

Summary
Chlamydia sp. are responsible for a wide range of diseases of significant clinical and public health importance. In this review, we highlight how recent cellular and functional genomic approaches have significantly increased our knowledge of the pathogenic mechanisms employed by these genetically intractable bacteria. As the extensive repertoire of chlamydial proteins that are translocated into the mammalian host are identified and characterized, a molecular understanding of how Chlamydiae co-opt host cellular functions and block innate immune pathways is beginning to emerge.
Introduction
Ocular and genital infections with Chlamydia trachomatis represent a significant public health concern because of their association with infectious blindness (trachoma) and adverse effects on female reproductive health (reviewed in (Schachter, 1999). Similarly, pulmonary infections with Chlamydophila pneumoniae are increasingly recognized as a common cause of community acquired pneumonias and a risk factor for chronic obstructive pulmonary disease, asthma and atherosclerosis (reviewed in (Blasi et al., 2009). A greater understanding of basic chlamydial biology is clearly important to develop vaccines, and identify new targets for drug intervention.
Chlamydiae undergo a distinct developmental cycle, converting between two morphologically and functionally discrete forms, the elementary body (EB) and the reticulate body (RB). The basic cycle follows this sequence: I) attachment and internalization, II) EB to RB differentiation, III) remodeling of the parasitophorous vacuole (“inclusion”) and bacterial replication, IV) inclusion expansion and transition of RB into EB, and V) release of bacteria from the host cell and infection of new target cells by EBs (Figure 1).
Figure 1. The Chlamydia trachomatis infectious cycle and modulation of host cell functions.
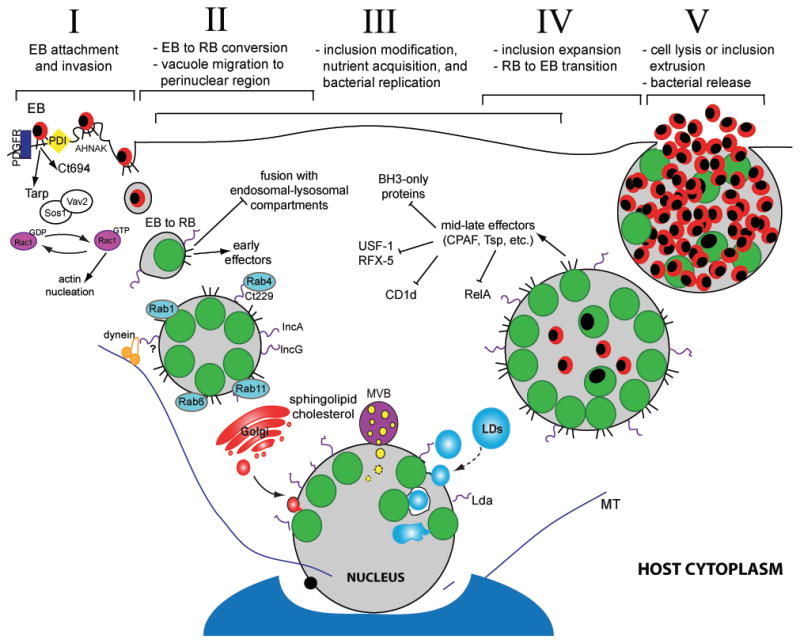
Chlamydia infection begins when an elementary body (EB) binds to the host cell surface (I). At this stage effector proteins injected into the host via the type III secretion system (T3SS) facilitate bacterial entry. Following endocytosis, EBs transition to reticulate bodies (RBs) (II). New effectors are secreted and the bacterial vacuole is modified by bacterial inclusion membrane proteins (Incs) to limit fusion with the host degradative compartments while promoting contact with other host organelles and factors, including Rab proteins. The inclusion interacts with the Golgi apparatus, multivesicular bodies (MVBs), and lipid droplets (LDs). LDs can be directly translocated into the inclusion lumen for nutrient delivery (III). During infection, host cell death and immune defenses are inhibited. Approximately midway through the infectious cycle, bacterial replication becomes asynchronous and RBs re-differentiate into EBs (IV). Late in the cycle, the inclusion is packed with EBs and fills almost the entire cell volume. Eventually, the inclusion and host cell rupture releasing infectious EBs into the extracellular space for reinfection (V).
Studies based on expression analysis (Nicholson et al., 2003, Belland et al., 2003), heterologous type III translocation systems (Subtil et al., 2005, Fields et al., 2003), function-based screens (Sisko et al., 2006), and bioinformatics (Samudrala et al., 2009) have yielded many candidate Chlamydia proteins that may participate in the manipulation of host cellular processes. Here we review the basic aspects of Chlamydia replication within infected cells and highlight recent findings that have significantly expanded our understanding of chlamydial pathogenesis.
