

Abstract
Transferases and hydrolases catalyze different chemical reactions and express different dynamic responses upon ligand binding. To insulate the ligand molecule from the surrounding water, transferases bury it inside the protein by closing the cleft, while hydrolases undergo a small conformational change and leave the ligand molecule exposed to the solvent. Despite these distinct ligand-binding modes, some transferases and hydrolases are homologous. To clarify how such different catalytic modes are possible with the same scaffold, we examined the solvent accessibility of ligand molecules for 15 SCOP superfamilies, each containing both transferase and hydrolase catalytic domains. In contrast to hydrolases, we found that nine superfamilies of transferases use two major strategies, oligomerization and domain fusion, to insulate the ligand molecules. The subunits and domains that were recruited by the transferases often act as a cover for the ligand molecule. The other strategies adopted by transferases to insulate the ligand molecule are the relocation of catalytic sites, the rearrangement of secondary structure elements, and the insertion of peripheral regions. These findings provide insights into how proteins have evolved and acquired distinct functions with a limited number of scaffolds.
Keywords: enzyme, evolution, superfamily, oligomerization, domain fusion
Introduction
Each enzyme catalyzes its specific chemical reaction on the unique three-dimensional (3D) structure to play essential roles in biological systems.1 However, this does not necessarily mean that onefold corresponds to only one function. During evolution, some enzymes were modified to acquire novel functions while keeping their folds unaltered. In other words, many homologous enzymes catalyze different chemical reactions.2–6 In fact, more than a quarter of the superfamilies containing enzymes in the SCOP (Structural Classification of Proteins) database7 catalyze multiple reactions, if enzyme reactions differing in the first three digits of the Enzyme Commission (EC) number8 are regarded as distinct. As the mechanisms to alter the enzymatic functions, Todd et al. proposed six kinds of modifications, and reported that functional diversities are commonly generated by gene duplication and incremental mutations.6
Not only 3D structures, but also conformational changes are important in molecular functions.9–12 Recently, we found that transferases and hydrolases undergo different motions upon ligand binding to achieve their functions.13 Transferases often exhibit large rigid-body domain motions upon ligand binding, to insulate the ligand molecule from the surrounding water. By contrast, hydrolases change their structures to a small extent and keep the ligand molecule exposed to water on the protein surface. The distinctive responses to ligand binding suggest that the key difference between transferases and hydrolases lies in the ligand binding mode: the ligand molecule is insulated from water in the former, while it is exposed in the latter.13
Although enzymatic functions evolve, it is extraordinary that such disparate ligand binding modes operate in the same scaffold. The ligand insulation in transferases frequently results from domain movement, while hydrolases often have a single domain structure,13 suggesting that, in the course of evolution, interconversion of the two enzymatic reactions in onefold is difficult. In this research, we scrutinized homologous pairs of transferases and hydrolases whose structures were assigned to the same SCOP superfamily from the viewpoint of ligand binding modes, or the insulation of ligand molecules,13 and elucidated how these different binding modes required for distinct enzymatic functions are accomplished within the same scaffold.
Results and Discussion
Strategies to insulate ligand molecules in transferases
In the SCOP database,7 15 superfamilies contain both types of catalytic domains categorized as transferases and hydrolases. Their oligomeric states were assessed by the UniProt,14 PQS,15 and PiQSi databases,16 as well as in the literature. Catalytic residues were identified by the UniProt annotations (Materials and Methods). In the analysis, we focused on how the representative transferases insulated the ligand molecules from the surrounding water, in comparison to the corresponding hydrolases. All results are provided in Table S1 of the Supporting Information, and summarized in Table 1. Figure 1 shows the ligand insulation strategies employed by the transferases in the 15 superfamilies. The two major strategies are oligomerization and domain fusion. This implies that the additional subunit or domain acts as a cover for the crevice containing the ligand molecule. These alterations are adopted in nine superfamilies. In three superfamilies, the insertion of a peripheral region, the relocation of the catalytic site, and the rearrangements of the secondary structures were observed. In total, the insulation mechanisms were identified in 12 superfamilies, while the mechanisms for the remaining three were not clearly interpretable (see Supporting Information). We will discuss each of the strategies in the following sections.
Table I.
