replication and spread by fluorescence microscopy. Omission of IPTG greatly reduced plaque size in each cell type (Fig. 4C).
The inducible virus had some technical advantages over the deletion mutant. Most importantly, vO3-HAi could be grown to high yields in the presence of inducer. Second, the consequence of O3 expression could be determined simply by adding or withholding IPTG, whereas the deletion mutant had to be compared with wild-type virus. There was, however, one major difference between the two types of mutants: whereas O3 was entirely absent from the deletion mutant, the stocks of vO3-HAi contained O3 because they were produced in the presence of IPTG unless otherwise mentioned. Consequently, omission of IPTG only prevented de novo synthesis of O3 following infection.
O3 is not required for VACV morphogenesis.
VACV morphogenesis involves a complex series of events that requires cleavage of the A17 membrane protein and several core proteins by the I7 protease (2, 5). Failure of cleavage is a sign of a defect in morphogenesis (22). Cells were infected with vO3-HAi in the presence and absence of IPTG or with the parent vT7LacOI, and the lysates were analyzed by Western blotting with antibodies to A17 and the A3 core protein. In each case, the lowest band represents the processed form of the protein and the upper band or bands represent precursor (Fig. 5A). The pattern of bands was similar regardless of whether IPTG was present or absent, suggesting that protein cleavage and morphogenesis were not prevented by diminished O3.
FIG. 5.

Proteolytic processing and morphogenesis of VACV in the absence of O3 expression. (A) Proteolytic processing of membrane and core proteins. BS-C-1 cells were infected with vO3-HAi in the presence or absence of 50 μM IPTG, the parental virus vT7LacOI, or vO3Δ. After 24 h, lysates were prepared and analyzed by SDS-PAGE and Western blotting with antibodies to the HA tag on O3, the membrane protein A17, the core protein A3, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control. Bands were visualized by chemiluminescence. (B) Electron microscopy of BS-C-1 cells infected with vO3-HAi in the absence of IPTG. After 20 h, ultrathin cell sections were prepared for viewing with a transmission electron microscope. IV, immature virions; WV, wrapped virions.
The deletion mutant, vO3Δ, was analyzed in parallel with vO3-HAi. For comparison, similar optical density (OD) units of purified virions were used. Although processing of A17 and A3 also occurred in the cells infected with vO3Δ, the amounts of protein were much smaller than in cells infected with vO3-HAi (Fig. 5A). As mentioned above, the vO3-HAi virions contained O3 but the vO3Δ virions did not. As will be shown below, the presence of O3 in the MV membrane is important for virus entry.
To further investigate morphogenesis, cells were infected with vO3-HAi in the presence or absence of IPTG and examined by transmission electron microscopy. Mature as well as immature forms of VACV were seen in the absence (Fig. 5B) as well as in the presence (not shown) of IPTG. Thus, both biochemical and microscopic studies suggested that O3 was not required for VACV morphogenesis.
O3 null mutants have low infectivity.
Since O3 did not affect the morphogenesis of VACV, a role of O3 in virion infectivity was examined. Virions were purified from cells infected with VACV WR and vO3Δ in the absence of IPTG, whereas vO3-HAi was made in the presence and absence of the inducer. The specific infectivity of the virions was determined by comparing the OD with the PFU. When the specific infectivity of virions was calculated, the amount of vO3Δ was 1,000-fold less than that of VACV WR, whereas the difference was 20-fold for vO3-HAi virus made in the absence of IPTG but similar to that of WR for vO3-HAi virus made in the presence of IPTG. We presume that the difference between the deletion and inducible mutant is due to trace amounts of O3 made during infection by the latter, even in the absence of IPTG, although this was not detectable by Western blotting.
Despite the differences in infectivity, purified negatively stained vO3Δ virions were indistinguishable from wild-type virions by electron microscopy (not shown). The protein compositions of purified virions were determined by silver staining of SDS-polyacrylamide gels (Fig. 6A) and by Western blotting with antibodies to two MV internal proteins (A3 and I7) and nine MV surface proteins, including several components of the EFC (Fig. 6B). No differences were found between the proteins of vO3-HAi made in the presence and absence of IPTG, except for the absence of O3 in the latter (O3 was not detected in VACV WR because of the lack of the epitope tag). vO3Δ appeared to have diminished amounts of A26 and A27; the slightly smaller amount of H2 was not reproduced in another experiment.