Attachment and entry
The EB, the infectious bacterial form, attaches to and is internalized by the host cell. Multiple bacterial adhesins and ligands have been proposed, and host receptor utilization depends on both the host cell type and chlamydial species (Dautry-Varsat et al., 2005). Bacterial factors such as glycosaminoglycan (GAG) (Menozzi et al., 2002), major outer membrane (MOMP) (Su et al., 1996), OmcB (Fadel et al., 2007), and PmpD (Wehrl et al., 2004) have been proposed as adhesins or ligands for receptor interactions. Previously proposed host receptors include heparan sulfate, mannose receptor, mannose 6-phosphate receptor, and estrogen receptor (reviewed in (Campbell et al., 2006). Recently, two distinct roles were uncovered for cell surface exposed protein disulfide isomerase (PDI) in internalization: a structural one involved in EB attachment, and an enzymatic one required for bacterial entry (Conant et al., 2007, Abromaitis et al., 2009). PDI is predicted to bind one or more cell surface receptors (Abromaitis et al., 2009) which may account for the severe EB adhesion and invasion defects in PDI-deficient cells (Fudyk et al., 2002). RNAi screening exposed platelet derived growth factor receptor (PDGFR) and Abelson (Abl) kinase as essential host factors for EB entry (Elwell et al., 2008). Multiple cell surface proteins likely act in concert –or redundantly- to initiate EB invasion.
The exact mechanism of endocytosis remains unclear due to conflicting studies using pharmacological inhibitors and dominant-negative constructs (reviewed in (Dautry-Varsat et al., 2005). These results reflect that multiple redundant strategies likely exist to ensure chlamydial entry, and the route is dependent on the Chlamydia species or features of the host cell type being invaded. The unifying feature of EB entry, regardless of cell type or species, is the Rac1-dependent actin remodeling at attachment sites (Carabeo et al., 2004). The early translocated bacterial effector protein, Tarp. Phosphorylation of residues in the N-terminal tyrosine-rich tandem repeats of Tarp by host Src (Jewett et al., 2008) and Abl (Elwell et al., 2008) kinases recruit the guanine nucleotide exchange factors (GEFs), Sos1 and Vav2, which activate Rac1 (Lane et al., 2008). Subsequently, Rac1 recruits WAVE2 and Abi-1 leading to Arp2/3 complex activation and actin reorganization (Carabeo et al., 2007). These findings have been confirmed and extended in two unbiased RNAi screens for host factors required in chlamydial entry and replication (Elwell et al., 2008, Derre et al., 2007). Interestingly, Tarp from other Chlamydiae is not phosphorylated suggesting that Rac GEF recruitment occurs by an alternative pathway in these species or is not essential to control host actin (Clifton et al., 2005). Alternatively, a WH2-like domain in the C-terminal portion of Tarp can directly bind actin subunits and promote nucleation (Jewett et al., 2006). Ct694 is another bacterial protein translocated into the host cytoplasm at C. trachomatis attachment sites. Ct694 can bind the host protein AHNAK, a large multi-domain actin-binding protein that associates with the plasma membrane (Hower et al., 2009). By analogy to other invasive pathogens, it would not be surprising if multiple chlamydial effectors participate in invasion. Targeted knockdown of endocytic factors also revealed clathrin as a host component important in C. trachomatis entry of non-phagocytic cells (Hybiske et al., 2007a).
Chlamydia developmental transitions
EBs are stable in the extracellular environment by virtue of extensively cross-linked outer-membrane proteins (Newhall et al., 1983). These disulfide bonds are reduced during internalization (Hackstadt et al., 1985) followed by nucleoid decondensation and initiation of bacterial transcription. Within 15 minutes, new bacterial proteins are already being produced and RNA expression can be detected as early as one hour (Plaunt et al., 1988, Belland et al., 2003). RBs replicate by binary fission inside inclusion boundaries. Roughly midway through infection, replication becomes asynchronous as RBs begin differentiating back into EBs. Similar to the early conversion of EBs to RBs, the events involved in RB to EB transition are unknown. Dissociation of the type III secretion apparatus from the inclusion membrane as RBs detach is one proposed trigger for this process (Wilson et al., 2006), but this appealing model remains to be tested.
Inclusion modification and nutrient acquisition
The nascent inclusion membrane initially resembles the host plasma membrane, but these components are promptly lost as bacterially-derived proteins are produced and inserted (Scidmore et al., 2003). Because many markers of the endomembrane system do not associate with the inclusion, it was assumed to be disengaged from classical vesicular trafficking pathways (reviewed in (Fields et al., 2002). New cell biological studies, however, have led to a reassessment of how Chlamydia exploits or “mimics” host machinery to maneuver through the host cell and establish interactions with host organelles.
Interestingly, a subset of recycling endosome and Golgi-related Rab GTPases (e.g. Rab1, 4, and 11) -essential host proteins that regulate organelle identity and membrane trafficking (Seabra et al., 2004)- associate with the inclusion membrane (Rzomp et al., 2003). Rab6 and Rab10 associate with inclusions in a species-specific manner suggesting that bacterial proteins which differ between species may be involved in Rab recruitment (Rzomp et al., 2003). Chlamydial inclusion membrane proteins (Inc) are likely candidates for these bacterial factors. In agreement, the C. trachomatis Inc protein Ct229 is a Rab4 binding partner (Rzomp et al., 2006), and the C. pneumoniae Inc, Cpn0585, interacts with Rab1, 10, and 11 (Cortes et al., 2007). Ectopic overexpression of Cpn0585 saturates Rab11 binding and inhibits inclusion development indicating a role for Rab-Inc interactions in chlamydial growth (Cortes et al., 2007). SNARE (soluble NSF-sensitive attachment receptor) proteins, which regulate host membrane fusion, are also targeted by Chlamydia. The Inc proteins IncA, Ct223, and Ct813 contain SNARE-like motifs, and IncA can bind to a subset of host SNAREs (Vamp3, Vamp7 and Vamp8) in vitro. Importantly, SNARE recruitment to the inclusion is impaired in IncA-deficient strains (Delevoye et al., 2008). Chlamydia also co-opts the minus-end directed motor protein dynein to facilitate transportation of the inclusion along microtubules to a perinuclear region near the microtubule-organizing center (MTOC) (Grieshaber et al., 2003).
