

Abstract
The structure of the Atu1476 protein from Agrobacterium tumefaciens was determined at 2 Å resolution. The crystal structure and biochemical characterization of this enzyme support the conclusion that this protein is an S-formylglutathione hydrolase (AtuSFGH). The three-dimensional structure of AtuSFGH contains the α/β hydrolase fold topology and exists as a homo-dimer. Contacts between the two monomers in the dimer are formed both by hydrogen bonds and salt bridges. Biochemical characterization reveals that AtuSFGH hydrolyzes C—O bonds with high affinity toward short to medium chain esters, unlike the other known SFGHs which have greater affinity toward shorter chained esters. A potential role for Cys54 in regulation of enzyme activity through S-glutathionylation is also proposed.
Keywords: X-ray crystallography, hydrolase, protein structure, S-formylglutathione
Introduction
S-formylglutathione hydrolase (SFGH) (3.1.2.12) is a glutathione thiol esterase that hydrolyzes S-formylglutathione to glutathione and formate. SFGH is part of the formaldehyde detoxification pathway conserved in eukaryotes and prokaryotes. Genomic sequence analyses have revealed the existence of putative SFGH-encoding genes in a variety of organisms. This has lead to the proposal that SFGH and the glutathione-dependent formaldehyde oxidation pathway are distributed throughout eukaryotes and prokaryotes. In eukaryotes, SFGHs have been purified and characterized from human liver,1Arabidopsis thaliana,2,3 and Saccharomyces cerevisiae.4 SFGH has also been characterized from Paracoccus denitrificans,5 described as a human esterase D homolog. Human esterase D was recently identified as S-formylglutathione hydrolase.6,7 Recently two E. coli SFGH proteins, FrmB (YaiM) and YeiG, were purified and characterized.8 Alignment of sequences from both prokaryotic and eukaryotic SFGHs together with site directed mutagenesis experiments have revealed that SFGHs are serine hydrolases containing a conserved Ser-Asp-His catalytic triad.8,9
Structures of SFGHs from eukaryotes have been reported,9,10 however no prokaryotic SFGH structure is available. In view of the wide distribution of SFGH in nature and their importance in the formaldehyde detoxification pathway, we have expressed, purified, and crystallized a putative prokaryotic S-formylglutathione hydrolase from Agrobacterium tumefaciens (AtuSFGH). AtuSFGH consists of 277 amino acid and shares 46 and 47% sequence identity to the E. coli S-formylglutathione hydrolase proteins FrmB (YaiM) and YeiG, respectively, and 44, 47, and 53% sequence identity to ScSFGH, AtSFGH, and HSFGH, respectively.
In this article, we present the three-dimensional crystal structure and biochemical characterization of AtuSFGH. AtuSFGH has the typical SFGH hydrolase fold with a Ser-Asp-His catalytic triad similar to that of ScSFGH and AraSFGH.
Results and Discussion
Overall structure of A. tumefaciens esterase
The overall structure of AtuSFGH consists of six molecules (A–F) packed as three dimers per asymmetric unit. Each monomer in the dimer is related to the other monomer by non-crystallographic twofold symmetry. The dimer formed by monomers A and B is slightly rotated and translated compared with the other two dimers. The contacts (PISA server: http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) between the two monomers in the dimer are formed both by hydrogen bonds from main chain and side chain atoms (five) and salt bridges (four), with a buried surface area of 1037.7 Å2 (average of the three dimers).
AtuSFGH is a single domain protein with dimensions of 48 Å × 42 Å × 29 Å. The six monomers are highly similar (r.m.s.d. of 0.20 Å for all 277 equivalent Cα atoms). The AtuSFGH structure shows the general α/β-hydrolase fold.11 It consists of a nine stranded central β-sheet with strand order 123546789 flanked on one side by three α-helixes and on the other site by eight α-helixes and two small 310 helixes [Fig. 1(A)]. The β sheet is mainly parallel with only the first and third strand antiparallel. As in other α/β-hydrolases, the β-sheet has a left-handed superhelical twist, with the first and last strand oriented almost perpendicular to each other. When compared with the general α/β-hydrolase fold described by Ollis et al.,11AtuSFGH contains one additional β strand (β1), five additional α-helixes (α1, α3, α4, α7, and α8), and two small 310 helices (η1 and η2). The active site is located on the C-terminal side of the β sheet, near the dimer contacts of each monomer and contains the classical serine hydrolase catalytic triad Ser147, His256, and Asp223 [Fig. 1(B)]. The active site cavity has dimensions of 19 Å × 12 Å × 9 Å, with Ser147 positioned in the middle of this cavity. Ser147 is found on the characteristic “nucleophilic elbow,” a sharp γ-like turn between strand β6 and helix α6.11 The narrow turn places Ser147 near the N-terminal end of helix α6, allowing charge stabilization through the helix-dipole moment.11 The sharp turn places Ser147 in a slightly unfavorable region of the Ramachandran plot with φ and ψ angles of 63° and −109° (average of six monomers), respectively, as in other α/β hydrolases.11
Figure 1.