FIG. 6.

Protein composition of O3 mutant virions. (A) SDS-PAGE. Virions were purified from cells infected with vT7LacOI, vO3Δ, and vO3-HAi in the presence and absence of IPTG; equal numbers of particles, determined by OD, were analyzed by SDS-PAGE. Bands were visualized by silver staining. The positions and masses of marker proteins are shown on the right. (B) Western blotting. Proteins from purified virions were resolved by SDS-PAGE as in panel A and analyzed by Western blotting. The protein targets of the antibodies are indicated on the right.
O3 is required for fusion of infected cells and entry of virions.
The localization of O3 to the MV membrane and the O3 mutant phenotype characterized thus far pointed to a possible role in virus entry and membrane fusion. Previous studies had shown that virions on the surface of infected cells mediate cell-cell fusion after the pH is briefly lowered (11, 18). The surface virions can either be progeny formed during virus infection or an inoculum virus added at a high multiplicity, which cause “fusion from within” and “fusion from without,” respectively. For the former, BS-C-1 cells were infected with vO3-HAi in the presence or absence of IPTG. After 18 h, cells were treated briefly with either pH 7.0 or pH 5.5 buffer, and the cells were incubated in regular medium for an additional 3 h to allow fusion. Cells treated with pH 7.0 buffer did not fuse, regardless of whether IPTG was added (data not shown). However, cells infected with vO3-HAi in the presence of IPTG and briefly treated with pH 5.5 buffer fused with neighboring cells to form syncytia, whereas no syncytia formed in the absence of inducer (Fig. 7A).
FIG. 7.

Requirement of O3 for syncytium formation. (A) Fusion from within. BS-C-1 cells were infected with vO3-HAi in the absence and presence of IPTG for 18 h at 37°C. The cells were exposed to pH 5.5 or pH 7 buffer (not shown) for 2 min and incubated for an additional 3 h at 37°C in the presence of regular medium. The cells were fixed, stained with DAPI (4′,6-diamidino-2-phenylindole), and examined by phase-contrast and fluorescence microscopy to visualize DAPI and GFP. (B) Fusion from without. BS-C-1 cells were infected with vO3-HAi in the absence and presence of IPTG, and O3− and O3+ virions were purified, respectively. BS-C-1 cells on coverslips were incubated with 200 PFU per cell of O3− or O3+ virions for 1 h at 4°C, exposed to pH 5.5 buffer for 2 min, and incubated for 3 h in regular medium containing 300 μg/ml of cycloheximide at 37°C. The cells were fixed, stained with DAPI and phalloidin (Alexa fluor 594; fluorescent conjugated), and examined by fluorescence microscopy.
For fusion from without, stocks of vO3-HAi made in the presence (O3+) and absence (O3−) of IPTG were purified and adsorbed to cells. After brief low-pH treatment, the cells were incubated at 37°C for 3 h in the presence of cycloheximide to prevent viral gene expression and cytopathic effects. Syncytium formation only occurred when the virions formed in the presence of IPTG and contained O3 (Fig. 7B). Taken together, these experiments indicated that O3 was required for virions to mediate cell-cell membrane fusion.
Syncytium formation is thought to mimic the fusion event that occurs during virus entry (28). To monitor virus entry, cells were infected with vO3-HAi made in the presence and absence of IPTG as above, except that the low-pH step was omitted. After 2 h, the infected cells were fixed and then stained with antibody to the L1 membrane protein to visualize virus at the cell surface and with antibody to the A4 core protein to image cytoplasmic cores. This method takes advantage of the inability of the anticore antibody to bind to cores surrounded by the viral membrane (47). Both O3+ and O3− virions were detected at the cell surface with the anti-L1 antibody, suggesting that the binding step was not grossly impaired. In contrast, cytoplasmic cores were nine times more abundant when cells were infected with O3+ virions than with O3− virions (Fig. 8).
FIG. 8.