Once established in this niche, the inclusion can selectively interact with organelles that provide factors essential for chlamydial development such as eukaryotic lipids, including sphingolipids (Hackstadt et al., 1996, Moore et al., 2008), cholesterol (Carabeo et al., 2003) and glycerophospholipids (Wylie et al., 1997). While sphingolipids and cholesterol isolated from bacteria are not modified, fatty acids in the Sn2-position of host glycerophospholipids are exchanged with Chlamydia-derived branched chain fatty acids (Wylie et al., 1997, Su et al., 2004). Lipid transport pathways to the inclusion remain largely undefined.
The Golgi apparatus is a likely source of lipids for the Chlamydia inclusion as it is abundant in the perinuclear region where the inclusion is nestled during development. A portion of Chlamydia-acquired sphingolipids are intercepted from exocytic Golgi vesicles(Hackstadt et al., 1996). Changes in Golgi architecture also affect inclusion development. Golgin-84, a component of the structural scaffold that maintains Golgi apparatus morphology, is cleaved during infection resulting in Golgi fragmentation to ministacks (Heuer et al., 2009). Chlamydial replication is significantly reduced when Golgi fragmentation is inhibited by expression of truncated golgin-84. Inversely, RNAi depletion of giantin and GPP130 stimulate Golgi fragmentation and enhance chlamydial replication. Fragmentation is hypothesized to boost delivery of Golgi-derived lipids to the inclusion as ceramide transport is delayed by inhibitors of golgin-84 cleavage (Heuer et al., 2009). Retrograde Golgi traffic during chlamydial infections is further highlighted by the identification of COPI components as important host factors in C. caviae infection (Derre et al., 2007).
Multivesicular bodies (MVBs) may represent an alternative lipid transport pathway. MVBs are endocytic organelles where proteins and lipids that are destined for degradation or recycling to the Golgi are sorted and processed (Woodman et al., 2008). MVB protein components including CD63, LBPA, and MLN64 localize to the lumen of the Chlamydia inclusion, and CD63-positive vesicles are observed within the inclusion. Furthermore, pharmacological inhibitors of MVB maturation result in decreased sphingolipid transport to the inclusion and inhibit bacterial replication. (Beatty, 2006)
Non-classical transport pathways may also be involved in lipid delivery to the inclusion. Lipid droplets (LDs), eukaryotic neutral lipid storage organelles, proliferate at the inclusion periphery (Kumar et al., 2006). Because LDs are delimited by a phospholipid monolayer, it is unclear how these organelles might interact, fuse and deliver components to the inclusion. Electron and live time-lapse microscopy surprisingly reveal that intact LDs are translocated into the inclusion lumen (Cocchiaro et al., 2008). These observations indicate that uptake of entire organelles – and their associated material – by the inclusion may be a general alternative strategy for nutrient acquisition that circumvents the need for membrane fusion.
Physical association of mitochondria with inclusions has long been observed (Matsumoto et al., 1991)., but a link between mitochondrial function and Chlamydia infection was lacking until recently. RNAi knockdown of the Tim-Tom complex, a mitochondrial protein import system, inhibits C. caviae infection, but not C. trachomatis (Derre et al., 2007). These findings are another illustration that the requirements for successful bacterial growth and survival differ between chlamydial strains.
Clearly, Chlamydia can access multiple sources of lipid precursors and nutrients within its host cell. Given its obligate intracellular lifestyle, any one path is unlikely to be essential for chlamydial survival. It would not be surprising if Chlamydia taps into multiple, redundant host membrane trafficking pathways as has been observed in other intracellular pathogens such as Legionella pneumophila (reviewed in (Isberg et al., 2009).
Evasion of host defenses
Chlamydiae manipulate signaling pathways to hinder the activation of innate immune responses that are detrimental to bacterial or host survival. Here we focus on the inhibition of apoptosis and the manipulation of NF-κB -mediated signaling.
Inhibition of apoptosis
The effect of Chlamydia infection on apoptotic signaling programs is complex. Chlamydia is postulated to regulate apoptosis in a temporal manner to prevent the host cell from dying too early in infection and induce host-cell death late in the cycle (reviewed in (Ying et al., 2007, Byrne et al., 2004). The importance of proper temporal regulation of host cell death is illustrated by the fact that chlamydial development is impaired if host cell apoptosis is induced prematurely (Ying et al., 2008).
