

Abstract
The signal recognition particle (SRP) is a ribonucleoprotein complex which is crucial for the delivery of proteins to cellular membranes. Among the six proteins of the eukaryotic SRP, the two largest, SRP68 and SRP72, form a stable SRP68/72 heterodimer of unknown structure which is required for SRP function. Fragments 68e′ (residues 530 to 620) and 72b′ (residues 1 to 166) participate in the SRP68/72 interface. Both polypeptides were expressed in Escherichia coli and assembled into a complex which was stable at high ionic strength. Disruption of 68e′/72b′ and SRP68/72 was achieved by denaturation using moderate concentrations of urea. The four predicted tetratricopeptide repeats (TPR1 to TPR4) of 72b′ were required for stable binding of 68e′. Site-directed mutagenesis suggested that they provide the structural framework for the binding of SRP68. Deleting the region between TPR3 and TPR4 (h120) also prevented the formation of a heterodimer, but this predicted alpha-helical region appeared to engage several of its amino acid residues directly at the interface with 68e′. A 39-residue polypeptide (68h, residues 570–605), rich in prolines and containing an invariant aspartic residue at position 585, was found to be active. Mutagenesis scanning of the central region of 68h demonstrated that D585 was solely responsible for the formation of the heterodimer. Coexpression experiments suggested that 72b′ protects 68h from proteolytic digestion consistent with the assertion that 68h is accommodated inside a groove formed by the superhelically arranged four TPRs of the N-terminal region of SRP72.
Keywords: signal recognition particle, SRP, protein sorting, protein–protein interactions
Introduction
Sorting of secretory proteins is a fundamental cellular function in which the signal recognition particle (SRP) plays a central role. The SRP is a stable cytosolic ribonucleoprotein particle which acts in concert with ribosomes and membrane components. SRP binds to the signal sequence when the nascent polypeptide emerges from the ribosome. This step delays translation of the protein and is followed by the formation of a complex between the ribosome-bound SRP and the membrane-resident SRP receptor. The SRP is released from its membrane-bound state upon GTP-hydrolysis to resume translation and complete translocation.1–4
The SRP-mediated protein sorting mechanism has been conserved in all domains of life. The mammalian SRP contains a 303-nucleotide SRP RNA and six proteins, named SRP9, SRP14, SRP19, SRP54, SRP68, and SRP72 according to their approximate molecular weights in kilo-daltons.5 SRP9/14 and the terminal SRP RNA regions form the small domain (also referred to as Alu domain) which functions in translation delay. The large or S-domain of the dumbbell shaped human SRP contains proteins SRP19, SRP54, and SRP68/72 and participates in signal peptide binding and GTP-hydrolysis.6–8 In contrast to the multicomponent mammalian SRP which delivers proteins to the endoplasmic reticulum (ER),9 the SRP of most bacteria contains only one protein, a homologue of the SRP54 protein (Ffh), and a 4.5S or 6S RNA.10 Proteins SRP68 and SRP72 have not been found in the bacterial and archaeal genomes and thus appear to be a property of the eukaryotic SRPs.10–12
GFP-labeled human SRP68 and SRP72 were detected in the nucleolus and cytoplasm of transfected rat fibroblast cells suggesting a role of the proteins in SRP assembly.13 Saccharomyces cerevisiae SRP68 was shown to be required for stable SRP expression.14 A certain combination of mutations in SRP72 and SRP RNA led to accumulation of SRP precursors in the nucleus, further supporting the idea that SRP72 is implicated in the assembly of the SRP.15 Nevertheless, in vitro experiments with elastase treated canine SRP demonstrated that the SRP68/P72 heterodimer contributes directly to the signal peptide recognition function in the assembled particle.16 Recently, depletion of SRP68 and SRP72 by RNAi silencing was shown to be lethal for Trypanosoma brucei.17 Transfection of HeLa cells with shRNA-expression plasmids directed against SRP72 led to a reduction of the SRP RNA levels.18
Binding of SRP68/72 is impaired by SRP RNA mutations throughout the large domain of the human SRP with the most pronounced effects caused by changes in helix 5 and helix 8.19 Consistent with these findings, chemical modification and footprinting experiments show that SRP68/72 brings helices 6 and 8 closer together to prepare the SRP RNA for the binding of the highly conserved SRP54 protein.20 Because SRP72 binds only to a small section of helix 5, most RNA-protein contacts are likely to be brought about by SRP68.
We have shown that a single conserved adenosine residue within the eukaryotic 5e motif is essential for the formation of a complex with human SRP72.21 This portion of the SRP RNA binds to a relatively small 56-residue region near the C-terminus of SRP72 into which the consensus sequence PDPXRWLPXXER is imbedded. In contrast, the expansive RNA binding region of human SRP68 engages amino acid residues from positions 52 to 252.22
Proteins SRP68 and SRP72 share the same phylogenetic distribution and are released from the SRP as a stable heterodimer.10,16 We identified a stretch of 94 amino acid residues near the C-terminus of SRP68 (68e′) which binds to ∼150 amino acids from the N-terminal region of SRP72 (72b′). This 72b region is located within a predicted tandem array of four tetratricopeptide (TPR)-like motifs suggested to form a superhelical structure which binds to the C-terminal region of SRP68 by unknown means.22 In the presented work, we explore this unusually stable protein–protein interaction and identify the amino acid residues required for the formation of the human SRP68/72 heterodimer. The results from these experiments not only yield first insights into the molecular details of an exceptionally stable protein–protein interaction, but also are expected to guide urgently needed high-resolution structural investigations.
Results
SRP68/72 regions which participate in the formation of a heterodimer
Previous investigation of the interactions between human SRP68 and SRP72 identified the C-terminal region of SRP68 encompassing the amino acid residues at positions 530 to 620 (fragment 68e′) as being solely responsible for the binding to SRP72.22 We mixed Thioredoxin and 6xHis-tagged 68e′ (Thx-H68e′) with recombinantly generated SRP72 fragments which collectively extended over the full-length human SRP72.23 Nickel affinity chromatography, followed by SDS-PAGE of the free and bound fragments, showed that 72b (amino acid residues 1 to 285) and 72b′ (1 to 163) exhibited a high affinity for Thx-H68e′. In contrast, fragments 72a′ (residues 167 to 448) and 72c (residues 447 to 665) did not bind to Thx-H68e′.22 A comparison of aligned representative SRP72 sequences confirmed the presence of four predicted tetratricopeptide (TPR)-like motifs in 72b′ as well as the conserved or invariant nature of A64, Y86, Y89, Q117, and Y120 (“Materials and Methods” section). An insertion of 20 residues between TPR3 and TPR4 was designated as h120 for its predicted alpha-helical character and the presence of the conserved tyrosine at position 120 [Fig. 1(A)]. The alignment of representative eukaryotic SRP68 sequences did not reveal any striking secondary structure features in the 68e′ region. A possible exception was a weakly predicted beta-turn at F583 which is immediately followed by a short alpha-helix embedded into proline-rich surroundings [Fig. 1(C)].
Figure 1.