Requirement of O3 for cell entry. O3− and O3+ virions, prepared as in Fig. 7B, were adsorbed to HeLa cells for 1 h at 4°C. The cells were washed and incubated for additional 2 h at 37°C in the presence of medium containing 300 μg/ml of cycloheximide. The cells were fixed, permeabilized, and stained with mouse anti-L1 (green) and rabbit anti-A4 (red) antibodies followed by fluorescent-tagged corresponding secondary antibodies. The images were obtained by confocal microscopy and processed with Imaris software.
Interaction of O3 with EFC proteins.
A characteristic of the proteins involved in entry is their association in a complex. Hence, it was of interest to check the association of O3 with previously characterized EFC proteins. Cells were infected with VACV WR as a control or vO3-HA, and cell lysates were incubated with anti-HA antibody affinity beads to pull-down O3 together with interacting proteins. The bound proteins were analyzed by Western blotting with specific antibodies to five EFC proteins (A28, L5, H2, A16, and A21) and one EFC-interacting protein (L1). As specificity controls, a membrane protein not involved with entry (D8) and a core protein (A3) were also analyzed. Significantly, each of the EFC proteins and the EFC-interacting protein, but neither of the control proteins, were specifically immunopurified with O3 (Fig. 9A).
FIG. 9.

Interaction of O3 with EFC proteins. (A) IP of proteins associated with O3-HA. HeLa cells were infected with 5 PFU per cell of VACV WR (vWR) or vO3-HA. After 24 h, the cells were lysed and incubated with agarose beads coupled to anti-HA antibody. The input, unbound, and eluted bound proteins were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and probed with antibodies to the proteins listed on the right. (B) Association of EFC proteins in the absence of O3. HeLa cells were infected with VACV WR and vO3-HAi-A28TAP with (+) or without (−) IPTG. After 24 h, the cells were lysed and incubated with agarose beads coupled to streptavidin. The input, unbound, and bound proteins associated with A28TAP were analyzed by Western blotting using antibodies to proteins indicated on the right.
A distinction between the integral components of the EFC and the so-called “EFC-associated” proteins is the exclusive role of the former in the assembly or stability of the complex (3, 4, 39). To determine the effect of repressing O3 expression, the A28R ORF in vO3-HAi was replaced with a copy containing tandem-affinity calmodulin and streptavidin binding peptide tags to form vO3-HAi-A28TAP. A cyan fluorescent protein expression cassette was coinserted downstream of A28 to facilitate isolation of the recombinant virus. Cells were infected with the recombinant virus vO3-HAi-A28TAP in the presence and absence of IPTG to regulate O3 expression or with VACV WR as a control. Western blot analysis of the cell lysates (input) indicated similar expression of each of the proteins regardless of the virus or presence of IPTG, except for A28, which was not detected from VACV WR because antibody to calmodulin binding peptide was used, and O3-HA, which was only detected when IPTG was present (Fig. 9B). The lysates were incubated with streptavidin-agarose beads to capture the tandem affinity purification (TAP)-tagged A28 and its interacting proteins. The specificity of the affinity purification was demonstrated by the absence of any proteins in the bound fraction from cells infected with VACV WR (Fig. 9B). In contrast, the affinity beads captured each of the EFC and associated proteins (but not the A3 core protein) from the cells infected with vO3-HAi-A28TAP in the presence of IPTG (Fig. 9B). Importantly, less A16, A21, L5, L1, and F9 was recovered from the cells infected with vO3-HAi-A28TAP when IPTG was omitted and O3 was not expressed. An exception was H2, which was recovered equally in the bound fractions from cells infected with vO3-HAi-A28TAP in the presence or absence of IPTG. However, this was expected because the interaction of A28 and H2 does not require any other viral proteins (29). Thus, O3 was classified as an integral component of the EFC.