Fifteen Superfamilies Analyzed in This Study and Strategies Employed by the Transferases to Insulate the Ligand Molecules
| Superfamily (sccsa) | Transferaseb (PDB code; oligomeric state) | Hydrolaseb (PDB code; oligomeric state) | Strategy |
|---|---|---|---|
| Glycoside hydrolase/deacetylase (c.6.2) | 4-alpha-glucanotransferase (1k1y; homo dimer) | Golgi alpha-mannosidase II (1qwn; homo dimer) | Unidentified |
| Class I glutamine amidotransferase-like (c.23.16) | Imidazole glycerol phosphate synthase subunit hisH (1gpw; hetero dimer) | Gamma-glutamyl hydrolase (1l9x; homo dimer) | Oligomerization |
| DHS-like NAD/FAD-binding domain (c.31.1) | Deoxyhypusine synthase (1rqd; homo tetramer) | Silent information regulator 2 (1m2k; monomer) | Oligomerization |
| Rhodanese/cell cycle control phosphatase (c.46.1) | Rhodanese (1orb; monomer) | M-phase inducer phosphatase 2 (1qb0; -) | Domain fusion |
| Ribonuclease H-like (c.55.3) | DNA polymerase III epsilon subunit (2ido; hetero dimer) | Oligoribonuclease (1yta; homo dimer) | Rearrangement of SSE |
| Zn-dependent exopeptidases (c.56.5) | Glutaminyl cyclase (2afw; homo hexamer) | Carboxypeptidase A (2ctc; monomer) | Oligomerization |
| Alpha/beta-hydrolases (c.69.1) | Antigen 85C (1dqy; monomer) | Carboxylesterase bioH (1m33; monomer) | Unidentified |
| Periplasmic binding protein-like II (c.94.1) | Thiaminase I (4thi; monomer) | Lactoferrin (1lcf; monomer) | Relocation of catalytic sites |
| NagB/RpiA/CoA transferase-like (c.124.1) | Succinyl-CoA:3-ketoacid CoA transferase (1ooy; homo dimer) | Glucosamine-6-phosphate deaminase (1ne7; homo hexamer) | Hybrid/Mixture |
| Cysteine proteinases (d.3.1) | Arylamine N-acetyltransferase (1w6f; homo dimer) | Cathepsin S (2h7j; monomer) | Insertion of peripheral |
| HIT-like (d.13.1) | Galactose-1-phosphate uridylyltransferase (1hxp; homo dimer) | Fragile histidine triad protein (5fit; homo dimer) | Oligomerization |
| Ribosomal protein S5 domain 2-like (d.14.1) | 4-diphosphocytidyl-2C-methyl-D- erythritol kinase (1oj4; homo dimer) | Lon protease (1rre; homo hexamer) | Domain fusion |
| Pentein (d.126.1) | l-arginine:glycine amidinotransferase (8jdw; homo dimer) | N-succinylarginine dihydrolase (1ynh; homo dimer) | Unidentified |
| Phospholipase D/nuclease (d.136.1) | Polyphosphate kinase (1xdp; homo tetramer) | Tyrosyl-DNA phosphodiesterase (1rff; monomer) | Hybrid/Mixture |
| N-terminal nucleophile aminohydrolases (d.153.1) | Glutamine phosphoriboxylpyrophosphate amidotransferase (1ecg; homo tetramer) | Penicillin V acylase (2pva; homo tetramer) | Domain fusion |
SCOP concise classification string.
For several superfamilies, we analyzed many protein structures. However, in this shortened Table, only a protein was shown.
Figure 1.
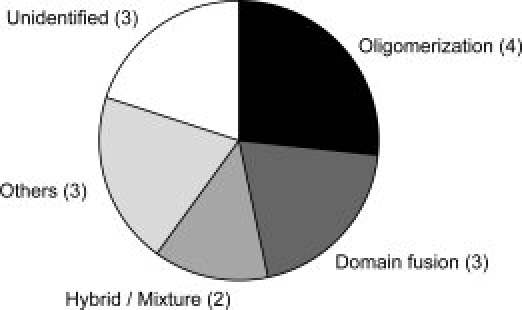
Strategies adopted by the transferases of 15 SCOP superfamilies to insulate ligand molecules. The two major strategies, “oligomerization” and “domain fusion,” were colored by black and dark gray, respectively. In the superfamilies classified into “hybrid/mixture” (gray), both oligomerization and domain fusion were found in the transferases. The three miner strategies, insertion of peripheral regions, relocation of catalytic sites, and rearrangement of secondary structures, were compiled into one strategy, “others” (light gray).
Oligomerization
In the four superfamilies, “DHS-like NAD/FAD-binding domain” [SCOP concise classification string (SCCS): c.31.1], “HIT-like” (d.13.1), “class I glutamine amidotransferase-like” (c.23.16) and “Zn-dependent exopeptidases” (c.56.5), the transferases insulate the ligand molecules by oligomerization. The “DHS-like NAD/FAD-binding domain” superfamily contains the transferase, deoxyhypusine synthase,17 and the hydrolase, silent information regulator 2,18 which are both single-domain proteins [Fig. 2(a)]. The difference between them is in the oligomeric state: The transferase forms a homotetramer, while the hydrolase is monomeric. The solvent accessibility of the ligand molecule in the transferase is reduced upon oligomerization [the right panel of Fig. 2(a)], and the complex structure clearly shows that the three additional subunits cover the ligand molecule [Fig. 2(a)].