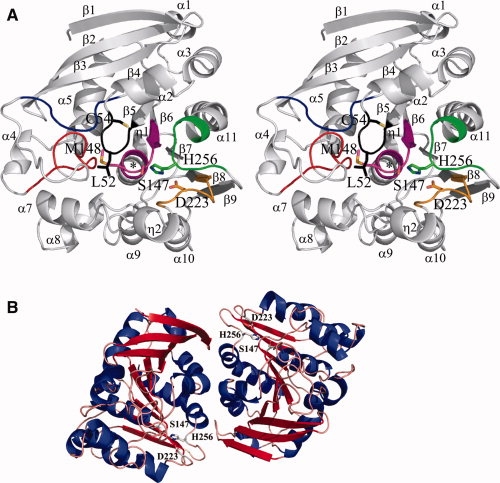
Crystal structure of AtuSFGH and superposition with eukaryotic SFGHs. (A) Stereo view of the monomer structure of AtuSFGH showing the conserved SFGHs sequence motifs. Motif I: Leu-Ser-Gly-Leu-Thr-Cys (black), Motif II: Pro-Asp-Thr-Ser-Pro-Arg-Gly (blue), Motif III: Gly-Xaa-Gly-Ala-Gly-Phe-Tyr-Xaa-Xaa-Ala-Thr (red), Motif IV: Ile-Xaa-Gly-His-Ser-Met-Gly-Gly-Xaa-Gly-Ala (magenta), Motif V: Asp-His-Ser-Tyr-Tyr-Phe (green), and new Motif VI: Xaa-Gly-Xaa-Xaa-Asp-Xaa-Phe (brown). Ser-Asp-His catalytic triad, the regulatory Cys and Leu/Met from the oxyanion hole are shown as ball-and-stick representations. The oxyanion hole is indicated with a black star. (B) The AtuSFGH dimer structure. The β-sheet is colored red and α-helixes are colored blue. The proposed active site residues are shown as ball-and-stick representations.
Structural comparison of AtuSFGH with known eukaryotic SFGH structures
A structure-based sequence alignment shows that the Ser-Asp-His catalytic triad and oxyanion hole residues are strictly conserved among the S-formylglutathione hydrolases family (Fig. 2). The structure-based sequence alignment reveals six consensus sequence motifs,8 all lining up the SFGHs active site [Figs. 1(A) and 2]. Structural superposition of AtuSFGH onto the eukaryotic SFGHs from human (hSFGH) and yeast (ScSFGH) shows that the three structures are very similar with root-mean-square difference (rmsd) of 1.3 Å for 272 Cα atoms for ScSFGH and rmsd of 1.0 Å for 271 Cα atoms for hSFGH. The three structures show the same relative arrangement of the two monomers. However, there are significant differences between AtuSFGH and hSFGH and ScSFGH in the nonconserved flexible loop regions at the N-terminus of the β sheet between strand β3 and β4, helix α5 and strand β6, helix α6 and β7, helix α9 and β8, helix α10 and β9 (Fig. 2). The differences in these loop regions are largely due to insertions in ScSFGH.
Figure 2.