Chlamydia blocks apoptosis primarily by inhibiting mitochondrial cytochrome c (cyt c) release (Fan et al., 1998). The Bcl-2 family of proteins, which includes anti-apoptotic Bcl-2 like proteins, pro-apoptotic BH3-only proteins, and Bax/Bak, regulates cyt c release (reviewed in (Hacker et al., 2007). Chlamydia induces degradation of BH3-only proteins (Ying et al., 2005, Dong et al., 2005) which likely leads to the observed reduction in Bax and Bak activation (Fischer et al., 2004) and subsequent block in Cyt c release. The secreted chlamydial protease-like activity factor (CPAF) has been implicated in degradation of BH3-only proteins (Pirbhai et al., 2006). However, cleavage of BH3-only proteins in cell lines engineered to express active recombinant CPAF occurs with kinetics distinct from canonical substrates and is prevented by the proteasome-specific inhibitor MG-132, suggesting that degradation occurs via a proteasome-dependent mechanism indirectly influenced by CPAF (Paschen et al., 2008). Interestingly, a recent study did not detect BH3-only protein cleavage during infection (Rajalingam et al., 2008).
Although its anti-apoptotic role is unclear, CPAF is emerging as a central immunoregulatory protein. CPAF downregulates MHC class I and II antigen presentation by degrading the transcription factors USF-1 and RFX-5 (Zhong et al., 2001). Similarly, CPAF also contributes to the degradation of CD1d, a MHC-like protein important in lipid antigen presentation (Kawana et al., 2007).
Since CPAF is synthesized mid-late in the infectious cycle (Belland et al., 2003), it is unlikely to participate in the anti-apoptotic effects observed early in infection. Other potential anti-apoptotic mechanisms include: stabilization of inhibitor of apoptosis (IAP) proteins (Rajalingam et al., 2006), and sequestration of pro-apoptotic phosphorylated BAD and protein kinase Cδ (PKCδ) at the inclusion (Tse et al., 2005, Verbeke et al., 2006). Sequestration of phosphorylated BAD is proposed to occur through interaction with 14-3-3β which is recruited to the inclusion membrane (Scidmore et al., 2001, Verbeke et al., 2006). PKCδ is similarly recruited away from its normal site of action on mitochondria by binding to diacylglycerol-rich membranes at the inclusion periphery (Tse et al., 2005). Increased expression of the anti-apoptotic protein Mcl-1 in infected cells has also been linked to activation of Raf/MEK/ERK (Rajalingam et al., 2008), a signaling cascade that affects inflammatory responses (Buchholz et al., 2007) and chlamydial lipid acquisition (Su et al., 2004). The molecular basis for these alternative anti-apoptotic pathways remains elusive; however, similar to membrane trafficking modulation, Chlamydia appears to have several redundant anti-apoptotic strategies.
Modulation of NF-kB signaling
Interference with NF-κB signaling is an emerging theme in chlamydial modulation of host immunity. The NF-κB transcription factor regulates several facets of host innate and adaptive immunity (Hayden et al., 2006). Recent findings present new mechanisms for NF-κB inhibition by chlamydial effectors.
The NF-κB subunits RelA (p65) and p50 form a heterodimeric complex that is translocated into the nucleus and acts as a transcriptional activator (Hayden et al., 2006). During Chlamydia infection, RelA is proteolyzed by the C. trachomatis Tsp-like protease (Ct441), and NF-κB nuclear translocation is blocked. Furthermore, ectopic expression of Tsp prevents TNF-α-induced NF-κB activation in human cells.(Lad et al., 2007)
Chlamydia may also block NF-κB activation by regulating ubiquitin-mediated protein degradation. In the canonical NF-κB activation pathway, nuclear translocation is dependent on the degradation of its inhibitor IκBα via ubiquitin-mediated proteolysis (Sun et al., 2008). Two C. trachomatis proteins, ChlaDub1 and ChlaDub2, are effectors with deubiquitinating and deneddylating activity (Misaghi et al., 2006). Ectopically expressed ChlaDub1 binds to IκBα and inhibits its ubiquitination thereby suppressing degradation and subsequent NF-κB activation (Negrate, 2008). Although C. pneumoniae lacks ChlaDub1, IL-17-induced NF-κB activation can be suppressed by Inc protein Cp0236 sequestration of NF-κB activator 1 (Act1) (Wolf et al., 2009). As more effector proteins are identified, additional steps in NF-κB activation will likely emerge as targets of inhibition by Chlamydiae.
Inclusion expansion
The developing inclusion must expand to accommodate increasing bacterial numbers. Inclusion growth is likely fueled by attainment of nutrients and lipid precursors from the host cell. Unlike other intracellular pathogens that remain individually surrounded by tight membrane compartments, the inclusion is a large, relatively spacious organelle. The structural integrity of the inclusion is preserved by a meshwork of host cytoskeletal structures primarily composed of F-actin and intermediate filaments (IFs). The Head domains of IFs surrounding the inclusion are progressively cleaved by CPAF generating structural changes in the filaments as the inclusion ages. The purpose of inclusion stabilization is unclear but may limit cytoplasmic exposure of lumenal contents. This would benefit the pathogen by reducing activation of innate immune responses via microbial pattern recognition receptors. (Kumar et al., 2008)
EBs exit and dissemination
At developmental cycle completion, EBs must exit the cell to initiate subsequent rounds of infection. Egress can occur via two discrete mechanisms. Cell lysis involves the sequential disruption of inclusion and cellular membranes by cysteine proteases, and the host cell is destroyed. Alternatively, the inclusion can remain membrane-bound and be pushed out, or “extruded”, from the host cell. This process is dependent on actin-polymerization and myosin, and the host cell is often left intact. (Hybiske et al., 2007b)
One chlamydial effector protein, Chlamydia protein associating with death domain, or CADD, can interact with TNF receptor death domains and induce Fas-related apoptosis upon ectopic expression (Stenner-Liewen et al., 2002). Yet, the biological relevance of this is unknown since host cell-death induction at the late developmental stages appears non-apoptotic (Ying et al., 2006). Interestingly, ectopic expression of CPAF in uninfected cells induces morphological changes that mimic phenotypes of Chlamydia-induced cell death suggesting it may contribute to this process in vivo (Paschen et al., 2008).