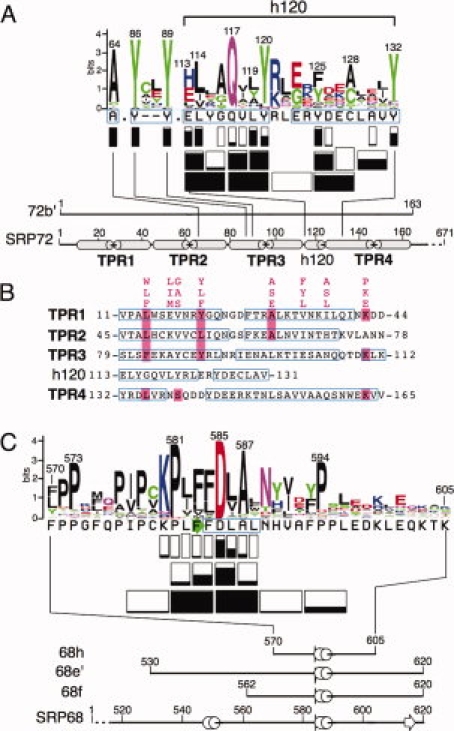
Properties of the interface between human SRP72 and SRP68. (A) The protein sequence logo of the 72b′ region was prepared using the interface at http://weblogo.berkeley.edu/logo.cgi. The height of each symbol is proportional to its frequency in the alignment of representative SRP72 sequences obtained as described in “Materials and Methods” section. Rectangles below each sequence indicate how many residues were altered. Negative effects of the ability to form complexes are shown by the extent of black filling and are listed in Table I. Not considered are the results obtained from competition experiments with A117–118 Q117A, and V118A [Fig. 4(D)]. Below the logo, amino acid positions are labeled in 20-residue increments according to full-length human SRP72. The termini of fragment 72b′ are numbered. Alpha helices, predicted from alignments of representative eukaryotic sequences (available at http://rnp.uthct.edu/rnp/SRPDB/srpprotein.html) are marked as gray cylinders. The positions of four tetratricopeptide repeats (TPR1 to TPR4) as well as a small predicted alpha-helix located between TPR3 and TPR4 (h120) are indicated. (B) Alignment of the four N-terminal TPR-like regions of human SRP72 and the h120 insertion between TPR3 and TPR4. The TPR consensus sequence24 and matching SRP72 residues are highlighted in pink. Amino acid residues are numbered according to full-length human SRP72.23 Regions predicted to be alpha-helical as determined by PSIPRED25 are shown in blue frames. (C) Protein logo of the 68h region with effects of mutant polypeptides depicted as in panel A. The termini of fragments 68e′, 68f, and 68h are numbered according to full-length human SRP68. Alpha helices and a beta turn, predicted from alignments of representative SRP68 sequences, are marked in blue and as a green arrow, respectively.
To measure the interactions between 68e′ and 72b′, we prepared GST-k72b′, a fusion protein of favorable solubility which contains a N-terminal Glutathione-S-transferase (GST) tag and a kinase site for radioactive labeling with [γ-32P]ATP. Sample aliquots were incubated with Ni-paramagnetic beads followed by measuring the bead-associated radioactivity in a scintillation counter (see “Materials and Methods” section). 0.5 to 1 nM of 32P-labeled GST-k72b′ were mixed with increasing known amounts of purified Thx-H68e′ until binding was saturated. The apparent association constant (K′a) was calculated by determining the concentration of unbound Thx-H68e′ that led to the formation of equal amounts of free and complexed GST-72b′. Assuming that one molecule of GST-k72b′ bound to one molecule of Thx-H68e′, the value of K′a was about 108 M−1 [Fig. 2(C)].
Figure 2.
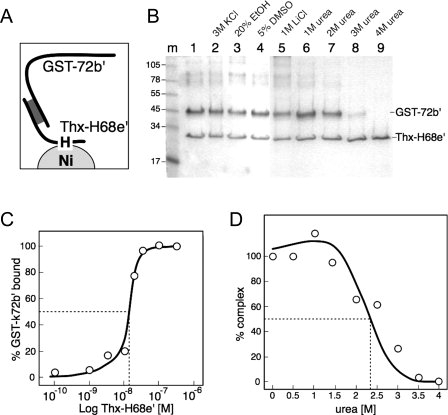
Binding of the GST-k72b′ fusion protein to the Trx-H68e′ polypeptide. (A) Depiction of the components in the assay. (B) Bacterial lysates overexpressing N-terminally GST-tagged 72b′ (GST-72b′) or Thioredoxin and His-tagged 68e′ (Thx-H68e′)22 were mixed to form complexes and subjected to different conditions as indicated. Ni-NTA magnetic agarose beads were added, concentrated in a magnetic separator, and the polypeptides that remained bound to beads were analyzed by SDS PAGE and Coomassie blue staining. Molecular masses in kDa of marker polypeptides are indicated in lane m. (C) Low amounts of 32P-labeled GST-k72b′ were mixed with increasing amounts of purified his-tagged Trx-68e′ (Thx-H68e′) as indicated, and the amount of radioactivity associated with the Ni-paramagnetic beads was measured by scintillation counting as described in “Materials and Methods” section. (D) Effect of increasing urea concentration on the disruption of the complex.
Disruption of the SRP68/72 interface
Human SRP68/72 had been shown to separate from the SRP RNA in a buffer containing 2 M KCl and remain heterodimeric.16 We incubated complexes between GST-72b′ and Thx-H68e′ at various potentially disruptive conditions, and measured the association of the polypeptides with Ni-NTA magnetic agarose beads. Figure 2(B) shows that the complex was stable at room temperature in buffers of elevated ionic strength (3 M KCl and 1 M LiCl, lanes 2 and 5), and remained intact in 20% ethanol and 5% DMSO (lanes 3 and 4). The intensities of the Coomassie blue stained GST-72b′ and Thx-H68e′ were stoichiometrically constant and consistent with the formation of 1:1 complexes. This was observed even when an excess of SRP72 was mixed in the initial preparation (not shown). Disruption of the complex occurred in urea at concentrations of 2 M and above, presumably due to the denaturation of GST-72b′, Thx-H68e′, or both polypeptides [Fig. 2(B), lanes 6 to 9]. Incubation at room temperature in 0.5 M increments of denaturant showed that equal amounts of free and complexed GST-72b′ were present at an urea concentration of ∼2.4 M [Fig. 2(D)].
As GST is known for its a tendency to form dimers in solution26 we measured the urea-dependent disruption of complexes composed of untagged 68e′ and 6xHis tagged 72b′ (H72b′) prepared as described in “Materials and Methods” section. Incubations were carried out at 4°C using increasing urea concentrations of up to 2 M followed by the recovery of the material which remained bound to Ni-NTA magnetic agarose beads [Fig. 3(A)]. Equal amounts of free and bound 68e′ were observed at an urea concentration of ∼0.9 M suggesting that the GST and Thioredoxin domains provide increased stability. When incubated in buffer with 1 M urea for up to 16 hours, the amount of complex decreased only slightly, suggesting negligible mild effects on stability during extended times of exposure to the denaturant [inserted graph of Fig. 3(A)].
Figure 3.
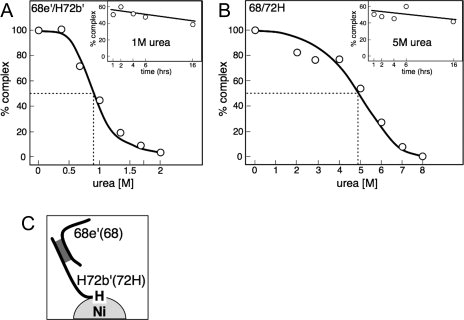
Effect of urea and incubation times on the stability of the SRP68/72 interaction. (A) Stability of 68e′/H72b′. Complexes were coexpressed in E. coli and subjected to increasing urea concentrations and incubation times. Polypeptides, either free or associated with Ni-NTA magnetic agarose beads, were visualized by SDS PAGE as described in “Materials and Methods” section. Dashed lines indicate the urea concentration that led to the formation of equal amounts of complex and free 68e′. The insert shows the stability of the complex over time when incubated in 1 M urea. (B) SRP68/72H was coexpressed in S. cerevisiae and purified as described in “Materials and Methods” section. Dashed lines indicate the urea concentration (∼5 M) that led to the formation of equal amounts of complexed and free polypeptides. The insert shows the stability of the 68/72H heterodimer over time when incubated in 5 M urea. (C) Depiction of the interacting components in the assay.