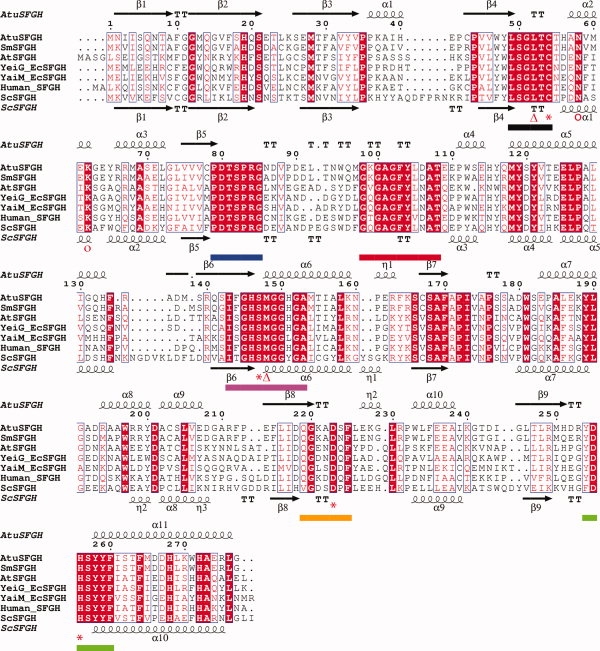
Sequence and structural homology of AtuSFGH. The sequence of AtuSFGH was aligned with five S-formylglutathion hydrolases (SFGHs). The sequences are from E. coli (FrmB (P51025) and YeiG (P33018)), A. thaliana (Q8LAS8), Sinorhizobium medical (A6U894), human esterase D (P10768), and S. cerevisiae ScSFGH (YJG8) (P40363). The multiple sequence alignment was guided by a structural alignment of AtuSFGH and ScSFGH (PDB code 1PV1) using SEQUOIA.12 Highly conserved residues are highlighted in red. The conserved cysteine residue and the residues contributing to the catalytic triad are marked with an asterisk below the alignment. The oxyanion hole residues are denoted with triangles. Proposed residues in substrate and product binding are marked with open circles. The secondary structure elements refer to the structure of AtuSFGH (above) and ScSFGH (below) the sequence alignment. The consensus sequence motifs are denoted with dotted lines below the alignment and have the same color as in Figure 1(A). The figure was prepared with ESPript.13
The architecture of the active site and oxyanion hole of AtuSFGH is similar to that of the eukaryotic SFGHs.9,10,14 The active site is formed by conserved secondary structure elements: helix α2, part of the N-terminus of helices α6, α7, and α11, the C-terminus of β-strands β4 and β6. Four consensus sequence motifs are fundamental for the architecture of the SFGH active site and contain the residues central for catalysis. These motifs are: motif-I at the loop region between β4-α2, motif-IV at β6-α6, motif-V at β9-α11 and the new motif-VI at β8–η2 [Figs. 1(A) and 2]. Most of the residues from the other two consensus motifs (motif II and motif III) are found in the loops between strand β5 and helix α4, and likely help to stabilize the fold of the active site [Figs. 1(A) and 2]. The LSGLTC hexapeptide (motif-I) makes a sharp turn at the back of the active site and is stabilized by hydrogen bonds between the main chain carbonyl oxygen of Gly51 and the main chain NH of Cys54 in AtuSFGH. This loop is held in position by conserved residues from motif-II and motif-III. The arrangement of the hexapeptide is essential for formation of the oxyanion hole, explaining its conservation within the SFGH family. This loop (motif-I) and the catalytic nucleophile Ser147 form a small cavity that would stabilize the negatively charged tetrahedral intermediate by the main chain amide of Met148 (Met162 in ScSFGH) and the main chain nitrogen atom of Leu52 (Leu58 in ScSFGH). Furthermore, the dipole of helix α6 would aid in stabilizing the tetrahedral intermediate.15 The conservation of the overall structures and the invariant active site motifs indicate the functional relationship of AtuSFGH with the other SGFH enzymes and would suggest a common catalytic mechanism.
Substrate specificity and kinetics
Carboxylesterase activity was determined by using p-nitrophenyl esters of various fatty acids (C2–C16). Arylesterase activity was determined by using α-naphthyl esters of lengths (C2–C10). Kinetic parameters for the hydrolysis of the various model esterase substrates are presented in Table I. AtuSFGH shows increasing activity against both p-nitrophenyl and α-naphthyl esters with increasing fatty acid chain length. The highest activity for the p-nitrophenyl esters is against p-nitrophenyl butyrate (8.40 μmol/min/mg protein), and for the α-naphthyl esters against α-naphthyl propionate (3.6 μmol/min/mg protein). Further increase in fatty acid chain length markedly decreases activity. AtuSFGH is unable to hydrolyse α-naphthyl dodecanoate, p-nitrophenyl caprate, p-nitrophenyl laurate, and p-nitrphenyl palmitate. The enzyme has more affinity toward C3 and C4 p-nitrophenyl esters than toward C3 and C4 α-naphthyl esters. The highest catalytic activity is against p-nitrophenyl butyrate. The maximal substrate specificity, as measured by the specificity constant kcat/Km was found for p-nitrophenyl butyrate. Thus, our experiments imply that AtuSFGH hydrolyses C—O bonds with high specificity toward medium chain esters (C3–C4).