Figure 2.
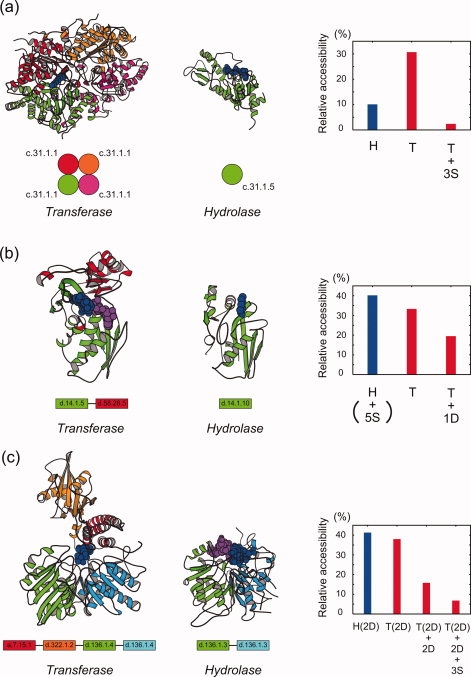
Structures of the representative transferases (left panels) adopting the three major strategies (a, oligomerization; b, domain fusion; c, hybrid), the corresponding structures of the hydrolases (middle panels) and the relative solvent accessibility of the ligand molecules (right panels). (a) The “DHS-like NAD/FAD-binding domain” (c.31.1) superfamily. The homotetrameric form of the transferase, deoxyhypusine synthase [Protein Data Bank (PDB) code: 1rqd, chain A, B, C, D],17 is shown in the left panel. The monomeric form of the hydrolase, silent information regulator 2 (PDB: 1m2k, chain A),18 is in the middle panel. Their ligand molecules are depicted by blue CPK models. Ribbon models were drawn by MOLSCRIPT.19 In the right panel, the relative solvent accessibility of the ligand molecules for the hydrolase and the transferase are shown by blue and red bars, respectively, where “S” stands for the addition of a subunit, and thus “+3S” means a change from monomer to homotetramer. The domain architectures are drawn schematically at the bottom. (b) The “ribosomal protein S5 domain 2-like” (d.14.1) superfamily. The structure of the transferase, 4-diphosphocytidyl-2C-methyl-d-erythritol kinase (PDB: 1oj4, chain A),20 is in the left panel, with the recruited domain in red. The structure of the hydrolase, Lon protease (PDB: 1rre, chain A),21 is in the middle panel. The relative solvent accessibility of the ligand molecules of the transferase decreases with the addition of a recruited domain (+1D) (right panel). (c) The “phospholipase D/nuclease” (d.136.1) superfamily. The monomeric form of the transferase, polyphosphate kinase (PDB: 1xdp, chain A),22 is shown in the left panel. The two recruited domains are colored red and orange, respectively. The structure of the hydrolase, tyrosyl-DNA phosphodiesterase (PDB: 1rff, chain A),23 is in the middle panel. The relative solvent accessibility of the ligand molecule of the transferase is reduced with the addition of the two recruited domains (+2D), and decreases further upon homotetramer formation (+3S) (right panel).
The hydrolase belonging to the “HIT-like” superfamily, fragile histidine triad protein,24 is a single-domain protein that forms a homodimer, in which the ligand molecule is located on the dimeric interface, but is fully exposed to water. The transferase in the same superfamily, galactose-1-phosphate uridylyltransferase,25 consists of two homologous domains resembling the dimeric form of the hydrolase. Although in this form the ligand molecule is still exposed on the surface, the transferase forms a homodimer to cover the ligand molecule. Another hydrolase in this superfamily, mRNA decapping enzyme,26 forms a homodimer. However, it cannot be simply compared with the transferase, because this hydrolase would be under allosteric control, to realize two different stages of the catalytic cycle on each protomer.26,27
The transferase in the “Zn-dependent exopeptidases” superfamily, glutaminyl cyclase,28 was also classified as those utilizing oligomerization. Although the accessibility of the ligand molecule is not necessarily low, the transferase forms oligomers, in which the subunit interfaces insulate the ligand molecule. On the other hand, in the corresponding hydrolases, carboxypeptidase A29 and carboxypeptidase D30 are monomeric.