We also investigated the full-length SRP68/72 complex, coexpressed in yeast and purified by Ni-NTA chromatography via the 6xHis tag attached to the C-terminus of SRP7220 (“Materials and Methods” section). In contrast to 68e′/H72b′, dissociation of 68/72H was observed over a wide range of urea concentrations. Equal amounts of free and complexed SRP68 were present at a relatively high urea concentration of ∼4.9 M. When incubated in a buffer containing 5M urea, the complex remained essentially stable for several hours [Fig. 3(B)]. In the light that 72b′ and 68e′ are the only regions which participate in the formation of a heterodimer this result suggests indirect stabilizing effects of regions which are located adjacent to the SRP68/72 interface.
Effects caused by alterations in GST-72b′
Three mutant derivatives of GST-72b′ were purified in attempting to reduce the size of the 72b′ fragment (residues 1 to 163) to a minimum while retaining its ability to bind to the 68e′ region. ΔTPR1 is a deletion of the N-terminal TPR (residues 11–44 removed), ΔTPR4 lacks residues 132–165, and Δh120 is a deletion of residues 113–131. The polypeptides were purified and used in a binding assay described in “Materials and Methods” section. The result is depicted in Figure 4(A). As expected, GST-k72b′ associated with the Ni-NTA magnetic agarose beads only when mixed with Thx-H68e′ [lanes 4 and 6 in Fig. 4(A)]. ΔTPR1, ΔTPR4, and Δh120 lost their ability to form a complex with Thx-H68e′ [Fig. 4(A), lanes 8, 10, and 12].
Figure 4.
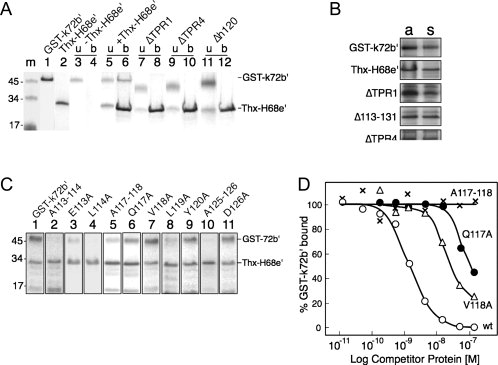
Effects of mutations in GST-72b′ on the formation of complexes with Thx-H68e′. (A) Purified GST-k72b′ or mutant polypeptides were incubated with Thx-H68e′, mixed with Ni-NTA magnetic agarose beads, and polypeptides were analyzed by SDS PAGE and Coomassie blue staining. Molecular mass markers in kDa are shown in lane m. Purified GST-k72b′ and Thx-H68e′ are in lanes 1 and 2, respectively. Lanes 3 and 4: unbound (u) and bound (b) GST-k72b′ in the absence of Thx-H68e′; lanes 5 and 6: in the presence of Thx-H68e′; lanes 7 and 8: deletion of the N-terminal tetratricopeptide (ΔTPR1); lanes 9 and 10: deletion of the forth tetratricopeptide (ΔTPR4); lanes 11 and 12: deletion of residues 113 to 131 (Δh120). (B) Aliquots of proteins (a) in E. coli cell lysates expressing wildtype or mutant polypeptides. Supernatants (s) after centrifugation of the same lysates analyzed by SDS PAGE. Equal intensities of the Coomassie blue stained bands from each polypeptide indicate it is completely soluble. (C) Binding activities of selected mutant derivatives of the GST72b′ polypeptide as indicated and analyzed as described in panel A showing the bead-bound polypeptides. (D) Binding of 32P-GST-k72b′ to Thx-H68e′ in the presence of unlabeled competitor proteins: GST-k72b′ (wt, open circles); A117–118 (marked x); Q117A (dots); and V118A (triangles). The amount of bound radioactive GST-k72b′ was determined by scintillation counting as described in “Materials and Methods” section.
Concerned about the possibility that the three deletion polypeptides are unavailable for binding in solution due to low solubility, lysates containing wildtype, ΔTPR1, ΔTPR4, or Δh120 were subjected to centrifugation under binding conditions. Aliquots of the supernatants and pellets where analyzed by SDS PAGE. Figure 4(B) illustrates that the solubilities of the mutant polypeptides were largely unaffected. Table I shows semiquantitatively the solubilities of all SRP72-derived mutant polypeptides used in this study.
Table I.
Activities of the GST-72b′ and H68h Mutant Derivatives
| SRP72 mutants | Activity (%) | Solubility | SRP68 mutants | Activity (%) |
|---|---|---|---|---|
| GST-k72b′ | 100 | +++ | H68h | 100 |
| ΔTPR1 (Δ11–44) | 0 | +++ | A577–580 | 97 ± 9 |
| A64L | 0 | + | K580A | 89 ± 11 |
| Y86A | 0 | + | A581–584 | 0 |
| Y89A | 9 ± 3 | + | A581–582 | 85 ± 10 |
| Δh120 (Δ113–131) | 0 | ++++ | P581A | 96 ± 3 |
| A113–116 | 0 | ++ | L582A | 108 ± 7 |
| A113–114 | 14 ± 2 | + | A583–584 | 56 ± 28 |
| E113A | 20 ± 6 | +++ | F583A | 94 ± 3 |
| L114A | 8 ± 6 | ++ | F584A | 97 ± 5 |
| A115–116 | 89 ± 20 | ++++ | A585–588 | 0 |
| G116A | 100 ± 1 | ++++ | A585–586 | 26 ± 7 |
| A117–120 | 0 | ++++ | D585A | 20 ± 10 |
| A117–118 | 14 ± 10 | +++ | L586A | 60 ± 9 |
| Q117A | 82 ± 6 | ++++ | A587–588 | 84 ± 17 |
| V118A | 98 ± 5 | +++ | A587L | 84 ± 2 |
| A119–120 | 0 | +++ | L588A | 102 ± 1 |
| L119A | 18 ± 3 | ++ | A589–592 | 96 ± 23 |
| Y120A | 47 ± 9 | ++++ | A593–596 | 77 ± 4 |
| A121–124 | 87 ± 15 | ++++ | ||
| A125–128 | 16 ± 6 | + | ||
| A125–126 | 15 ± 3 | +++ | ||
| Y125A | 21 ± 5 | +++ | ||
| D126A | 96 ± 6 | +++ | ||
| A127–128 | 120 ± 28 | +++ | ||
| A129–131 | 42 ± 10 | +++ | ||
| A130L | 95 ± 12 | +++ | ||
| Y132A | 34 ± 9 | +++ | ||
| ΔTPR4 (Δ132–165) | 0 | +++ |
Binding activities of the GST-72b′ mutant polypeptides were determined by measuring the protein amounts associated with Ni-NTA magnetic agarose beads which had been primed with Thx-H68e′ relative to the binding activity of GST-k72b′. The activities of mutants of H68h were determined in coexpression experiments with GST-k72b′ followed by incubation of the E. coli lysates with Ni-NTA magnetic agarose beads or GST Sepharose. The variable solubilities of the GST72b′ derivatives was assessed by centrifugation followed by SDS PAGE of polypeptides in the pellets and supernatants as indicated by ++++ (80–100%), +++ (50–80%), ++ (20–50%), and + (5–20%). Under the conditions used, H68h and all its mutant derivates exhibited favorable solubilities.
Altering the conserved residues at positions 64, 86, or 89 yielded polypeptides with the solubility reduced to 20% or less. Nevertheless, sufficient material could be isolated from the supernatants of the centrifugated lysates to carry out meaningful binding experiments. The polypeptides in the soluble fractions of A64L, Y86A, and Y89A were incapable to bind to Thx-H68e′. Changing the conserved tyrosine at position 132 to an alanine yielded a soluble protein with a moderate (34% ± 9%) binding activity (see Table I).