Perspectives
Despite the absence of classical genetic tools and the lack of a cell free culture system, functional and comparative genomic approaches, combined with high throughput RNAi screens have shed new light on the biology of these ancient pathogens. With the recent exciting reports of efficient DNA exchange among Chlamydiae in experimental systems (DeMars et al., 2008) and successful electroporation of recombinant DNA (Binet et al., 2009), research progress will likely accelerate. Unlike other host-pathogen systems where cellular microbiology had to catch up with genetics, Chlamydia investigators have a large set of well-defined phenotypes awaiting the identification of mutants.
Acknowledgments
We thank H.A. Saka for comments on the manuscript. We apologize to those investigators whose work we were unable to cite because of space constraints. Work in our laboratory is supported by the National Institutes of Health and the Burroughs Wellcome Trust Fund Program in the Pathogenesis of Infectious Diseases. J.L.C. was supported by an American Heart Association Predoctoral Fellowship.
References
- Abromaitis S, Stephens RS. Attachment and entry of Chlamydia have distinct requirements for host protein disulfide isomerase. PLoS Pathog. 2009;5:e1000357. doi: 10.1371/journal.ppat.1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty WL. Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J Cell Sci. 2006;119:350–359. doi: 10.1242/jcs.02733. [DOI] [PubMed] [Google Scholar]
- Belland RJ, Zhong G, Crane DD, Hogan D, Sturdevant D, Sharma J, et al. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proceedings of the National Academy of Sciences. 2003;100:8478–8483. doi: 10.1073/pnas.1331135100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binet R, Maurelli AT. Transformation and isolation of allelic exchange mutants of Chlamydia psittaci using recombinant DNA introduced by electroporation. Proc Natl Acad Sci U S A. 2009;106:292–297. doi: 10.1073/pnas.0806768106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi F, Tarsia P, Aliberti S. Chlamydophila pneumoniae. Clin Microbiol Infect. 2009;15:29–35. doi: 10.1111/j.1469-0691.2008.02130.x. [DOI] [PubMed] [Google Scholar]
- Buchholz KR, Stephens RS. The extracellular signal-regulated kinase/mitogen-activated protein kinase pathway induces the inflammatory factor interleukin-8 following Chlamydia trachomatis infection. Infect Immun. 2007;75:5924–5929. doi: 10.1128/IAI.01029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne GI, Ojcius DM. Chlamydia and apoptosis: life and death decisions of an intracellular pathogen. Nat Rev Microbiol. 2004;2:802–808. doi: 10.1038/nrmicro1007. [DOI] [PubMed] [Google Scholar]
- Campbell LA, Kuo C-C. Interactions of Chlamydia with the Host Cells that Mediate Attachment and Uptake. In: Bavoil PM, Wyrick PB, editors. Chlamydia: genomics and pathogenesis. Wymondham, U.K: Horizon Bioscience; 2006. pp. 505–522. [Google Scholar]
- Carabeo RA, Dooley CA, Grieshaber SS, Hackstadt T. Rac interacts with Abi-1 and WAVE2 to promote an Arp2/3-dependent actin recruitment during chlamydial invasion. Cell Microbiol. 2007;9:2278–2288. doi: 10.1111/j.1462-5822.2007.00958.x. [DOI] [PubMed] [Google Scholar]
- Carabeo RA, Grieshaber SS, Hasenkrug A, Dooley C, Hackstadt T. Requirement for the Rac GTPase in Chlamydia trachomatis invasion of non-phagocytic cells. Traffic. 2004;5:418–425. doi: 10.1111/j.1398-9219.2004.00184.x. [DOI] [PubMed] [Google Scholar]
- Carabeo RA, Mead DJ, Hackstadt T. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proc Natl Acad Sci U S A. 2003;100:6771–6776. doi: 10.1073/pnas.1131289100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton DR, Dooley CA, Grieshaber SS, Carabeo RA, Fields KA, Hackstadt T. Tyrosine phosphorylation of the chlamydial effector protein Tarp is species specific and not required for recruitment of actin. Infect Immun. 2005;73:3860–3868. doi: 10.1128/IAI.73.7.3860-3868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchiaro JL, Kumar Y, Fischer ER, Hackstadt T, Valdivia RH. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci U S A. 2008;105:9379–9384. doi: 10.1073/pnas.0712241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant CG, Stephens RS. Chlamydia attachment to mammalian cells requires protein disulfide isomerase. Cell Microbiol. 2007;9:222–232. doi: 10.1111/j.1462-5822.2006.00783.x. [DOI] [PubMed] [Google Scholar]
- Cortes C, Rzomp KA, Tvinnereim A, Scidmore MA, Wizel B. Chlamydia pneumoniae inclusion membrane protein Cpn0585 interacts with multiple Rab GTPases. Infect Immun. 2007;75:5586–5596. doi: 10.1128/IAI.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautry-Varsat A, Subtil A, Hackstadt T. Recent insights into the mechanisms of Chlamydia entry. Cell Microbiol. 2005;7:1714–1722. doi: 10.1111/j.1462-5822.2005.00627.x. [DOI] [PubMed] [Google Scholar]