In the “class I glutamine amidotransferase-like” superfamily, 3D structures of transferases with the ligand molecules bound at the catalytic residues have not been determined yet. In this case, we estimated the accessibility to the catalytic region, defined by catalytic residues and their neighbors, instead of the accessibility of the ligand molecule (See Materials and Methods in detail). The accessibility of the catalytic region in the transferase, imidazole glycerol phosphate synthase subunit hisH,31 decreased upon oligomerization. This means that the ligand molecules in the transferase are likely to be insulated by the recruited subunit. On the contrary, the accessibility of the ligand molecule in the hydrolase, gamma-glutamyl hydrolase,32 was unchanged upon oligomerization.
Domain fusion
In the three superfamilies, “ribosomal protein S5 domain 2-like” (d.14.1), “rhodanese/cell cycle control phosphatase” (c.46.1), and “N-terminal nucleophile aminohydrolases” (d.153.1), the transferases insulate the ligand molecules by recruiting additional domains. The “ribosomal protein S5 domain 2-like” superfamily contains the hydrolase, Lon protease.21 Although the N-terminus of the catalytic domain is linked with two noncatalytic domains, whose structures were determined independently, these domains are likely to be on the opposite side of the catalytic sites.21 On the other hand, the transferase, 4-diphosphocytidyl-2C-methyl-d-erythritol kinase,20 has an additional domain near the catalytic site, and the solvent accessibility of the ligand molecules is lower [Fig. 2(b)]. In the Supporting Information section, we discuss the influence of the missing part of the chain in the crystal structure, as shown in the case of this superfamily, and concluded that it may not contribute to the insulation of the ligand molecule to a large extent. Throughout this work, we focused only on the regions with structures that have been determined.
In the “rhodanese/cell cycle control phosphatase” superfamily, the hydrolase33 is a single-domain protein, while the corresponding transferase34 contains an additional domain at the N-terminus, which covers the ligand molecule. The transferases35,36 in the “N-terminal nucleophile aminohydrolases” superfamily insulate the ligand molecules by the additional domains at the C-termini. The hydrolase, penicillin V acylase,37 is a single-domain protein and forms a homo tetramer, but the recruited subunits do not affect the accessibility of the ligand molecule.
Hybrid/mixture
In the “phospholipase D/nuclease” (d.136.1) superfamily, both oligomerization and domain fusion are adopted in the transferase. The hydrolase belonging to the superfamily, tyrosyl-DNA phosphodiesterase,23 is a monomeric protein consisting of two homologous domains. In contrast, the transferase, polyphosphate kinase,22 contains two additional domains at the N- terminus in addition to the two homologous catalytic domains, and forms a homotetramer. The domain architectures and the accessibilities of the ligand molecules shown in Fig. 2(c) indicate that the two additional domains and the three subunits concertedly function to insulate the ligand molecules.
In the “NagB/RpiA/CoA transferase-like” (c.124.1) superfamily, the two transferases adopt two different strategies for the insulation, that is, domain fusion in succinyl-CoA:3-ketoacid CoA transferase38 and oligomerization in glutaconate CoA transferase.39 Succinyl-CoA:3-ketoacid CoA transferase consists of two domains, and the N-terminal domain covers the catalytic site on the C-terminal domain. Glutaconate CoA transferase is an octamer of single-domain subunits, and their interfaces cover the ligand molecule. Interestingly, the two domains of succinyl-CoA:3-ketoacid CoA transferase are homologous to the two subunits of glutaconate CoA transferase, respectively.
Other strategies
The transferases in the “cysteine proteinases” (d.3.1), “ribonuclease H-like” (c.55.3) and “periplasmic binding protein-like II” (c.94.1) superfamilies each employ unique strategies to insulate the ligand molecules. In the “cysteine proteinases” superfamily, the transferase, arylamine N-acetyltransferase,40 uses a peripheral region to insulate the ligand molecule. The counterpart of the hydrolase, cathepsin S,41 lacks the corresponding peripheral region [Fig. 3(a)]. In the “ribonuclease H-like” superfamily, a subtle rearrangement of the helices plays a key role to insulate the ligand molecule. In the transferase, DNA polymerase III epsilon subunit,42 two helices (residues 59–67 and 144–152) interact with the ligand molecule to shield it, whereas the corresponding two helices are far from the ligand molecule in the hydrolase, oligoribonuclease [Fig. 3(b)].
Figure 3.