Alanine-scanning was used to investigate the h120 insertion between TPR3 and TPR4 suspected to be required for the specific binding to SRP68. Changes to alanines were made in groups of four or three adjacent residues, and binding of the mutant GST-72b′ derivatives to Thx-H68e′ was examined. The A121–124 and A129–131 mutant polypeptides remained active to a significant degree (87% ± 15% and 42% ± 10%, respectively). In contrast, the activities of A113–116, A117–120, and A125–128 were reduced to 16% or less. Mutant polypeptide A117–120 had a favorably solubility but was inactive. Conversely, although the solubility of A125–128 was low, some binding (16% ± 6%) was observed. This suggested that binding could be observed unimpeded even when the solubility of the mutant polypeptides was poor (Table I).
Of the dialanine substitutions, no effects on binding were observed with A115–116 and A127–128. Activities were nearly completely lost in polypeptides carrying the A113–114, A117–118, A119–120, or A125–126 mutations [Fig. 4(C), summarized in Figure 1A and Table I]. Analysis of the corresponding single residue changes showed that both E113 and the moderately conserved L114 were required. Also L119 and Y120 were found to be important for the formation of a complex. Y120A retained a binding activity of 47% ± 9% despite the highly conserved nature of this tyrosine. With respect to positions 125 and 126, only Y125 was found to be somewhat important (21% ± 5%).
The A117–118 double mutant was largely inactive (14% ± 10%) but changes of individual residues yielded polypeptides with substantial binding capabilities (82% ± 6% for Q117A, 98% ± 5% for V118A). Because Q117 stood out as an invariant residue in the alignment of representative SRP72 sequences, this result was surprising. To investigate the specificity of the interaction between GST-72b′ and Thx-H68e′ we carried out competition experiments using a small fixed amount of 32P-labeled GST-k72b′ and 8nM of Thx-H68e′ in the presence of increasing amounts of unlabeled competitor proteins. Competition effects were compared at the half-saturation point of the complex [Fig. 2(C)]. The concentration of the Q117A mutant polypeptide required to reduce binding to 50% was nearly two orders of magnitude higher than for wildtype GST-k72b′. Compared to Q117A, stronger competition was observed with the V118A change, but it was still significantly weaker than the competition with GST-k72b′. Within the limits of the assay, the A117–118 double-mutation polypeptide was unable to compete. We conclude that the reason for the invariance of the glutamine at position 117 of human SRP72 is due to its important contribution in the formation of a complex with the 68e′ region.
A 39-amino acid SRP68-derived peptide binds to 72b′
Smaller derivatives of fragment 68e′ (residues 530 to 620) containing the previously identified patch of conserved residues at 580 to 587 were expressed in E. coli.21 Lysates containing polypeptide 68f (residues 562 to 620) expressed from a Kanamycin resistant plasmid (“Materials and Methods” section) were mixed with 6xHis-tagged 72b′ (H72b′) and samples were analyzed using the assay depicted in Figure 3(C). Although yields were low, active binding was observed by colocalization of the H72b′ and 68f polypeptides with the Ni-NTA beads (not shown). Complexes of higher yield were obtained by cotransforming the Kanamycin resistant 68f construct with the Ampicillin resistant H72b′-encoding plasmid [Fig. 5(A), lane 2].
Figure 5.
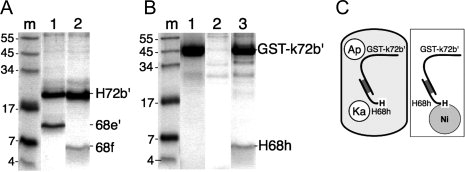
Binding of 68f and H68h to the 72b′ region. (A) Recombinant fragments 68e′ (residues 530 to 620 of human SRP68) and 68f (562 to 620) were coexpressed with his-tagged 72b′ in E. coli, lysates were incubated with Ni-NTA Superflow beads, and the bound polypeptides were analyzed by SDS PAGE. Lane m: molecular mass marker in kDa; lane 1: H72b′ coexpressed with 68e′; lane 2: H72b′ coexpressed with 68f. (B) Similar to (A), but using His-tagged 68h (H68h, residues 570 to 605) in coexpression with GST-k72b′. Lane 1: purified GST-k72′; lane 2: lack of binding of GST-k72′ to Ni-NTA Superflow beads; lane 3: lysate coexpressing GST-k72b′ and H68h. (C) Depiction of the coexpression of GST-k72b′ and H68h inside an E. coli cell (left); Ap is for Ampicillin, Ka is for Kanamycin resistance. Purification of complexes on Ni-NTA beads (right).
The N-terminally 6xHis-tagged 68h peptide (H68h) contained the 39 amino acid residues at positions 570 to 605 and was encoded by a Kanamycin resistance plasmid. Different E. coli host strains and growth conditions were explored to express H68h, but all attempts to isolate intact H68h proved unsuccessful. However, cotransformation with the Ampicillin resistant GST-k72b′-encoding plasmid [depicted in Fig. 5(C)] demonstrated that H68h was translated and capable to form a stable heterodimer [Fig. 5(B), lane 3].
Alanine-scanning mutagenesis of the central region of H68h
The 68h region is proline-rich and contains near its center the three conserved amino acid residues K580, P581, A587 as well as the invariant D585. Introducing five consecutive tetraalanine blocks within the central region of 68h showed that the alterations of A581–584 and A585–588 polypeptides abolished binding to GST-k72b′. In contrast, mutations in A577–580, A589–592, and A593–596 reduced the binding activities slightly or not at all. Of the dialanine changes, binding was significantly reduced in A585–586 (26% ± 7%), A583–584 (56% ± 28%), and less so in A581–582 and A587–588 (see Table I).
We took advantage of the fact that H68h was proteolyzed during expression in E. coli unless it had an opportunity to form a complex with the coexpressed 72b′ region. Aliquots of the lysates were mixed with Ni-NTA beads as indicated in Figure 5(C) and the bead-bound polypeptides were analyzed by SDS PAGE. Figure 6(A) demonstrates that H68h formed a protected complex with GST-k72b′. As expected, ΔTPR1 and ΔTPR4, and to some extent the Δh120 polypeptide, did not bind to H68h.
Figure 6.
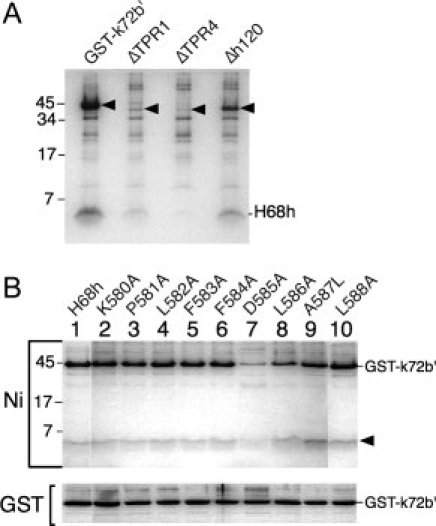
Binding of coexpressed mutated polypeptides. (A) Protection of H68h by GST-k72b′. Arrowheads indicate the migrations of GST-k72b′, ΔTPR1, ΔTPR4, and Δh120 polypeptide. Lysates coexpressing H68h were incubated with Ni-NTA beads and the bound polypeptides were separated by SDS PAGE. (B) Amino acid residues in H68h required for binding to GST-k72b′. Analysis of lysates coexpressing GST-k72b′ with H68h or H68h mutant derivatives (as indicated) on Ni-NTA beads (Ni) or glutathione Sepharose 4B (GST). The arrowhead points to the 5.2 kDa wildtype or mutant H68h polypeptides.