Table I.
Kinetic Analysis of wt. AtuSFGH with Various Substrates
| Km (M) | Vmax (moles min−1 mg−1) | kcat (s−1) | kcat/Km (M−1 s−1) | |
|---|---|---|---|---|
| pNP-acetate | 0.48 ± 0.045 | 3.66 ± 0.11 | 1.95 | 4.07 × 103 |
| pNP-propionate | 0.05 ± 0.006 | 3.46 ± 0.12 | 1.84 | 3.85 × 104 |
| pNP-butyrate | 0.11 ± 0.007 | 8.40 ± 0.14 | 4.48 | 4.08 × 104 |
| pNP-caproate | 0.02 ± 0.002 | 1.26 ± 0.033 | 0.67 | 3.96 × 104 |
| α-naphthyl propionate | 0.35 ± 0.027 | 3.57 ± 0.19 | 1.90 | 5.54 × 103 |
| α-naphtyl butyrate | 0.44 ± 0.040 | 3.24 ± 0.21 | 1.73 | 3.93 × 103 |
AtuSFGH shares 46% and 47% sequence identity to the E. coli SFGHs FrmB (YaiM) and YeiG, respectively. FrmB and YeiG have high carboxylesterase activity against p-nitrophenyl esters of fatty acids (C2–C6) and α-naphthyl acetate.8 The highest hydrolytic activity of these E. coli proteins is toward S-formylglutathione, an intermediate of the glutathione-dependent pathway of formaldehyde detoxification.8 These E. coli proteins are able to hydrolyse two types of bonds: C—O and C—S.8 The eukaryotic SFGHs from yeast,4A. thaliana,3 and human1 show comparable kinetics to the E. coli SFGHs.8 Unlike the other SFGHs, AtuSFGH has greater affinity and catalytic efficiency toward C3 and C4 p-nitrophenyl than do the other members of this family. AtuSFGH is also active toward C3 and C4 α-naphthyl esters, whereas the E. coli proteins are only active toward α-naphthyl acetate. Comparison of AtuSFGH, ScSFGH, and hSFGH acyl binding pockets show that the acyl binding pockets of ScSFGH and hSFGH are shallower than the pocket in AtuSFGH, preventing binding of C3 and C4 esters. This is in agreement with kinetic studies that demonstrate that ScSFGH and hSFGH prefer short chain esters.4
Mutation of Cys54 and Ser147
Ser147 and Cys54 are absolutely conserved in the SFGHs of bacteria, yeast, animals, and plants. The role of Cys54 and Ser147 were assessed by mutagenesis and enzyme kinetics. The S147A mutant was unable to hydrolyse α-naphthyl butyrate, in agreement with Ser147 being the nucleophilic residue in the catalytic mechanism of AtuSFGH. The conserved Cys54 from motif-I is positioned on the edge of the active site cavity, 8.9 Å from the catalytic Ser147 [Fig. 1(C)]. This residue has been proposed to serve a gate-keeping function in regulating access to the active site via disulfide formation with glutathione.8,9 The C54S mutant showed a fourfold decrease in α-naphthyl butyrate activity (data not shown). As with AtSFGH3,9 and the E. coli SFGHs FrmB and YeiG,8 the activity of wild type AtuSFGH was inhibited by the thiol reacting agent N-ethylmaleimide (NEM), however, the C54S mutant is insensitive to NEM (data not shown). The effect of the thiol inhibitors on the activity of SFGHs can be explained by the binding of the inhibitor molecule to the conserved cysteine residue located in the active site of the enzyme (Cys54 in AtuSFGH), consequently, blocking the binding of substrate to the active site of SFGHs. SFGH produces glutathione as a reaction product; under high levels of glutathione in the cell, this cysteine would likely become susceptible particularly to S-glutathionylation and would serve as an auto-regulatory mechanism in these organisms.