DISCUSSION
In the original annotation of the VACV Copenhagen genome (17), only ORFs of 65 or more amino acids were annotated. Ten additional smaller ORFs that do not appear to be disrupted genes are annotated in the WR strain (www.poxvirus.org/), and a few of these, including O3L, remained uncharacterized. A homolog of the VACV O3L ORF was first described for molluscum contagiosum virus (37) and myxoma virus (6). We identified ORFs varying from 29 to 48 amino acids at the same gene locus in all chordopoxvirus genomes sequenced to date. Although the amino acid identities of these ORFs are low, the uniform presence of a putative N-terminal hydrophobic domain along with the common gene location and length provide compelling evidence that these are orthologs. It was intriguing, therefore, to determine a role for this small protein.
We demonstrated that the O3L ORF was expressed and that synthesis was dependent on viral DNA replication and occurred late in the virus growth cycle. Although O3 had not been identified by mass spectroscopy of purified virions (8, 35, 53), probably because of its hydrophobicity and small size, an association with MVs was shown here by Western blotting. The extractability of O3 with detergent further suggested membrane localization, consistent with the N-terminal hydrophobic domain. In view of its conservation, late expression, and membrane localization, it seemed likely that O3 would have an important role in morphogenesis or virus entry. Although we could delete the O3L ORF, the plaques formed by the mutant were tiny and there was a 3-log reduction in production of infectious virus. A mutant virus with regulated O3 expression had a similar phenotype when inducer was omitted. A role for O3 in morphogenesis was not supported either by biochemical studies that showed processing of membrane and core proteins or by visualization of assembly of virions by electron microscopy. Moreover, purified O3-deficient virions appeared normal and had only minor differences in protein content as analyzed by SDS-PAGE and Western blotting. A defect in entry of O3-deficient virus was indicated by a severe reduction in the number of cores in infected cells; furthermore, O3-deficient virus could not mediate cell fusion triggered by low pH treatment.
The phenotype of O3-deficient virions was similar to that of virions deficient in other EFC proteins, except that a deletion mutant had not previously been isolated. A defining characteristic of EFC components is their association in a multiprotein complex. The interaction of O3 with several EFC proteins was demonstrated using recombinant viruses with an epitope or affinity tag at the C terminus of O3. Moreover, the formation or stabilization of the EFC was impaired when O3 was not expressed. However, EFC formation was not completely abrogated, possibly accounting for the low infectivity of O3 null mutants.
Except for its small size, O3 closely resembles other EFC proteins. Five of the eight previously recognized EFC proteins (A21, A28, G3, H2, and L5) have a single transmembrane domain at or very close to the N terminus, whereas the other three (A16, G9, and J5) have one near the C terminus. The helical hydrophobic sequence of O3 extends from amino acids 4 to 22, exactly the same as that of A21 and G3, whereas those of L5 and H2 are from amino acids 30 to 48 and 32 to 50, respectively. The long C-terminal segments of A21, A28, G3, H2, and L5 extend into the cytoplasm, whereas the long N-terminal segments of A16, G9, and J5 are cytoplasmic. It is likely that O3 has a topology similar to that of the other EFC components with N-terminal transmembrane domains, although the C-terminal segment of O3 would be only 13 amino acids long. Apparently, O3 associates with the other EFC proteins through the transmembrane domain since a C-terminal truncation mutant was still capable of transcomplementation of an O3 null mutant (P. S. Satheshkumar, unpublished data).
Poxviruses are assembled in virus factories, and the surface proteins extend into the cytoplasm rather than the lumen of the endoplasmic reticulum (27). For this reason, poxviruses encode their own cytoplasmic redox system exclusively for intramolecular disulfide bond formation (41). Indeed, all of the EFC polypeptides except G3, which has only one cysteine, require the viral redox system for intramolecular disulfide formation (32, 33, 38, 40, 41, 42). O3 has one cysteine that is not engaged in intermolecular disulfide bonds, as shown by SDS-PAGE in the absence of reducing agent. Predictions of signal peptides and glycosylation sites that are valid for proteins that traffic through the secretory pathway are not useful for MV membrane proteins. For example, the EFC proteins A21, A28, and G3 are predicted to have cleavable signal peptides but shortened proteins have not been detected and the EFC proteins A16, A21, A28, A16, G9, J5, and L5 have 1 to 5 Asn-X-Ser/Thr motifs but are not normally glycosylated. Similarly, the significance of a prediction by SignalP (Center for Biological Sequence Analysis) of a signal peptidase cleavage site and the presence of a putative N-glycosylation site in O3 are suspect. Nevertheless, we cannot rule out the possibility that a portion of O3 traffics through the secretory pathway. The small 13- or 14-amino-acid size of the putative secreted peptide makes analysis difficult. We can, however, dismiss the importance of such a pathway for the role of O3 in virus entry because the entry-proficient O3+ and entry-defective O3− MVs were both purified from the cytoplasm and not the medium; furthermore, a C-terminal truncated ORF that does not even encode the hypothetical secreted peptide was able to complement an O3-null mutant as stated above. Therefore, if a secreted peptide were made, it would have another function.