Individual residues at positions 580 to 588 were changed to alanine codons in the Kanamycin resistant H68h expression plasmid. (An exception was the change to leucine of the naturally occurring alanine at position 587.) Wildtype and mutant plasmids were cotransformed with the GST-k72b′ expression plasmid. Protein production was induced by adding IPTG, aliquots of the lysates were mixed with Ni-NTA beads [Fig. 5(C)] or glutathione Sepharose, and the bound polypeptides were analyzed by SDS PAGE as described in “Materials and Methods” section. The results shown in Figure 6(B) and listed in Table I demonstrate that a change of the invariant D585 to alanine reduced the binding activity to 20% ± 10%. Less pronounced effects were observed with L586A (binding of 60% ± 9%) and A587L (84% ± 2%). Altering the conserved residues K580 and P581 had insignificant effects on the formation of a complex.
Discussion
Although the genes for SRP68 and SRP72 are located on different chromosomes, the proteins exist and have been studied predominantly in their heterodimeric form. Conditions of even higher ionic strength (3 M KCl) than used earlier to isolate canine SRP68/72 yielded stable GST-72b′/Thx-H68e′ complexes.16 Attempts to distinguish between electrostatic and hydrophobic effects of this strong protein–protein interaction were unsuccessful.
SRP68 and SRP72 could be separated by disruption of hydrogen bonds and unfolding using moderate concentrations of urea.27 The investigated complexes likely shared identical interactions, but the urea concentration needed for the separation of the bound polypeptides varied from 0.9 M to 4.9 M. These differences might be due to stabilizing effects caused by GST, Thioredoxin, or regions in SRP68 and SRP72 when positioned near the protein–protein interface. For example, the predicted TPR5 and TPR6, although not essential for binding, might indirectly add stability to the adjacent 72b′ region.23 It is also possible that full-length SRP68/72 is particularly resistant to denaturation as it was coexpressed and isolated from yeast as a complex. In contrast, the GST-72b′/Thx-H68e′ and 68e′/H72b′ complexes were assembled in vitro from their components and may have had fewer opportunities to stabilize.
Due to its modular character, the deletion of the N-terminal TPR1 or the C-terminal TPR4 from the active 72b′ region was expected to yield properly folded polypeptides. Indeed, the favorable solubility of the deletion mutant proteins indicated that no disruptions of the overall structure had occurred (Table I). The protein with the deletion of h120 was also soluble, suggesting that removal of this predicted alpha-helix allows proper folding. Although likely to be correctly folded, the ΔTPR1 and ΔTPR4 polypeptides were incapable of binding to 68e′ suggesting that at least four superhelically arranged TPRs are needed to form a groove which is sufficiently large to accommodate SRP68 residues 570 to 605 of the H68h polypeptide. This mode of interaction has been observed in other TPR-mediated protein–protein complexes24 and is consistent with the observed proteolytic resistance of H68h when coexpressed with GST-k72b′ [Fig. 5(B)].
Changes of the residues at positions 64, 86, and 89 in TPR2 and TPR3 yielded inactive polypeptides of poor solubility [Fig. 1(A) and Table I]. Consistent with these findings, A64 and Y89 are part of the TPR consensus sequence and likely to be required for maintaining properly folded TPRs in a superhelical arrangement.28 Mutations at these consensus position have been reported to result in a disruption of the function in several other TPR-containing proteins.29 The contribution of the conserved Y86 to folding is less well understood but this residue occupies an important eighth position of TPR3 [Fig. 1(B)]. The TPRs of SRP72 lack conserved residues expected to decorate the superhelical scaffold and engage another protein. Therefore, the four TPRs function primarily in maintaining the structural framework for the binding of SRP68.
Lack of knowledge about the structure of the predicted h120 alpha-helix makes it difficult to ascertain its role on the molecular level. The presence of the invariant Q117 and the conserved Y120, as well as the uniqueness of h120 within the repeated TPR framework, suggests a direct engagement of this region with SRP68 [Fig. 1(A)]. Although we cannot exclude the possibility that h120 modulates the conformation of the predicted TPR superhelix, this appears to be unlikely as SRP68 engages via a single residue [D585, Fig. 1(C)].
Our approach for identification of the amino acid residues which maintain the SRP68/72 interface provided important insights but was limited by its semiquantitative character. This became evident when the A117–118 double mutant was shown to be inactive, whereas alterations of its individual residues yielded essentially active polypeptides. As predicted from the invariance of Q117, the Q117A mutant polypeptide did not compete in followup experiments. Due to the lack of high-resolution data for the SRP68/72 interface, we refrained from carrying out elaborate competition experiments for all the potentially involved residues.
Within SRP68, the 68h region displays the highest density of proline residues. Several other protein–protein interactions involve proline-rich amino acid sequences.30 Three highly conserved amino acid residues present themselves in the 580–588 region, but when changed did not abolish the binding of the mutant peptides to GST-k72b′. Only the invariant D585 proved to be important [Fig. 1(C)]. Given the complexity of the interface with respect to the SRP72 portion [Fig. 1(A)], it is premature to speculate about how this aspartic acid residue might engage the 72b′ region.
We have identified the amino acid residues required for the formation of SRP68/72 and demonstrated that coexpression can be used to conveniently monitor the formation of protein–protein complexes if at least one unbound form is proteolytically sensitive. By having achieved a reduction of the size of the SRP68/72 interface to its minimum, new opportunities for solving the high-resolution structure of this interesting protein–protein interaction are likely to emerge.
Materials and Methods
Purification of full-length human SRP68/72H
Using the Yeast Transformation System 2 kit (Clontech) competent yeast BCY123 cells were prepared by the Lithium acetate method and were transformed with pRS68–72His (kindly provided by K. Nagai, MRC, Cambridge, UK). This plasmid encodes both full-length human SRP68 and C-terminally His-tagged human SRP72. Transformants were selected by growth for 3 to 4 days at 30°C on -Ura plates prepared from minimal synthetic defined (SD) agar base supplemented with -Ura DO (Clontech). Two colonies were transferred into 10 mL of -Ura medium supplemented with 2% glucose, Adenine hemisulfate (Sigma) and DL-Tyrosine (ICN Biochemicals) followed by shaking at 30°C for 16 to 17 hours. The culture was added to 100 mL of supplemented -Ura medium with Tyrosine, Adenine hemisulfate, and containing 2% d-Raffinose pentahydrate (Sigma). Shaking was continued for 7 to 8 hours until the A600 reached ∼0.8. A 20% d-Galactose (MP Biomedicals) solution was added to a final concentration of two percent and incubation was continued for 16 to 17 hours. Cells were harvested by centrifugation in a 50 mL conical tube, washed with 100 mL of ice-cold sterile deionized water, and the pelleted cells were stored at −70°C.
One gram of frozen cells were resuspended in 400 μL of CelLytic Y Cell Lysis reagent (Sigma) containing 10 mM 2-ME. The cell suspension was mixed with 150 μL of a protease inhibitor, equivalent to one tenth of one EDTA-free complete mini protease inhibitor cocktail tablet (Roche). Four grams of acid washed glass beads (425 to 600 μm in diameter, Sigma) were added to the sample, followed by vortexing in the cold at maximum speed, eight times for 30 s with interruptions by placing the sample for 1 min on ice. The lysed cells were mixed with 2 mL of 75 mM Na-phosphate, pH 7.4, 750 mM NaCl, 15 mM Imidazole, 5.5 mM 2-ME, 15% glycerol, and 0.75 M urea. The glass beads were pelleted by centrifugation at 2,000 rpm in a Sorvall HB1000B rotor for 5 min at 4°C. The removed supernatant (∼3 mL) was incubated on ice for 30 min., transferred to a centrifuge tube (Beckman N0. 349622), and subjected to a high-speed centrifugation (50,000 rpm, 105,000 g, Beckman TLA-100.3) for 1 hour at 4°C. The supernatant from the high-speed spin was added to a 500 μL suspension of Ni-NTA Superflow beads (Qiagen) which had been equilibrated in 50 mM Na-phosphate, pH 7.4, 500 mM NaCl, 10 mM Imidazole, 5 mM 2-ME, 10% glycerol, and 0.5 M urea (binding buffer). The sample was subjected to slow rotation for 1 hour at 4°C and then loaded stepwise onto a 0.5 mL spin-column (Evergreen Scientific). The column was washed with 5 mL of binding buffer containing 30 mM Imidazole. SRP68/72 was eluted stepwise with binding buffer containing 100 to 300 mM Imidazole. Aliquots of the eluate fractions were analyzed by SDS PAGE on 8% Tris-tricine gels, pooled appropriately, and dialyzed at 4°C against 20 mM Na-phosphate, pH 7.4, 300 mM NaCl, 5 mM 2-ME, and 30% glycerol. The SRP68/72 preparation was stored at −20°C. The protein concentration was determined by SDS-PAGE of sample aliquots and Coomassie blue staining using a standard curve generated with known amounts of lysozyme.