Materials and Methods
Purification and crystallization
AtuSFGH was expressed and purified as previously described.16 The initial crystallization condition was determined with an optimized sparse crystallization matrix17 at room temperature using the sitting drop-vapor diffusion technique. The optimized crystallization condition consisted of 0.1M Bis-Tris pH 6.5, 0.2M MgCl2, and 25% PEG3350. Crystals were cryoprotected using Paratone-N oil and were flash-frozen for diffraction data collection.
Data collection and processing
X-ray data were collected at 100 K with an in-house apparatus consisting of a Rigaku 007 microfocus rotating anode X-ray generator with VariMax optics and an R-AXIS IV++ image-plate detector (Rigaku/MSC). Diffraction data to 2.0 Å was collected and processed and scaled using DENZO and SCALEPACK.18 Table IIa shows the data collection statistics. Based on calculations of the Matthews coefficient Vm,19 the AtuSFGH crystal contains six molecules per asymmetric unit with a solvent content of 45%. Examination of the self-rotation function clearly indicates the presence of non-crystallographic symmetry.
Table II.
Data Collection Statistics and Refinement Statistics
| a. Data collection statistics | |
| Space group | P1 |
| Unit cell dimensions | a = 66.43 Å, b = 68.95 Å, c = 95.65 Å |
| α = 92.41°, β = 99.79°, γ = 98.43° | |
| Resolution range (Å) | 25.0–2.02 (2.09–2.02) |
| Observed reflections | 153,093 |
| Unique reflections | 102,062 |
| Completeness (%) | 95 (79.9) |
| I/σ(I) | 23.2 (4.3) |
| Rmerge | 5.8% (28.8%) |
| b. Refinement statistics | |
| Resolution range (Å) | 25.0–2.02 |
| Rwork/Rfree (%)a | 18.1/21.8 |
| No. of protein atoms | 13,226 |
| No. of water molecules | 670 |
| No. of Mg2+ ions | 6 |
| No. of Cl− ions | 12 |
| Average B factor (Å2) | |
| Protein | 26.9 |
| Water | 26.9 |
| Mg2+ | 33.6 |
| Cl− | 29.2 |
| Rms deviations | |
| Bond length (Å) | 0.009 |
| Bond angles (°) | 1.21 |
| Residues in regions of Ramachandran plot | |
| Most favored (%) | 88.0 |
| Additionally allowed (%) | 11.4 |
| Generously allowed (%) | 0.6 |
Rwork = Σhkl||Fobs| − |Fcalc||/Σhkl |Fobs|, where the crystallographic R-factor was calculated with 95% of the data used in the refinement; Rfree = the crystallographic R-factor based on 5% of the data withheld from the refinement for cross validation.
Structure determination and refinement
The structure was solved by molecular replacement using the program MrBump20 with MolRep21 implemented in CCP4. A molecular replacement solution was found using the crystal structure of ScSFGH (PDB code = 1PV1) as the search model, which shows 44% sequence identity to AtuSFGH. The initial model after MolRep and rigid body refinement (Refmac5) as part of MrBUMP had a Rwork = 34.9% and Rfree = 40.0%. Initial refinement was carried out in CNS22 using simulated annealing, torsion angle dynamics with start temperature 1500 K. Further refinement was carried out with Refmac5.23 NCS restraints were used during refinement and lowered toward the end of refinement. The last round of refinement was carried out without NCS restraints. Model building was performed in Coot.24 Water molecules were added using ARP/wARP25 after the Rfree had decreased to below 30%. The R factors converge to a final crystallographic Rwork = 18.1% and Rfree = 21.8%. The final refinement statistics are summarized in Table IIb.
Kinetic and inhibition assays
Enzymatic assays were carried out in triplicate using 2–5 μg mL−1 of purified enzyme in buffer Tris-HCl, pH 8.0. Enzymatic activity toward carboxylesters was measured by continuous reading of the p-nitrophenol released by enzymatic action at 400 nm (ɛ = 16,300M−1 cm−1) using p-nitrophenyl esters of various fatty acids (C2–C16) as substrates. Aryl esterase activity was evaluated by measuring the release of α-naphthol at 310 nm (ɛ = 3000M−1 cm−1) from α-naphthyl esters (α-naphthyl-acetate, α-naphthyl-propionate, and α-naphthyl-dodecanoate). The enzymatic activity was performed according to the protocols and procedures previously described.8 The kinetic parameters were determined by nonlinear curve fitting using Microcal Origin version 8 (OriginLab).