In conclusion, O3 is the newest and smallest member of the EFC and also the smallest poxvirus protein for which a role has been determined.
Acknowledgments
We thank Andrea Weisberg and Catherine Cotter in our laboratory for electron microscopy and cell culture, respectively, and Lily Koo and Steven Becker from the National Institute of Allergy and Infectious Diseases biological imaging facility for help with confocal microscopy analysis. Antibodies were kindly provided by G. Cohen and R. Eisenberg, University of Pennsylvania; Mariano Esteban, CSIC, Madrid; and Geoffrey Smith, Imperial College, London.
This research was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Published ahead of print on 7 October 2009.
REFERENCES
- 1.Alexander, W. A., B. Moss, and T. R. Fuerst. 1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA polymerase and the Escherichia coli lac repressor. J. Virol. 66:2934-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ansarah-Sobrinho, C., and B. Moss. 2004. Role of the I7 protein in proteolytic processing of vaccinia virus membrane and core components. J. Virol. 78:6335-6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bisht, H., A. S. Weisberg, and B. Moss. 2008. Vaccinia virus L1 protein is required for cell entry and membrane fusion. J. Virol. 82:8687-8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown, E., T. G. Senkevich, and B. Moss. 2006. Vaccinia virus F9 virion membrane protein is required for entry but not virus assembly, in contrast to the related L1 protein. J. Virol. 80:9455-9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrd, C. M., and D. E. Hruby. 2005. A conditional-lethal vaccinia virus mutant demonstrates that the I7L gene product is required for virion morphogenesis. Virol. J. 2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cameron, C., S. Hota-Mitchell, L. Chen, J. Barrett, J. X. Cao, C. Macaulay, D. Willer, D. Evans, and G. McFadden. 1999. The complete DNA sequence of myxoma virus. Virology 264:298-318. [DOI] [PubMed] [Google Scholar]
- 7.Carter, G. C., M. Law, M. Hollinshead, and G. L. Smith. 2005. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 86:1279-1290. [DOI] [PubMed] [Google Scholar]
- 8.Chung, C.-S., C.-H. Chen, M.-Y. Ho, C.-Y. Huang, C.-L. Liao, and W. Chang. 2006. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 80:2127-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davison, A. J., and B. Moss. 1989. The structure of vaccinia virus late promoters. J. Mol. Biol. 210:771-784. [DOI] [PubMed] [Google Scholar]
- 10.Demkowicz, W. E., J. S. Maa, and M. Esteban. 1992. Identification and characterization of vaccinia virus genes encoding proteins that are highly antigenic in animals and are immunodominant in vaccinated humans. J. Virol. 66:386-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doms, R. W., R. Blumenthal, and B. Moss. 1990. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol. 64:4884-4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Earl, P. L., N. Cooper, L. S. Wyatt, B. Moss, and M. W. Carroll. 1998. Preparation of cell cultures and vaccinia virus stocks, p. 16.16.1-16.16.3. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. John Wiley and Sons, New York, NY. [Google Scholar]
- 13.Earl, P. L., and B. Moss. 1998. Characterization of recombinant vaccinia viruses and their products, p. 16.18.1-16.18.11. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. Greene Publishing Associates & Wiley Interscience, New York, NY. [Google Scholar]
- 14.Earl, P. L., B. Moss, L. S. Wyatt, and M. W. Carroll. 1998. Generation of recombinant vaccinia viruses, p. 16.17.1-16.17.19. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.), Current protocols in molecular biology, vol. 2. Greene Publishing Associates & Wiley Interscience, New York, NY. [Google Scholar]