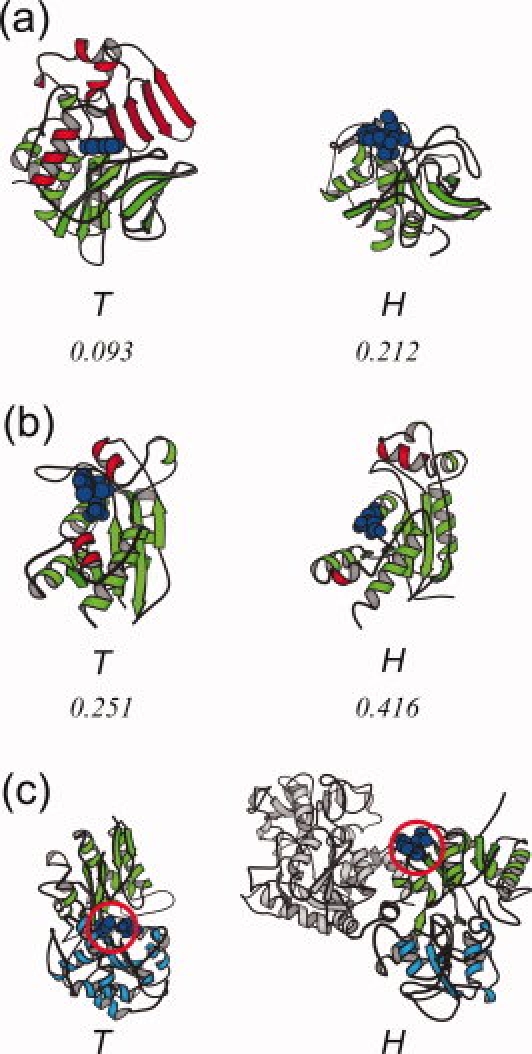
The structures of the transferases adopting strategies other than those in Figure 2, in comparison with the structures of the hydrolases. (a) The structures of the transferase, arylamine N-acetyltransferase (PDB: 1w6f, chain A),40 and the hydrolase, cathepsin S (PDB: 2h7j, chain A),41 belonging to the “cysteine proteinases” (d.3.1) superfamily. The peripheral region (186–275) inserted within the transferase (red) covers the ligand molecule (blue). The values at the bottom indicate the relative solvent accessibility of the ligand molecule. (b) The structures of the transferase, DNA polymerase III epsilon subunit (PDB: 2ido, chain A),42 and the hydrolase, oligoribonuclease (PDB: 1yta, chain A), of the “ribonuclease H-like” (c.55.3) superfamily. The two helices shown in red (59–67, 144–152) in the transferase cover the ligand molecules. (c) The structures of the transferase, thiaminase I (PDB: 4thi, chain A),43 and the hydrolase, lactoferrin (PDB: 1lcf, chain A),44 of the “periplasmic binding protein-like II” (c.94.1) superfamily. In the transferase, the catalytic sites (C113 and E241, blue CPK) are in the middle (red circle) of the two sub-domains (9–113 and 270–354 in green, 114–269 and 355–370 in cyan). On the other hand, in the hydrolase, the catalytic residues are located at K73 and S259, shown in the red circle. The peripheral domain is colored gray. The relative accessibility for the hydrolase is not given, because the ligand-bound form is not available.
In the “periplasmic binding protein-like II” superfamily, the catalytic site of the transferase is located at a completely different position from that of the hydrolase. In the transferase, thiaminase I,43 the ligand molecule is packed at the cleft between two sub-domains, while the hydrolase, lactoferrin,44 is supposed to bind the ligand on the surface of one sub-domain, located far from the cleft [Fig. 3(c)]. The other domain in the hydrolase does not affect the insulation.
Materials and Methods
Dataset construction
Proteins in the SCOP 1.737 and UniProt 13.3 databases14 were combined with 95% sequence identity to integrate the SCOP classification and the UniProt annotation of the EC number,8 the catalytic residues and the oligomeric state. Fifteen SCOP superfamilies, each containing both kinds of catalytic domains, transferase and hydrolase, were identified. Complex structures were also assessed by the PQS database15 to confirm the UniProt annotations. In the case of a discrepancy between UniProt and PQS, we referred to the PiQSi database16 and the literature. Homologous proteins were grouped together if they shared the domain architectures, oligomeric states, and EC numbers, and were further classified with the criterion of 2.5 Å RMSD for Cα atoms, using MATRAS.45 The representative proteins were selected by referring to the resolution, the number of missing residues and the ligand molecules (Table S1). In this study, we excluded a superfamily containing both transferases and hydrolases, “(trans)glycosidases” (c.1.8), because it was reported that the two selected transferases also have hydrolase activity.46,47
Accessibility analysis
Accessibilities of the ligand molecules were calculated by NACCESS.48 When there is no ligand molecule at the catalytic residues, we calculated the accessibility of the catalytic region, instead of the accessibility of the ligand molecule. Here, the catalytic region means the set of catalytic residues and their neighboring residues located within 4.5 Å distance from any atoms in the catalytic residues. The accessibility of catalytic region was calculated for three transferases and a hydrolase denoted in the parenthese of ACC column in Table S1.