For inhibition assays, purified wild type AtuSFGH and the Cys54Ser AtuSFGH mutant were incubated in 20 mM Hepes pH 7.4 with thiol inhibitor N-ethylmaleimide (0–1 mM) for 30 min at 22°C. The residual activity with NEM inhibitor was assayed by measuring the release of α-naphthol from the substrate α-naphthyl-butyrate (1 mM) at 310 nm.
Site-directed mutagenesis
Site-directed mutagenesis of the AtuSFGH mutants (S147A and C54S) was performed using the QuikChange™ site-directed mutagenesis kit (http://www.jbc. org/cgi/redirect-inline?ad = Stratagene) according to the manufacturer's protocol. The Atu open reading frame cloned on p15TV-Lic expression vector was used as a template. The sense primers used were: S147A: 5′-G TCC ATT TTC GGC CAT GCC ATG GGC GGT CAT GGT G-3′ and C54S: 5′-G TAT CTC TCC GGC CTC ACC GCC ACC CAT GCC AAT GTC ATG-3′, and anti-sense primers were : S147A 5′-C ACC ATG ACC GCC CAT GGC ATG GCC GAA AAT GGA C-3′ and C54S 5′-C ATG ACA TTG GCA TGG GTG GCG GTG AGG CCG GAG AGA TAC-3′, respectively. The standard PCR mixture contained 50 ng of template DNA and 15 pmole of each primer. PCR amplifications were carried out in a GeneAmp PCR PTC100 System. The specific mutations were verified by DNA sequencing.
Sequence alignment
A Psi-BLAST search26 with the AtuSFGH sequence revealed that AtuSFGH is well conserved, having sequence identity of 44–86% with several S-formylglutathione hydrolases (SFGH) and putative S-formylglutathione hydrolases from bacteria, yeast, mammals, and plants. The highest sequence identity (86%) is with a putative SFGH from Rhizobium (Sinorhizobium) meliloti (R01391) and SFGH (Smed_1021) from Sinorhizobium medicae (strain WSM419) (Ensifer medicae). Other SFGH proteins with high-sequence identity are (69%) SFGH from Ochrobactrum anthropi (strain ATCC 49188), (60%) SFGH from Sphingomonas wittichii RW1, (53%) SFGH (Esterase D) [ESD] from Homo sapiens, (49%) SFGH [fghA] from Paracoccus denitrificans, (47%) SFGH (AtSFGH) (Esterase D) from A. thaliana (Mouse-ear Cress), (42%) YJG8, a SFGH from S. cerevisiae (Baker's yeast), and the recently characterized SFGHs from E. coli proteins FrmB alias YaiM (46%) and YeiG (47%). Structure-based sequence alignments were performed with SEQUOIA.12
Structural analysis
All models were validated using MOLPROBITY27 and the ADIT validation server at RCSB-Rutgers (http://deposit.pdb.org/validate/). For structural analyses the following programs were used: superpositions by LSQMAN28 and DALI.29 Figures were prepared with PYMOL (http://www.pymol.org).
Acknowledgments
Atomic coordinates have been deposited in the Protein 3E4D Data Bank (PDB) with the PDB accession code.