Preparation of GST-72b′ and GST-k72b′
The coding sequence of human SRP72 (GenBank accession no. 076094) was assembled using standard molecular biology techniques from clones identified in a lambda-gt10 cDNA library of human HepG2 hepatoma cells as well as a 191-bp HindIII fragment excised from EST H07969 (Genome Systems) as described.13 For bacterial expression, the gene was inserted into pGEX-2TK (Pharmacia) to yield pGST-72, encoding N-terminally GST tagged human SRP72. A derivative, pGST-72b′ (amino acid residues 1 to 163, see Supporting Information), was constructed using suitable restriction enzymes in combination with PCR and ligation of the amplified DNA fragments. Competent E. coli DH5-alpha cells (Life Technologies) were transformed with the ligation mixtures and grown on LB plates containing 100 μg/mL Ampicillin. Plasmids were prepared on a small scale. Clones were chosen after restriction mapping and according to their ability to express polypeptides of the expected size. Plasmid DNAs were purified by CsCl density gradient centrifugation. Commercial providers were used to verify the sequence. pGST-k72b′, containing a site for radioactive labeling of the fusion protein by protein kinase and [γ-32P]ATP was constructed by the insertion of a synthetic DNA adaptor into the NcoI site of pGST-72b′.
For overexpression and purification of GST-tagged 72b′, competent E. coli Rosetta pLysS (Novagen) cells were transformed with pGST-72b′ or pGST-k72b′ and incubated over night at 37°C on LB agar plates containing 200 μg/mL Ampicillin and 34 μg/mL Chloramphenicol. Several colonies were transferred into 50 mL of LB containing antibiotics. Cells were grown by shaking at 37°C until the A600 reached 0.2, at which time IPTG was added to a final concentration of 1 mM. The cultures were incubated with continuous shaking at 37°C for 2 hours. Cells were harvested by centrifugation and frozen at −70°C. Frozen cells were resuspended in 6.5 mL of ice-cold 50 mM Na-phosphate, pH 8.0, 200 mM NaCl, 5 mM EDTA, 5 mM DTT (lysis buffer) and sonicated, using a Sonic Dismembrator Model 300 (Fisher Scientific) at a setting of 35 percent, three times for 15 s with 15 s intervals on ice. The lysate was subjected at 4°C to a low-speed centrifugation at 15,000 rpm using a Sorvall SS34 rotor for 20 min. The supernatant of the low-speed spin (1.6 mL) was mixed with 100 μL of a glutathione Sepharose 4B suspension (GE Healthcare Life Sciences) equilibrated in 50 mM Na-phosphate, pH 6.5, 50 mM NaCl, 5 mM EDTA, 5 mM DTT (equilibration buffer) followed by rotation of the sample at room temperature for 1 hour. The Sepharose beads were washed twice for 30 min with 500 μL of equilibration buffer. The protein was eluted by the addition of 200 μL of 50 mM Na-phosphate, pH 7.5, 500 mM NaCl, 0.1% Tween-20, 5 mM 2-ME, and 20 mM Glutathione (reduced form). For the preparation of untagged polypeptides, the Sepharose-bound GST fusion proteins was digested overnight at room temperature with 0.43 NIH-units of thrombin (Sigma) in 400 μL of 50 mM Na-phosphate (pH 8.0), 50 mM NaCl, 1 mM MgCl2, 10% glycerol, eluted, and stored on ice until further use within one month.
His-tagged 72b′ (H72b′) construction and purification
pH72b′, a plasmid encoding a tag of six histidines and a TEV protease site at the N-terminus of 72b′ (amino acid residues 1 to 166 of human SRP72, see Supporting Information) was constructed by the ligation of restricted and annealed synthetic oligonucleotides into pET (Novagen). The H72b′ polypeptide was expressed at room temperature in E. coli Rosetta (DE3) pLysS cells by transformation with pH72b′, selection on Ampicillin/Chloramphenicol plates, and induction of a liquid culture with 0.5 mM IPTG for 4 hours at an A600 of 0.2. Cells were lysed in 50 mM Na-phosphate, pH 7.4, 500 mM NaCl, 5 mM 2-ME, 10 mM Imidazole, 5 M urea, and kept for 30 min. on ice. The lysate was subjected to a high-speed centrifugation (Beckman TLA-100.3 rotor, 50K rpm for 1 hour at 4°C). The supernatant (∼3 mL) was harvested, mixed with 400 μL of Ni-NTA Superflow beads (Qiagen) by slow rotation for 1 hour min. at 4°C, and loaded stepwise into a 0.5 mL column (Evergreen). The column was washed with phosphate buffer to remove the urea in one molar increments. The H72b′ protein was eluted in buffer containing 50 mM Na-phosphate, pH 7.4, 170 mM NaCl, 5 mM 2-ME, and 150 mM Imidazole. In the described experiments, the Ampicillin resistant H72b′ plasmid was used in coexpression experiments with the Kanamycin resistant 68f-encoding plasmid.
Isolation of SRP68 fragments containing the SRP72 binding site
Thx-H68e′, a Thioredoxin and His-tagged 68e′ fragment of human SRP68 (residues 530 to 620) was expressed and purified as described previously.21 Plasmids p68e′ and p68f for the expression of untagged polypeptides 68e′ (residues 530 to 620) and 68f (562 to 620) respectively were generated by PCR from the pThx-H68e′ followed by cloning of the amplified restricted DNAs into Kanamycin resistant pET28a (Novagen). His-tagged 68h (H68h, residues 570 to 605) was assembled by ligation of two complementary synthetic oligonucleotides compatible with the NcoI and EcoRI restrictions sites of pET28a.
Binding of GST-tagged 72b′ to Thx-H68e′
10 mg of Rosetta (DE3) pLysS cells expressing GST-72b′ were lysed by sonication in 1.7 mL of 50 mM Na-phosphate, pH 7.4, 150 mM NaCl, 1 mM EDTA. Similarly, cells expressing Thx-H68e′ were lysed separately in 50 mM Na-phosphate, pH 7.4, 200 mM NaCl. Lysates were subjected to centrifugation in a microfuge for 20 min. and the pellets were discarded. The GST-72b′ lysate was mixed with 130 μL of equilibrated glutathione Sepharose 4B suspension (Pharmacia) and the protein was bound to the beads by slow rotation for 1 hour at 4°C. Following a brief centrifugation, the supernatant was removed, 1.5 mL of Thx-H68e′-containing lysate were added to the GST-72b′-charged beads, and the sample was rotated for 2 hours at 4°C The beads were washed twice with 1 mL of 50 mM Na-phosphate, pH 7.4, 150 mM NaCl, and 1 mM 2-ME. The GST-72b′/Thx-H68e′ complex was eluted with 200 μL of 50 mM Na-phosphate, pH 7.4, 200 mM NaCl, 20 mM Glutathione (reduced form, Sigma).