- 15.Fogg, C. N., J. L. Americo, P. L. Earl, W. Resch, L. Aldaz-Carroll, R. J. Eisenberg, G. H. Cohen, and B. Moss. 2008. Disparity between in vitro neutralization of vaccinia virus by antibody to the A27 protein and protection of mice against intranasal challenge. J. Virol. 82:8022-8029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foo, C. H., H. Lou, J. C. Whitbeck, M. Ponce-de-Leon, D. Atanasiu, R. J. Eisenberg, and G. H. Cohen. 2009. Vaccinia virus L1 binds to cell surfaces and blocks virus entry independently of glycosaminoglycans. Virology 385:368-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goebel, S. J., G. P. Johnson, M. E. Perkus, S. W. Davis, J. P. Winslow, and E. Paoletti. 1990. The complete DNA sequence of vaccinia virus. Virology 179:247-266, 517-563. [DOI] [PubMed] [Google Scholar]
- 18.Gong, S. C., C. F. Lai, and M. Esteban. 1990. Vaccinia virus induces cell fusion at acid pH and this activity is mediated by the N-terminus of the 14-kDa virus envelope protein. Virology 178:81-91. [DOI] [PubMed] [Google Scholar]
- 19.Howard, A. R., T. G. Senkevich, and B. Moss. 2008. Vaccinia virus A26 and A27 proteins form a stable complex tethered to mature virions by association with the A17 transmembrane protein. J. Virol. 82:12384-12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang, C.-Y., T.-Y. Lu, C.-H. Bair, Y.-S. Chang, J.-K. Jwo, and W. Chang. 2008. A novel cellular protein, VPEF, facilitates vaccinia virus penetration into HeLa cells through fluid phase endocytosis. J. Virol. 82:7988-7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Izmailyan, R. A., C.-Y. Huang, S. Mohammad, S. N. Isaacs, and W. Chang. 2006. The envelope G3L protein is essential for entry of vaccinia virus into host cells. J. Virol. 80:8402-8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katz, E., and B. Moss. 1970. Formation of a vaccinia virus structural polypeptide from a higher molecular weight precursor: inhibition by rifampicin. Proc. Natl. Acad. Sci. USA 6:677-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kochan, G., D. Escors, J. M. Gonzalez, J. M. Casasnovas, and M. Esteban. 2008. Membrane cell fusion activity of the vaccinia virus A17-A27 protein complex. Cell. Microbiol. 10:1149-1164. [DOI] [PubMed] [Google Scholar]
- 24.Law, M., G. C. Carter, K. L. Roberts, M. Hollinshead, and G. L. Smith. 2006. Ligand-induced and non-fusogenic dissolution of a viral membrane. Proc. Natl. Acad. Sci. USA 103:5989-5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lustig, S., C. Fogg, J. C. Whitbeck, R. J. Eisenberg, G. H. Cohen, and B. Moss. 2005. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J. Virol. 79:13454-13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mercer, J., and A. Helenius. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531-535. [DOI] [PubMed] [Google Scholar]
- 27.Moss, B. 2007. Poxviridae: the viruses and their replication, p. 2905-2946. In D. M. Knipe and P. M. Howley (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 28.Moss, B. 2006. Poxvirus entry and membrane fusion. Virology 344:48-54. [DOI] [PubMed] [Google Scholar]
- 29.Nelson, G. E., J. R. Sisler, D. Chandran, and B. Moss. 2008. Vaccinia virus entry/fusion complex subunit A28 is a target of neutralizing and protective antibodies. Virology 380:394-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson, G. E., T. R. Wagenaar, and B. Moss. 2008. A conserved sequence within the H2 subunit of the vaccinia virus entry/fusion complex is important for interaction with the A28 subunit and infectivity. J. Virol. 82:6244-6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nichols, R. J., E. Stanitsa, B. Unger, and P. Traktman. 2008. The vaccinia virus gene I2L encodes a membrane protein with an essential role in virion entry. J. Virol. 82:10247-10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ojeda, S., A. Domi, and B. Moss. 2006. Vaccinia virus G9 protein is an essential component of the poxvirus entry-fusion complex. J. Virol. 80:9822-9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ojeda, S., T. G. Senkevich, and B. Moss. 2006. Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene. J. Virol. 80:51-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parkinson, J. E., and G. L. Smith. 1994. Vaccinia virus gene A36R encodes a Mr 43-50 K protein on the surface of extracellular enveloped virus. Virology 204:376-390. [DOI] [PubMed] [Google Scholar]