References
- 1.Uotila L, Koivusal M. Purification and properties of S-formylglutathione hydrolase from human liver. J Biol Chem. 1974;249:7664–7672. [PubMed] [Google Scholar]
- 2.Haslam R, Rust S, Pallett K, Cole D, Coleman J. Cloning and characterisation of S-formylglutathione hydrolase from Arabidopsis thaliana: a pathway for formaldehyde detoxification. Plant Physiol Biochem. 2002;40:281–288. [Google Scholar]
- 3.Kordic S, Cummins I, Edwards R. Cloning and characterization of an S-formylglutathione hydrolase from Arabidopsis thaliana. Arch Biochem Biophys. 2002;399:232–238. doi: 10.1006/abbi.2002.2772. [DOI] [PubMed] [Google Scholar]
- 4.Degrassi G, Uotila L, Klima R, Venturi V. Purification and properties of an esterase from the yeast Saccharomyces cerevisiae and identification of the encoding gene. Appl Environ Microbiol. 1999;65:3470–3472. doi: 10.1128/aem.65.8.3470-3472.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harms N, Ras J, Reijnders WNM, van Spanning RJM, Stouthamer AH. S-formylglutathione hydrolase of Paracoccus denitrificans is homologous to human esterase D: a universal pathway for formaldehyde detoxification? J Bacteriol. 1996;178:6296–6299. doi: 10.1128/jb.178.21.6296-6299.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Apeshiotis F, Bender K. Evidence that S-formylglutathione hydrolase and esterase-D polymorphisms are identical. Human Genet. 1986;74:176–177. doi: 10.1007/BF00282086. [DOI] [PubMed] [Google Scholar]
- 7.Eiberg H, Mohr J. Identity of the polymorphisms for esterase-D and S-formylglutathione hydrolase in red-blood-cells. Human Genet. 1986;74:174–175. doi: 10.1007/BF00282085. [DOI] [PubMed] [Google Scholar]
- 8.Gonzales CF, Proudfoot M, Brown G, Korniyenko Y, Mori H, Savchenko AV, Yakunin AF. Molecular basis for formaldehyde detoxification. Characterization of two S-formylglutathione hydrolases from Escherichia coli, FrmB and YeiG. J Biol Chem. 2007;281:14514–14522. doi: 10.1074/jbc.M600996200. [DOI] [PubMed] [Google Scholar]
- 9.Cummins I, McAuley K, Fordham-Skelton A, Schwoerer R, Steel PG, Davis BG, Edwards R. Unique regulation of the active site of the serine esterase S-formylglutathione hydrolase. J Mol Biol. 2006;359:422–432. doi: 10.1016/j.jmb.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 10.Legler PN, Kumaran D, Swaminathan S, Studier FW, Millard CB. Structural characterization and reversal of the natural organophosphate resistance of a D-type esterase. Saccharomyces cerevisiae S-formylglutathione hydrolase. Biochemistry. 2008;47:9592–9601. doi: 10.1021/bi8010016. [DOI] [PubMed] [Google Scholar]
- 11.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, Goldman A. The alpha/beta-hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 12.Bruns CM, Hubatsch I, Ridderstrom M, Mannervik B, Tainer JA. Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J Mol Biol. 1999;288:427–439. doi: 10.1006/jmbi.1999.2697. [DOI] [PubMed] [Google Scholar]
- 13.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 14.Wu D, Li Y, Song G, Zhang D, Shaw N, Liu Z. Crystal structure of human esterase D: a potential genetic marker of retinoblastoma. FASEB J. 2009;23:1441–1446. doi: 10.1096/fj.08-125286. [DOI] [PubMed] [Google Scholar]
- 15.Hol WG. The role of the a-helix dipole in protein function and structure. Prog Biophys Mol Biol. 1985;45:149–195. doi: 10.1016/0079-6107(85)90001-x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang RG, Skarina T, Katz JE, Beasley S, Khachatryan A, Vyas S, Arrowsmith CH, Clarke S, Edwards A, Joachimiak A, Savchenko A. Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase. Structure. 2001;9:1095–1106. doi: 10.1016/s0969-2126(01)00675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimber MS, Vallee F, Houston S, Necakov A, Skarina T, Evdokimova E, Beasley S, Christendat D, Savchenko A, Arrowsmith CH, Vedadi M, Gerstein M, Edwards AM. Data mining crystallization databases: knowledge-based approaches to optimize protein crystal screens. Proteins. 2003;51:562–568. doi: 10.1002/prot.10340. [DOI] [PubMed] [Google Scholar]
- 18.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular crystallography, Part A. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 19.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 20.Keegan RM, Winn MD. Automated search-model discovery and preparation for structure solution by molecular replacement. Acta Crystallogr D. 2007;63:447–457. doi: 10.1107/S0907444907002661. [DOI] [PubMed] [Google Scholar]
- 21.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 2007;30:1022–1025. [Google Scholar]
- 22.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 23.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 24.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.Morris RJ, Perrakis A, Lamzin VS. ARP/wARP and automatic interpretation of protein electron density maps. Macromolecular crystallography, Part D. Methods Enzymol. 2003;374:229–244. doi: 10.1016/S0076-6879(03)74011-7. [DOI] [PubMed] [Google Scholar]
- 26.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis IW, Murray LW, Richardson JS, Richardson DC. MolProbity: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleywegt GJ. Experimental assessment of differences between related protein crystal structures. Acta Crystallogr D. 1999;55:1878–1884. doi: 10.1107/s0907444999010495. [DOI] [PubMed] [Google Scholar]
- 29.Holm L, Sander C. Dali—a network tool for protein-structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]