- Delevoye C, Nilges M, Dehoux P, Paumet F, Perrinet S, Dautry-Varsat A, Subtil A. SNARE protein mimicry by an intracellular bacterium. PLoS Pathog. 2008;4:e1000022. doi: 10.1371/journal.ppat.1000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMars R, Weinfurter J. Interstrain gene transfer in Chlamydia trachomatis in vitro: mechanism and significance. J Bacteriol. 2008;190:1605–1614. doi: 10.1128/JB.01592-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derre I, Pypaert M, Dautry-Varsat A, Agaisse H. RNAi screen in Drosophila cells reveals the involvement of the Tom complex in Chlamydia infection. PLoS Pathog. 2007;3:1446–1458. doi: 10.1371/journal.ppat.0030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F, Pirbhai M, Xiao Y, Zhong Y, Wu Y, Zhong G. Degradation of the proapoptotic proteins Bik, Puma, and Bim with Bcl-2 domain 3 homology in Chlamydia trachomatis-infected cells. Infect Immun. 2005;73:1861–1864. doi: 10.1128/IAI.73.3.1861-1864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Ceesay A, Kim JH, Kalman D, Engel JN. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog. 2008;4:e1000021. doi: 10.1371/journal.ppat.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadel S, Eley A. Chlamydia trachomatis OmcB protein is a surface-exposed glycosaminoglycan-dependent adhesin. J Med Microbiol. 2007;56:15–22. doi: 10.1099/jmm.0.46801-0. [DOI] [PubMed] [Google Scholar]
- Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, et al. Inhibition of apoptosis in Chlamydia-infected Cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med. 1998;187:487–496. doi: 10.1084/jem.187.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields KA, Hackstadt T. The chlamydial inclusion: escape from the endocytic pathway. Annu Rev Cell Dev Biol. 2002;18:221–245. doi: 10.1146/annurev.cellbio.18.012502.105845. [DOI] [PubMed] [Google Scholar]
- Fields KA, Mead DJ, Dooley CA, Hackstadt T. Chlamydia trachomatis type III secretion: evidence for a functional apparatus during early-cycle development. Mol Microbiol. 2003;48:671–683. doi: 10.1046/j.1365-2958.2003.03462.x. [DOI] [PubMed] [Google Scholar]
- Fischer SF, Harlander T, Vier J, Hacker G. Protection against CD95-induced apoptosis by chlamydial infection at a mitochondrial step. Infect Immun. 2004;72:1107–1115. doi: 10.1128/IAI.72.2.1107-1115.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fudyk T, Olinger L, Stephens RS. Selection of mutant cell lines resistant to infection by Chlamydia spp [corrected] Infect Immun. 2002;70:6444–6447. doi: 10.1128/IAI.70.11.6444-6447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshaber SS, Grieshaber NA, Hackstadt T. Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J Cell Sci. 2003;116:3793–3802. doi: 10.1242/jcs.00695. [DOI] [PubMed] [Google Scholar]
- Hacker G, Weber A. BH3-only proteins trigger cytochrome c release, but how? Arch Biochem Biophys. 2007;462:150–155. doi: 10.1016/j.abb.2006.12.022. [DOI] [PubMed] [Google Scholar]
- Hackstadt T, Rockey DD, Heinzen RA, Scidmore MA. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J. 1996;15:964–977. [PMC free article] [PubMed] [Google Scholar]
- Hackstadt T, Todd WJ, Caldwell HD. Disulfide-mediated interactions of the chlamydial major outer membrane protein: role in the differentiation of chlamydiae? J Bacteriol. 1985;161:25–31. doi: 10.1128/jb.161.1.25-31.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- Heuer D, Lipinski AR, Machuy N, Karlas A, Wehrens A, Siedler F, et al. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature. 2009;457:731–735. doi: 10.1038/nature07578. [DOI] [PubMed] [Google Scholar]
- Hower S, Wolf K, Fields KA. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early-cycle development. Mol Microbiol. 2009 doi: 10.1111/j.1365-2958.2009.06732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS. Mechanisms of Chlamydia trachomatis entry into nonphagocytic cells. Infect Immun. 2007a;75:3925–3934. doi: 10.1128/IAI.00106-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci U S A. 2007b;104:11430–11435. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isberg RR, O’Connor TJ, Heidtman M. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat Rev Microbiol. 2009;7:13–24. doi: 10.1038/nrmicro1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett TJ, Dooley CA, Mead DJ, Hackstadt T. Chlamydia trachomatis tarp is phosphorylated by src family tyrosine kinases. Biochem Biophys Res Commun. 2008;371:339–344. doi: 10.1016/j.bbrc.2008.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. Chlamydial TARP is a bacterial nucleator of actin. Proc Natl Acad Sci U S A. 2006;103:15599–15604. doi: 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawana K, Quayle AJ, Ficarra M, Ibana JA, Shen L, Kawana Y, et al. CD1d degradation in Chlamydia trachomatis-infected epithelial cells is the result of both cellular and chlamydial proteasomal activity. J Biol Chem. 2007;282:7368–7375. doi: 10.1074/jbc.M610754200. [DOI] [PubMed] [Google Scholar]
- Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol. 2006;16:1646–1651. doi: 10.1016/j.cub.2006.06.060. [DOI] [PubMed] [Google Scholar]
- Kumar Y, Valdivia RH. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe. 2008;4:159–169. doi: 10.1016/j.chom.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lad SP, Li J, da Silva Correia J, Pan Q, Gadwal S, Ulevitch RJ, Li E. Cleavage of p65/RelA of the NF-kappaB pathway by Chlamydia. Proc Natl Acad Sci U S A. 2007;104:2933–2938. doi: 10.1073/pnas.0608393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane BJ, Mutchler C, Al Khodor S, Grieshaber SS, Carabeo RA. Chlamydial entry involves TARP binding of guanine nucleotide exchange factors. PLoS Pathog. 2008;4:e1000014. doi: 10.1371/journal.ppat.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Bessho H, Uehira K, Suda T. Morphological studies of the association of mitochondria with chlamydial inclusions and the fusion of chlamydial inclusions. J Electron Microsc (Tokyo) 1991;40:356–363. [PubMed] [Google Scholar]
- Menozzi FD, Pethe K, Bifani P, Soncin F, Brennan MJ, Locht C. Enhanced bacterial virulence through exploitation of host glycosaminoglycans. Mol Microbiol. 2002;43:1379–1386. doi: 10.1046/j.1365-2958.2002.02841.x. [DOI] [PubMed] [Google Scholar]
- Misaghi S, Balsara ZR, Catic A, Spooner E, Ploegh HL, Starnbach MN. Chlamydia trachomatis-derived deubiquitinating enzymes in mammalian cells during infection. Mol Microbiol. 2006;61:142–150. doi: 10.1111/j.1365-2958.2006.05199.x. [DOI] [PubMed] [Google Scholar]
- Moore ER, Fischer ER, Mead DJ, Hackstadt T. The chlamydial inclusion preferentially intercepts basolaterally directed sphingomyelin-containing exocytic vacuoles. Traffic. 2008;9:2130–2140. doi: 10.1111/j.1600-0854.2008.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrate G, Krieg A, Faustin B, Loeffler M, Godzik A, Krajewski S, Reed JC. ChlaDub1 of Chlamydia trachomatis suppresses NF-kB activation and inhibits IkBa ubiquitination and degradation. Cellular Microbiology. 2008;10:1879–1892. doi: 10.1111/j.1462-5822.2008.01178.x. [DOI] [PubMed] [Google Scholar]
- Newhall WJ, Jones RB. Disulfide-linked oligomers of the major outer membrane protein of chlamydiae. J Bacteriol. 1983;154:998–1001. doi: 10.1128/jb.154.2.998-1001.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson TL, Olinger L, Chong K, Schoolnik G, Stephens RS. Global stage-specific gene regulation during the developmental cycle of Chlamydia trachomatis. J Bacteriol. 2003;185:3179–3189. doi: 10.1128/JB.185.10.3179-3189.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen SA, Christian JG, Vier J, Schmidt F, Walch A, Ojcius DM, Hacker G. Cytopathicity of Chlamydia is largely reproduced by expression of a single chlamydial protease. J Cell Biol. 2008;182:117–127. doi: 10.1083/jcb.200804023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirbhai M, Dong F, Zhong Y, Pan KZ, Zhong G. The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J Biol Chem. 2006;281:31495–31501. doi: 10.1074/jbc.M602796200. [DOI] [PubMed] [Google Scholar]
- Plaunt MR, Hatch TP. Protein synthesis early in the developmental cycle of Chlamydia psittaci. Infect Immun. 1988;56:3021–3025. doi: 10.1128/iai.56.12.3021-3025.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalingam K, Sharma M, Lohmann C, Oswald M, Thieck O, Froelich CJ, Rudel T. Mcl-1 is a key regulator of apoptosis resistance in Chlamydia trachomatis-infected cells. PLoS ONE. 2008;3:e3102. doi: 10.1371/journal.pone.0003102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalingam K, Sharma M, Paland N, Hurwitz R, Thieck O, Oswald M, et al. IAP-IAP complexes required for apoptosis resistance of C. trachomatis-infected cells. PLoS Pathog. 2006;2:e114. doi: 10.1371/journal.ppat.0020114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzomp KA, Moorhead AR, Scidmore MA. The GTPase Rab4 interacts with Chlamydia trachomatis inclusion membrane protein CT229. Infect Immun. 2006;74:5362–5373. doi: 10.1128/IAI.00539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzomp KA, Scholtes LD, Briggs BJ, Whittaker GR, Scidmore MA. Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect Immun. 2003;71:5855–5870. doi: 10.1128/IAI.71.10.5855-5870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samudrala R, Heffron F, McDermott JE. Accurate prediction of secreted substrates and identification of a conserved putative secretion signal for type III secretion systems. PLoS Pathog. 2009;5:e1000375. doi: 10.1371/journal.ppat.1000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachter J. Infection and Disease Epidemiology. In: Stephens RS, editor. Chlamydia: Intracellular Biology, Pathogenesis and Immunity. Washington DC: American Society for Microbiology Press; 1999. pp. 139–170. [Google Scholar]