- 35.Resch, W., K. K. Hixson, R. J. Moore, M. S. Lipton, and B. Moss. 2007. Protein composition of the vaccinia virus mature virion. Virology 358:233-247. [DOI] [PubMed] [Google Scholar]
- 36.Satheshkumar, P. S., A. Weisberg, and B. Moss. 2009. Vaccinia virus H7 protein contributes to the formation of crescent membrane precursors of immature virions. J. Virol. 83:8439-8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Senkevich, T. G., E. V. Koonin, J. J. Bugert, G. Darai, and B. Moss. 1997. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology 233:19-42. [DOI] [PubMed] [Google Scholar]
- 38.Senkevich, T. G., and B. Moss. 2005. Vaccinia virus H2 protein is an essential component of a complex involved in virus entry and cell-cell fusion. J. Virol. 79:4744-4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Senkevich, T. G., S. Ojeda, A. Townsley, G. E. Nelson, and B. Moss. 2005. Poxvirus multiprotein entry-fusion complex. Proc. Natl. Acad. Sci. USA 102:18572-18577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Senkevich, T. G., B. M. Ward, and B. Moss. 2004. Vaccinia virus A28L gene encodes an essential protein component of the virion membrane with intramolecular disulfide bonds formed by the viral cytoplasmic redox pathway. J. Virol. 78:2348-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Senkevich, T. G., C. L. White, E. V. Koonin, and B. Moss. 2002. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc. Natl. Acad. Sci. USA 99:6667-6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Townsley, A., T. G. Senkevich, and B. Moss. 2005. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is required for cell entry and cell-cell fusion. J. Virol. 79:10988-10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Townsley, A. C., T. G. Senkevich, and B. Moss. 2005. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J. Virol. 79:9458-9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Townsley, A. C., A. S. Weisberg, T. R. Wagenaar, and B. Moss. 2006. Vaccinia virus entry into cells via a low pH-dependent endosomal pathway. J. Virol. 80:8899-8908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turner, P. C., and R. W. Moyer. 2006. The cowpox virus fusion regulator proteins SPI-3 and hemagglutinin interact in infected and uninfected cells. Virology 347:88-99. [DOI] [PubMed] [Google Scholar]
- 46.Turner, P. C., and R. W. Moyer. 2008. The vaccinia virus fusion inhibitor proteins SPI-3 (K2) and HA (A56) expressed by infected cells reduce the entry of superinfecting virus. Virology 380:226-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanderplasschen, A., M. Hollinshead, and G. L. Smith. 1998. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J. Gen. Virol. 79:877-887. [DOI] [PubMed] [Google Scholar]
- 48.Wagenaar, T. R., and B. Moss. 2007. Association of vaccinia virus fusion regulatory proteins with the multicomponent entry/fusion complex. J. Virol. 81:6286-6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagenaar, T. R., and B. Moss. 2009. Expression of the A56 and K2 proteins is sufficient to inhibit vaccinia virus entry and cell fusion. J. Virol. 83:1546-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wagenaar, T. R., S. Ojeda, and B. Moss. 2008. Vaccinia virus A56/K2 fusion regulatory protein interacts with the A16 and G9 subunits of the entry fusion complex. J. Virol. 82:5153-5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolffe, E. J., D. M. Moore, P. J. Peters, and B. Moss. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797-2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolffe, E. J., S. Vijaya, and B. Moss. 1995. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 211:53-63. [DOI] [PubMed] [Google Scholar]
- 53.Yoder, J. D., T. S. Chen, C. R. Gagnier, S. Vemulapalli, C. S. Maier, and D. E. Hruby. 2006. Pox proteomics: mass spectrometry analysis and identification of Vaccinia virion proteins. Virol. J. 3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]