To explore the binding conditions, 15 μL (ca. 1.8 μg) of GST-72b′/Thx-H68e′ were diluted 10-fold with 50 mM Na-phosphate, pH 7.4, 300 mM NaCl, 0.1% Tween 20, 10 mM Imidazole buffer supplemented with KCl, ethanol, DMSO, LiCL, or urea as shown in Results. Samples were incubated at room temperature for 10 min., 20 μL of a 5% suspension of Ni-NTA Magnetic Agarose beads (Qiagen) were added followed by incubating at room temperature for one hour with occasional mixing by pipetting. The beads were concentrated in a magnetic separator, the unbound polypeptides were removed, and the beads were washed by adding 100 μL of the appropriate buffer followed by separation in the magnetic field and removal of the unbound material. The bead-bound proteins were eluted by adding 20 μL of 50 mM Na-phosphate pH 8.0, 300 mM NaCl, 0.1% Tween 20, and 250 mM Imidazole, mixed with 6 μL of twice-concentrated SDS loading buffer (100 mM Tris-HCl pH 6.8, 4% SDS, 0.2% Bromophenol blue, 20% glycerol). Samples were subjected to electrophoresis on 10% polyacrylamide SDS Tricine gels followed by staining of the polypeptides with Coomassie blue.
Assay of the binding activities of mutated GST-72b′
GST-k72b′ or mutant derivatives of GST-72b′ were purified on glutathione Sepharose 4B beads, and Thx-H68e′ was purified on Ni-NTA Superflow beads as described above. The concentrations of the polypeptides were determined by SDS PAGE followed by Coomassie blue staining with known amounts of lysozyme for reference. 0.3 to 0.4 μM of Thx-H68e′ were mixed at room temperature in 100 μL of binding buffer (50 mM Na-phosphate pH 7.4, 300 mM NaCl, 0.1% Tween 20, and 1 mM 2-ME) with up to 0.18 μM of GST-72b′ or its mutant derivatives. Samples were kept over night on ice. 20 μL of Ni-NTA Magnetic Agarose beads (Qiagen) were added, samples were incubated at room temperature for 1 hour with occasional mixing, and exposed to the magnetic field. Unbound polypeptides were pooled with polypeptides in a wash with 100 μL binding buffer and were analyzed in parallel with the bead-bound proteins as described above.
Isolation of the 68e′/H72b′ complex
Competent E. coli BL21(DE3) cells were cotransformed with p68e and pH72b′, and colonies were isolated after growth overnight at 37°C on LB agar plates containing Ampicillin and 50 μg/mL Kanamycin. Eight colonies were resuspended in 10 mL of LB containing antibiotics and the culture was shaken for 1 hour at 37°C. IPTG was added to a final concentration of 0.8 mM and growth was continued by shaking at room temperature for seven hours. Cells from a 5 mL culture were harvested by centrifugation, frozen at −70°C, and resuspended in 50 mM Na-phosphate, pH 7.4, 500 mM NaCl, 20 mM Imidazole, 5 mM 2-ME, 0.5 M urea, and protease inhibitor cocktail followed by sonication, high-speed centrifugation, and binding of the complex to Ni-NTA Superflow beads as described above. The column was washed with 50 mM Na-phosphate, pH 7.4, 500 mM NaCl, 30 mM Imidazole, 5 mM 2-ME. The 68e′/H72b′ complex was eluted with 200 μL of 50 mM Na-phosphate, pH 7.4, 500 mM NaCl, 5 mM 2-ME, 1% Tween 20, and 150 mM Imidazole.
For assessing the stability of the SRP68/72H and 68e′/H72b′ complexes, aliquots of the preparations were incubated in 100 μL of 50 mM Na-phosphate, pH 7.4, 300 mM NaCl, 0.1% Tween 20, 1 mM 2-ME, and 15 mM Imidazole containing up to 8 M urea followed by incubation at 4°C for up to 16 hours. The disruption of the heterodimers was monitored by incubating the samples with 20 μL of a 5% suspension of Ni-NTA Magnetic Agarose beads followed by SDS PAGE as described earlier.
Site-directed mutagenesis of GST-72b′ and H68h
Plasmids encoding mutant derivatives of GST-72b′ were assembled by carrying out two PCR reactions for each construct with the appropriate mutagenic primers and the flanking primers ATA GCA TGG CCT TTG CAG GGC TGG C and GCT TAC AGA CAA GCT GTG ACC GTC in a Rapid Cycler (Idaho Technology) for 35 cycles at an annealing temperature of 40°C. The two PCR products were purified by agarose gel electrophoresis and mixed to amplify the full-length gene using only the flanking primers. The amplified DNA was digested with NcoI and EcoRI, purified, and ligated to the pGST-72b′ vector DNA which had been restricted with EcoRI and BamHI to remove the wildtype insert. Competent E. coli DH5α cells were transformed, individual colonies were screened by restriction mapping, and the mutant plasmid sequences were verified. Mutations in H68h were generated by complete gene synthesis using two complementary mutant synthetic oligonucleotides compatible with the NcoI and EcoRI restrictions sites of pET28a.
Radioactive labeling of GST-k72b′
The glutathione Sepharose 4B-bound protein was washed twice with 1 mL of 20 mM HEPES, pH 8.0, 50 mM NaCl, 1 mM MgCl2 followed by elution in HEPES buffer containing 150 mM NaCl, 1 mM MgCl2, and 20 mM glutathione. A 30-μL protein labeling reaction was carried out in 20 mM HEPES, pH 7.9, 5 mM DTT, 10 mM MgCl2 containing 0.5 μg of purified GST-k72b′ polypeptide, 40 units of the catalytic subunit of protein kinase A from bovine heart (Sigma), and [γ-32P]ATP (3000 Ci/mmol, PE Biosystems). The sample was incubated at room temperature for 30 min. and then stored at −70°C. For binding in a reaction volume of 100 μL, ∼1 nM of 32P-labeled GST-k72b′ were mixed with increasing amounts of purified Thx-H68e′. Aliquots of binding mixtures were incubated with Ni-paramagnetic beads and the bead-associated radioactivity in each sample was measured by Cerenkov radioation counting in a Beckman Coulter LS6500 Scintillation Counter.
Binding experiments with mutated GST-72b′
Polypeptides with mutations in GST-72b′ were purified as described above for wildtype GST-72b′22. Protein concentrations were determined by SDS PAGE using known amounts of lysozyme. 100 μL binding reactions were assembled at room temperature in 50 mM Na-phosphate, pH 7.4, 300 mM NaCl, 0.1% Tween 20, 1 mM 2-ME, 15 mM Imidazole (binding buffer) containing up to 0.83 μg of wildtype GST-k72b′ or mutant polypeptide and a fixed amount (0.8 to 1.0 μg; 0.3 to 0.4 μM) of purified Trx-H68e′. Samples were incubated over night at 4°C, mixed with Ni-NTA Magnetic Agarose beads and processed as described above.
For the competition experiments 8 nM of Thx-H68e′, 1 nM of 32P-labeled GST-k72b′, and increasing amounts of the various competitor GST-72b′ mutant polypeptides were incubated for one hour on ice. Ni-NTA Magnetic Agarose beads were added, and incubation was continued on ice with gentle mixing every 15 min. Unbound polypeptides were removed in the magnetic field, the beads were washed with binding buffer, and the bound material was eluted in 250 mM imidazol buffer and its radioactivity was measured as described above.
Comparative sequence analysis of SRP68 and SRP72
Sets of representative sequences were used as input to a series of PERL scripts to identify SRP68 and SRP72 homologs in the NCBI databases.31 Sequences were aligned using MUSCLE32 followed by manual adjustments in Jalview.33 The updated full and representative SRP68 and SRP72 alignments are available at http://rnp.uthct.edu/rnp/SRPDB/srpprotein.html. Consensus secondary structures were predicted with using Jpred 3.34 Protein sequence logos were prepared using the interface at http://weblogo.berkeley.edu/logo.cgi.