- Scidmore MA, Fischer ER, Hackstadt T. Restricted fusion of Chlamydia trachomatis vesicles with endocytic compartments during the initial stages of infection. Infect Immun. 2003;71:973–984. doi: 10.1128/IAI.71.2.973-984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore MA, Hackstadt T. Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol. 2001;39:1638–1650. doi: 10.1046/j.1365-2958.2001.02355.x. [DOI] [PubMed] [Google Scholar]
- Seabra MC, Wasmeier C. Controlling the location and activation of Rab GTPases. Curr Opin Cell Biol. 2004;16:451–457. doi: 10.1016/j.ceb.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Sisko JL, Spaeth K, Kumar Y, Valdivia RH. Multifunctional analysis of Chlamydia-specific genes in a yeast expression system. Molecular Microbiology. 2006;60:51–66. doi: 10.1111/j.1365-2958.2006.05074.x. [DOI] [PubMed] [Google Scholar]
- Stenner-Liewen F, Liewen H, Zapata JM, Pawlowski K, Godzik A, Reed JC. CADD, a Chlamydia protein that interacts with death receptors. J Biol Chem. 2002;277:9633–9636. doi: 10.1074/jbc.C100693200. [DOI] [PubMed] [Google Scholar]
- Su H, McClarty G, Dong F, Hatch GM, Pan ZK, Zhong G. Activation of Raf/MEK/ERK/cPLA2 signaling pathway is essential for chlamydial acquisition of host glycerophospholipids. J Biol Chem. 2004;279:9409–9416. doi: 10.1074/jbc.M312008200. [DOI] [PubMed] [Google Scholar]
- Su H, Raymond L, Rockey DD, Fischer E, Hackstadt T, Caldwell HD. A recombinant Chlamydia trachomatis major outer membrane protein binds to heparan sulfate receptors on epithelial cells. Proc Natl Acad Sci U S A. 1996;93:11143–11148. doi: 10.1073/pnas.93.20.11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtil A, Delevoye C, Balana ME, Tastevin L, Perrinet S, Dautry-Varsat A. A directed screen for chlamydial proteins secreted by a type III mechanism identifies a translocated protein and numerous other new candidates. Mol Microbiol. 2005;56:1636–1647. doi: 10.1111/j.1365-2958.2005.04647.x. [DOI] [PubMed] [Google Scholar]
- Sun SC, Ley SC. New insights into NF-kappaB regulation and function. Trends Immunol. 2008;29:469–478. doi: 10.1016/j.it.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse SM, Mason D, Botelho RJ, Chiu B, Reyland M, Hanada K, et al. Accumulation of diacylglycerol in the Chlamydia inclusion vacuole: possible role in the inhibition of host cell apoptosis. J Biol Chem. 2005;280:25210–25215. doi: 10.1074/jbc.M501980200. [DOI] [PubMed] [Google Scholar]
- Verbeke P, Welter-Stahl L, Ying S, Hansen J, Hacker G, Darville T, Ojcius DM. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog. 2006;2:e45. doi: 10.1371/journal.ppat.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrl W, Brinkmann V, Jungblut PR, Meyer TF, Szczepek AJ. From the inside out--processing of the Chlamydial autotransporter PmpD and its role in bacterial adhesion and activation of human host cells. Mol Microbiol. 2004;51:319–334. doi: 10.1046/j.1365-2958.2003.03838.x. [DOI] [PubMed] [Google Scholar]
- Wilson DP, Timms P, McElwain DL, Bavoil PM. Type III secretion, contact-dependent model for the intracellular development of Chlamydia. Bull Math Biol. 2006;68:161–178. doi: 10.1007/s11538-005-9024-1. [DOI] [PubMed] [Google Scholar]
- Wolf K, Plano GV, Fields KA. A protein secreted by the respiratory pathogen Chlamydia pneumoniae impairs IL-17 signaling via interaction with human Act1. Cell Microbiol. 2009 doi: 10.1111/j.1462-5822.2009.01290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman PG, Futter CE. Multivesicular bodies: co-ordinated progression to maturity. Curr Opin Cell Biol. 2008;20:408–414. doi: 10.1016/j.ceb.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie JL, Hatch GM, McClarty G. Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. J Bacteriol. 1997;179:7233–7242. doi: 10.1128/jb.179.23.7233-7242.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Fischer SF, Pettengill M, Conte D, Paschen SA, Ojcius DM, Hacker G. Characterization of host cell death induced by Chlamydia trachomatis. Infect Immun. 2006;74:6057–6066. doi: 10.1128/IAI.00760-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Pettengill M, Latham ER, Walch A, Ojcius DM, Hacker G. Premature apoptosis of Chlamydia-infected cells disrupts chlamydial development. J Infect Dis. 2008;198:1536–1544. doi: 10.1086/592755. [DOI] [PubMed] [Google Scholar]
- Ying S, Pettengill M, Ojcius DM, Hacker G. Host-cell survival and death during Chlamydia infection. Curr Immunol Rev. 2007;3:31–40. doi: 10.2174/157339507779802179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Seiffert BM, Hacker G, Fischer SF. Broad degradation of proapoptotic proteins with the conserved Bcl-2 homology domain 3 during infection with Chlamydia trachomatis. Infect Immun. 2005;73:1399–1403. doi: 10.1128/IAI.73.3.1399-1403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, Fan P, Ji H, Dong F, Huang Y. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J Exp Med. 2001;193:935–942. doi: 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]