Conclusions
We investigated 15 SCOP superfamilies, each containing both transferases and hydrolases, and revealed that oligomerization and domain fusion play important roles in the insulation of the ligand molecule required for the transferase activity. These findings provide insights into how proteins evolved to acquire distinct functions2–6,49 with a limited number of scaffolds.50
Acknowledgments
The authors thank Keiichi Homma for critical reading of the manuscript.
Glossary
Abbreviations
- 3D
three-dimensional
- EC number
Enzyme Commission number
- PDB
Protein Data Bank
- PQS
Protein Quaternary Structure file server
- SCOP
Structural Classification of Proteins
- SCCS
SCOP concise classification string
References
- 1.Branden C, Tooze J. Introduction to protein structure. 2nd ed. New York: Garland Publishing; 1998. [Google Scholar]
- 2.Glasner ME, Gerlt JA, Babbitt PC. Evolution of enzyme superfamilies. Curr Opin Chem Biol. 2006;10:492–497. doi: 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 3.Hegyi H, Gerstein M. The relationship between protein structure and function: a comprehensive survey with application to the yeast genome. J Mol Biol. 1999;288:147–164. doi: 10.1006/jmbi.1999.2661. [DOI] [PubMed] [Google Scholar]
- 4.Martin AC, Orengo CA, Hutchinson EG, Jones S, Karmirantzou M, Laskowski RA, Mitchell JB, Taroni C, Thornton JM. Protein folds and functions. Structure. 1998;6:875–884. doi: 10.1016/s0969-2126(98)00089-6. [DOI] [PubMed] [Google Scholar]
- 5.Nagano N, Orengo CA, Thornton JM. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J Mol Biol. 2002;321:741–765. doi: 10.1016/s0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- 6.Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol. 2001;307:1113–1143. doi: 10.1006/jmbi.2001.4513. [DOI] [PubMed] [Google Scholar]
- 7.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 8.Webb EC. Enzyme nomenclature. Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology. New York: Academic Press; 1992. [Google Scholar]
- 9.Gerstein M, Lesk AM, Chothia C. Structural mechanisms for domain movements in proteins. Biochemistry. 1994;33:6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 10.Hammes GG. Multiple conformational changes in enzyme catalysis. Biochemistry. 2002;41:8221–8228. doi: 10.1021/bi0260839. [DOI] [PubMed] [Google Scholar]
- 11.Joseph D, Petsko GA, Karplus M. Anatomy of a conformational change: hinged “lid” motion of the triosephosphate isomerase loop. Science. 1990;249:1425–1428. doi: 10.1126/science.2402636. [DOI] [PubMed] [Google Scholar]
- 12.Koshland DE. Correlation of structure and function in enzyme action. Science. 1963;142:1533–1541. doi: 10.1126/science.142.3599.1533. [DOI] [PubMed] [Google Scholar]
- 13.Koike R, Amemiya T, Ota M, Kidera A. Protein structural change upon ligand binding correlates with enzymatic reaction mechanism. J Mol Biol. 2008;379:397–401. doi: 10.1016/j.jmb.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 14.The UniProt Consortium. The Universal Protein Resource (UniProt) Nucl Acids Res. 2009;37:D169–D174. doi: 10.1093/nar/gkn664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henrick K, Thornton JM. PQS: a protein quaternary structure file server. Trends Biochem Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 16.Levy ED. PiQSi: protein quaternary structure investigation. Structure. 2007;15:1364–1367. doi: 10.1016/j.str.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 17.Umland TC, Wolff EC, Park MH, Davies DR. A new crystal structure of deoxyhypusine synthase reveals the configuration of the active enzyme and of an enzyme.NAD.inhibitor ternary complex. J Biol Chem. 2004;279:28697–28705. doi: 10.1074/jbc.M404095200. [DOI] [PubMed] [Google Scholar]
- 18.Chang JH, Kim HC, Hwang KY, Lee JW, Jackson SP, Bell SD, Cho Y. Structural basis for the NAD-dependent deacetylase mechanism of Sir2. J Biol Chem. 2002;277:34489–93448. doi: 10.1074/jbc.M205460200. [DOI] [PubMed] [Google Scholar]