Acknowledgments
The authors thank Kiyoshi Nagai, MRC, Cambridge, UK, for providing pRS68–72His and Elena Menichelli, The Scripps Research Institute, La Jolla, CA, for offering helpful suggestions during the expression of human SRP68/72 in yeast.
Glossary
Abbreviations:
- 2-ME
2-mercapto ethanol
- DMSO
dimethyl sulfoxide
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- IPTG
isopropyl-beta-d-thiogalactopyranoside
- LB
Luria-Bertani medium
- Ni-NTA
nickel nitrilotriacetic acid
- PAGE
polyacrylamide gel electrophoresis
- PCR
polymerase chain reaction
- SDS
sodium dodecyl sulfate
- SRP
signal recognition particle
- TEV
tobacco etch virus
- TPR
tetratricopeptide repeat
- Tricine
N-tris(hydroxymethyl)methylglycine.
References
- 1.Doudna JA, Batey RT. Structural insights into the signal recognition particle. Annu Rev Biochem. 2004;73:539–557. doi: 10.1146/annurev.biochem.73.011303.074048. [DOI] [PubMed] [Google Scholar]
- 2.Halic M, Beckmann R. The signal recognition particle and its interactions during protein targeting. Curr Opin Struct Biol. 2005;15:116–125. doi: 10.1016/j.sbi.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 3.Luirink J, Sinning I. SRP-mediated protein targeting: structure and function revisited. Biochim Biophys Acta. 2004;1694:17–35. doi: 10.1016/j.bbamcr.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Shan SO, Walter P. Co-translational protein targeting by the signal recognition particle. FEBS Lett. 2005;579:921–926. doi: 10.1016/j.febslet.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 5.Walter P, Blobel G. Disassembly and reconstitution of signal recognition particle. Cell. 1983;34:525–533. doi: 10.1016/0092-8674(83)90385-9. [DOI] [PubMed] [Google Scholar]
- 6.Andrews DW, Walter P, Ottensmeyer FP. Evidence for an extended 7SL RNA structure in the signal recognition particle. EMBO J. 1987;6:3471–3477. doi: 10.1002/j.1460-2075.1987.tb02671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weichenrieder O, Wild K, Strub K, Cusack S. Structure and assembly of the Alu domain of the mammalian signal recognition particle. Nature. 2000;408:167–173. doi: 10.1038/35041507. [DOI] [PubMed] [Google Scholar]
- 8.Halic M, Becker T, Pool MR, Spahn CM, Grassucci RA, Frank J, Beckmann R. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature. 2004;427:808–814. doi: 10.1038/nature02342. [DOI] [PubMed] [Google Scholar]
- 9.Walter P, Ibrahimi I, Blobel G. Translocation of proteins across the endoplasmic reticulum: signal recognition protein (srp) binds to in-vitro assembled polysomes synthesizing secretory protein. J Cell Biol. 1981;91:545–550. doi: 10.1083/jcb.91.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersen ES, Rosenblad MA, Larsen N, Westergaard JC, Burks J, Wower IK, Wower J, Gorodkin J, Samuelsson T, Zwieb C. The tmRDB and SRPDB resources. Nucl Acids Res. 2006;34:D163–D168. doi: 10.1093/nar/gkj142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zwieb C, Eichler J. Getting on target: the archaeal signal recognition particle. Archaea. 2002;1:27–34. doi: 10.1155/2002/729649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zwieb C, Van Nues RW, Rosenblad MA, Brown JD, Samuelsson T. A nomenclature for all signal recognition particle RNAs. RNA. 2005;11:7–13. doi: 10.1261/rna.7203605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Politz JC, Yarovoi S, Kilroy SM, Gowda K, Zwieb C, Pederson T. Signal recognition particle components in the nucleolus. Proc Natl Acad Sci USA. 2000;97:55–60. doi: 10.1073/pnas.97.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown JD, Hann BC, Medzihradszky KF, Niwa M, Burlingame AL, Walter P. Subunits of the Saccharomyces cerevisiae signal recognition particle required for its functional expression. EMBO J. 1994;13:4390–4400. doi: 10.1002/j.1460-2075.1994.tb06759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Nues RW, Leung E, Mcdonald JC, Dantuluru I, Brown JD. Roles for Srp72p in assembly, nuclear export and function of the signal recognition particle. RNA Biol. 2008;5:73–83. doi: 10.4161/rna.5.2.6042. [DOI] [PubMed] [Google Scholar]
- 16.Scoulica E, Krause E, Meese K, Dobberstein B. Disassembly and domain structure of the proteins in the signal-recognition particle. Eur J Biochem. 1987;163:519–528. doi: 10.1111/j.1432-1033.1987.tb10899.x. [DOI] [PubMed] [Google Scholar]
- 17.Lustig Y, Goldshmidt H, Uliel S, Michaeli S. The Trypanosoma brucei signal recognition particle lacks the Alu-domain-binding proteins: purification and functional analysis of its binding proteins by RNAi. J Cell Sci. 2005;118:4551–4562. doi: 10.1242/jcs.02578. [DOI] [PubMed] [Google Scholar]
- 18.Lakkaraju AK, Luyet PP, Parone P, Falguieres T, Strub K. Inefficient targeting to the endoplasmic reticulum by the signal recognition particle elicits selective defects in post-ER membrane trafficking. Exp Cell Res. 2007;313:834–847. doi: 10.1016/j.yexcr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 19.Yin J, Iakhiaeva E, Menichelli E, Zwieb C. Identification of the RNA binding regions of SRP68/72 and SRP72 by systematic mutagenesis of human SRP RNA. RNA Biol. 2007;4:154–159. doi: 10.4161/rna.4.3.5428. [DOI] [PubMed] [Google Scholar]
- 20.Menichelli E, Isel C, Oubridge C, Nagai K. Protein-induced conformational changes of RNA during the assembly of human signal recognition particle. J Mol Biol. 2007;367:187–203. doi: 10.1016/j.jmb.2006.12.056. [DOI] [PubMed] [Google Scholar]
- 21.Iakhiaeva E, Wower J, Wower IK, Zwieb C. The 5e motif of eukaryotic signal recognition particle RNA contains a conserved adenosine for the binding of SRP72. RNA. 2008;14:1143–1153. doi: 10.1261/rna.979508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iakhiaeva E, Bhuiyan SH, Yin J, Zwieb C. Protein SRP68 of human signal recognition particle: identification of the RNA and SRP72 binding domains. Protein Sci. 2006;12:467–468. doi: 10.1110/ps.051861406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iakhiaeva E, Yin J, Zwieb C. Identification of an RNA-binding domain in human SRP72. J Mol Biol. 2005;345:659–666. doi: 10.1016/j.jmb.2004.10.087. [DOI] [PubMed] [Google Scholar]
- 24.Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 25.Mcguffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 26.Wilce MC, Feil SC, Board PG, Parker MW. Crystallization and preliminary x-ray diffraction studies of a glutathione S-transferase from the Australian sheep blowfly, Lucilia cuprina. J Mol Biol. 1994;236:1407–1409. doi: 10.1016/0022-2836(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 27.Bennion BJ, Daggett V. The molecular basis for the chemical denaturation of proteins by urea. Proc Natl Acad Sci USA. 2003;100:5142–5147. doi: 10.1073/pnas.0930122100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein–protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 29.Das AK, Cohen PW, Barford D. The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein–protein interactions. EMBO J. 1998;17:1192–1199. doi: 10.1093/emboj/17.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zarrinpar A, Bhattacharyya RP, Lim WA. The structure and function of proline recognition domains. Sci STKE 2003: 2003:RE8. doi: 10.1126/stke.2003.179.re8. [DOI] [PubMed] [Google Scholar]
- 31.Mcginnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucl Acids Res. 2004;32:W20–5. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 34.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucl Acids Res. 2008;36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]