- 19.Kraulis PJ. MOLSCRIPT. A program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24:946–950. [Google Scholar]
- 20.Miallau L, Alphey MS, Kemp LE, Leonard GA, Mcsweeney SM, Hecht S, Bacher A, Eisenreich W, Rohdich F, Hunter WN. Biosynthesis of isoprenoids: crystal structure of 4-diphosphocytidyl-2C-methyl-D-erythritol kinase. Proc Natl Acad Sci USA. 2003;100:9173–9178. doi: 10.1073/pnas.1533425100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Botos I, Melnikov EE, Cherry S, Tropea JE, Khalatova AG, Rasulova F, Dauter Z, Maurizi MR, Rotanova TV, Wlodawer A. The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser-Lys dyad in the active site. J Biol Chem. 2004;279:8140–8148. doi: 10.1074/jbc.M312243200. [DOI] [PubMed] [Google Scholar]
- 22.Zhu Y, Huang W, Lee SS, Xu W. Crystal structure of a polyphosphate kinase and its implications for polyphosphate synthesis. EMBO Rep. 2005;6:681–687. doi: 10.1038/sj.embor.7400448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davies DR, Interthal H, Champoux JJ, Hol WG. Explorations of peptide and oligonucleotide binding sites of tyrosyl-DNA phosphodiesterase using vanadate complexes. J Med Chem. 2004;47:829–837. doi: 10.1021/jm030487x. [DOI] [PubMed] [Google Scholar]
- 24.Lima CD, Klein MG, Hendrickson WA. Structure-based analysis of catalysis and substrate definition in the HIT protein family. Science. 1997;278:286–290. doi: 10.1126/science.278.5336.286. [DOI] [PubMed] [Google Scholar]
- 25.Wedekind JE, Frey PA, Rayment I. Three-dimensional structure of galactose-1-phosphate uridylyltransferase from Escherichia coli at 1.8 A resolution. Biochemistry. 1995;34:11049–11061. doi: 10.1021/bi00035a010. [DOI] [PubMed] [Google Scholar]
- 26.Gu M, Fabrega C, Liu SW, Liu H, Kiledjian M, Lima CD. Insights into the structure, mechanism, and regulation of scavenger mRNA decapping activity. Mol Cell. 2004;14:67–80. doi: 10.1016/s1097-2765(04)00180-7. [DOI] [PubMed] [Google Scholar]
- 27.Chen N, Walsh MA, Liu Y, Parker R, Song H. Crystal structures of human DcpS in ligand-free and m7GDP-bound forms suggest a dynamic mechanism for scavenger mRNA decapping. J Mol Biol. 2005;347:707–718. doi: 10.1016/j.jmb.2005.01.062. [DOI] [PubMed] [Google Scholar]
- 28.Huang KF, Liu YL, Cheng WJ, Ko TP, Wang AH. Crystal structures of human glutaminyl cyclase, an enzyme responsible for protein N-terminal pyroglutamate formation. Proc Natl Acad Sci USA. 2005;102:13117–13122. doi: 10.1073/pnas.0504184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teplyakov A, Wilson KS, Orioli P, Mangani S. High-resolution structure of the complex between carboxypeptidase A and L-phenyl lactate. Acta Crystallogr. 1993;49:534–540. doi: 10.1107/S0907444993007267. [DOI] [PubMed] [Google Scholar]
- 30.Aloy P, Companys V, Vendrell J, Aviles FX, Fricker LD, Coll M, Gomis-Ruth FX. The crystal structure of the inhibitor-complexed carboxypeptidase D domain II and the modeling of regulatory carboxypeptidases. J Biol Chem. 2001;276:16177–16184. doi: 10.1074/jbc.M011457200. [DOI] [PubMed] [Google Scholar]
- 31.Douangamath A, Walker M, Beismann-Driemeyer S, Vega-Fernandez MC, Sterner R, Wilmanns M. Structural evidence for ammonia tunneling across the (beta alpha)(8) barrel of the imidazole glycerol phosphate synthase bienzyme complex. Structure. 2002;10:185–193. doi: 10.1016/s0969-2126(02)00702-5. [DOI] [PubMed] [Google Scholar]
- 32.Li H, Ryan TJ, Chave KJ, Van Roey P. Three-dimensional structure of human gamma-glutamyl hydrolase. A class I glatamine amidotransferase adapted for a complex substate. J Biol Chem. 2002;277:24522–24529. doi: 10.1074/jbc.M202020200. [DOI] [PubMed] [Google Scholar]
- 33.Reynolds RA, Yem AW, Wolfe CL, Deibel MR, Chidester CG, Watenpaugh KD. Crystal structure of the catalytic subunit of Cdc25B required for G2/M phase transition of the cell cycle. J Mol Biol. 1999;293:559–568. doi: 10.1006/jmbi.1999.3168. [DOI] [PubMed] [Google Scholar]
- 34.Gliubich F, Gazerro M, Zanotti G, Delbono S, Bombieri G, Berni R. Active site structural features for chemically modified forms of rhodanese. J Biol Chem. 1996;271:21054–21061. doi: 10.1074/jbc.271.35.21054. [DOI] [PubMed] [Google Scholar]
- 35.Kim JH, Krahn JM, Tomchick DR, Smith JL, Zalkin H. Structure and function of the glutamine phosphoribosylpyrophosphate amidotransferase glutamine site and communication with the phosphoribosylpyrophosphate site. J Biol Chem. 1996;271:15549–15557. doi: 10.1074/jbc.271.26.15549. [DOI] [PubMed] [Google Scholar]
- 36.Mouilleron S, Badet-Denisot MA, Golinelli-Pimpaneau B. Glutamine binding opens the ammonia channel and activates glucosamine-6P synthase. J Biol Chem. 2006;281:4404–4412. doi: 10.1074/jbc.M511689200. [DOI] [PubMed] [Google Scholar]
- 37.Suresh CG, Pundle AV, Sivaraman H, Rao KN, Brannigan JA, Mcvey CE, Verma CS, Dauter Z, Dodson EJ, Dodson GG. Penicillin V acylase crystal structure reveals new Ntn-hydrolase family members. Nat Struct Biol. 1999;6:414–416. doi: 10.1038/8213. [DOI] [PubMed] [Google Scholar]
- 38.Coros AM, Swenson L, Wolodko WT, Fraser ME. Structure of the CoA transferase from pig heart to 1.7 A resolution. Acta Cryst. 2004;60:1717–1725. doi: 10.1107/S0907444904017974. [DOI] [PubMed] [Google Scholar]
- 39.Jacob U, Mack M, Clausen T, Huber R, Buckel W, Messerschmidt A. Glutaconate CoA-transferase from Acidaminococcus fermentans: the crystal structure reveals homology with other CoA-transferases. Structure. 1997;5:415–426. doi: 10.1016/s0969-2126(97)00198-6. [DOI] [PubMed] [Google Scholar]
- 40.Sandy J, Holton S, Fullam E, Sim E, Noble M. Binding of the anti-tubercular drug isoniazid to the arylamine N-acetyltransferase protein from Mycobacterium smegmatis. Protein Sci. 2005;14:775–782. doi: 10.1110/ps.041163505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patterson AW, Wood WJ, Hornsby M, Lesley S, Spraggon G, Ellman JA. Identification of selective, nonpeptidic nitrile inhibitors of cathepsin s using the substrate activity screening method. J Med Chem. 2006;49:6298–6307. doi: 10.1021/jm060701s. [DOI] [PubMed] [Google Scholar]
- 42.Kirby TW, Harvey S, Derose EF, Chalov S, Chikova AK, Perrino FW, Schaaper RM, London RE, Pedersen LC. Structure of the Escherichia coli DNA polymerase III epsilon-HOT proofreading complex. J Biol Chem. 2006;281:38466–38471. doi: 10.1074/jbc.M606917200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campobasso N, Costello CA, Kinsland C, Begley TP, Ealick SE. Crystal structure of thiaminase-I from Bacillus thiaminolyticus at 2.0 Å resolution. Biochemistry. 1998;37:15981–15989. doi: 10.1021/bi981673l. [DOI] [PubMed] [Google Scholar]
- 44.Smith CA, Anderson BF, Baker HM, Baker EN. Structure of copper- and oxalate-substituted human lactoferrin at 2.0 Å resolution. Acta Cryst. 1994;50:302–316. doi: 10.1107/S0907444994000491. [DOI] [PubMed] [Google Scholar]
- 45.Kawabata T. MATRAS: a program for protein 3D structure comparison. Nucl Acids Res. 2003;31:3367–3369. doi: 10.1093/nar/gkg581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skov LK, Mirza O, Henriksen A, De Montalk GP, Remaud-Simeon M, Sarcabal P, Willemot RM, Monsan P, Gajhede M. Amylosucrase, a glucan-synthesizing enzyme from the alpha-amylase family. J Biol Chem. 2001;276:25273–25278. doi: 10.1074/jbc.M010998200. [DOI] [PubMed] [Google Scholar]
- 47.Uitdehaag JC, Kalk KH, Van Der Veen BA, Dijkhuizen L, Dijkstra BW. The cyclization mechanism of cyclodextrin glycosyltransferase (CGTase) as revealed by a gamma-cyclodextrin-CGTase complex at 1.8-Å resolution. J Biol Chem. 1999;274:34868–34876. doi: 10.1074/jbc.274.49.34868. [DOI] [PubMed] [Google Scholar]
- 48.Hubbard SJ, Thornton JM. 1993. NACCESS. Department of Biochemistry and Molecular Biology, University College, London.
- 49.Bashton M, Chothia C. The generation of new protein functions by the combination of domains. Structure. 2007;15:85–99. doi: 10.1016/j.str.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 50.Chothia C. Proteins. One thousand families for the molecular biologist. Nature. 1992;357:543–544. doi: 10.1038/357543a0. [DOI] [PubMed] [Google Scholar]