

Abstract
Genetic crosses have been employed to study various traits of rodent malaria parasites and to locate loci that contribute to drug resistance, immune protection, and disease virulence. Compared with human malaria parasites, genetic crossing of rodent malaria parasites is more easily performed; however, genotyping methods using microsatellites (MS) or large-scale single nucleotide polymorphisms (SNPs) that have been widely used in typing Plasmodium falciparum are not available for rodent malaria species. Here we report a genome-wide search of the Plasmodium yoelii yoelii (P. yoelii) genome for simple sequence repeats (SSRs) and the identification of nearly 600 polymorphic microsatellite (MS) markers for typing the genomes of P. yoelii and Plasmodium berghei. The MS markers are randomly distributed across the 14 physical chromosomes assembled from genome sequences of three rodent malaria species, although some variations in the numbers of MS expected according to chromosome size exist. The majority of the MS markers are AT-rich repeats, similar to those found in the P. falciparum genome. The MS markers provide an important resource for genotyping, lay a foundation for developing linkage maps, and will greatly facilitate genetic studies of P. yoelii.
Keywords: Rodent malaria parasite, Simple sequence repeat (SSR), Genetic markers, Genotyping, MS
1. Introduction
Rodent malaria species are excellent models for studying Plasmodium phenotypes. Disease phenotypes are the result of complex interactions between malaria parasites and their hosts, and genetic variations in both systems can influence a disease outcome. Thus, the option to use inbred mice when studying parasite virulence factors, which minimize the variations in host genetic background, is a significant advantage. Genetic crosses of rodent malaria parasites using inbred mice can be performed to map genes that contribute to a disease phenotype. The availability of large numbers of inbred mice and the simplicity in housing rodent hosts make rodent malaria parasites an attractive system for studying malaria disease phenotypes. Additionally, an elegant selection strategy termed linkage group selection (LGS) has made cloning progeny of rodent malaria crosses unnecessary, greatly facilitating genetic mapping selectable traits [1].
Various methods have been developed for genotyping malaria parasites, including isoenzyme electrophoresis, restriction fragment length polymorphism, amplified fragment length polymorphism (AFLP), microsatellite (MS), and single nucleotide polymorphism (SNP) [2–11]. AFLP has been successfully employed to type rodent malaria parasites, leading to the identification of genetic loci and candidate genes involved in drug resistance, immune protection, and other traits [1, 5, 6, 12, 13]. Although SNP mapping and high throughput parallel sequencing represent the future choices for genotyping large numbers of polymorphisms in malaria parasites, MS analysis can still be very useful for many genetic studies. In particular, for rodent malaria parasite traits, genetic mapping is likely to be performed using progeny from genetic crosses, as population-based association studies are unlikely unless efforts are made to survey and collect additional isolates from African thicket rats. Genetic mapping using progeny from a genetic cross may not require a large number of genetic markers. For initial mapping, a relatively small number of progeny (< 100) is generally obtained from a cross because of the time-consuming process in cloning independent recombinant progeny; and the number of recombination events from the progeny will be limited. A few hundreds of genetic markers should be able to detect the majority of recombination events in the progeny; and MS can still be effective for typing progeny that requires only relative small numbers of markers. Use of high-density array or parallel sequencing will be more efficient if large numbers of genetic markers (thousands or more) are to be interrogated. However, a high-density microarray genotyping chip typically costs several hundreds of dollars plus reagent and labor expenses. It can be time consuming to analyze the large volume of data collected from a high-density array or high throughput parallel sequencing. Additionally, performing array hybridization or parallel sequencing typically require expensive equipment or special core facilities. Compared with two alleles for most SNPs, a MS is usually highly polymorphic with multiple alleles in a parasite population, which can be useful for typing closely related parasites such as parasite clones originally deriving from a single parasite. For an example, Plasmodium yoelii yoelii 17X lethal (for simplicity, P. yoelii will be used for Plasmodium yoelii yoelii), P. yoelii 17X nonlethal, and P. yoelii 312 derive from a common ancestor after its initial isolation from an African rat and have a very similar genome; however, some changes have occurred in their genomes after being propagated in different laboratories over the years. MS with different allele sizes can be identified (see below) and used to type these parasites.
Here we report screening the P. yoelii genome [14] for SSRs and developing nearly 600 polymorphic MS markers for typing P. yoelii and Plasmodium berghei. The MS markers were assigned to the composite chromosome maps that were assembled from genome sequences of three rodent malaria species [15]. These MS markers provide a foundation for developing linkage maps for P. yoelii and will play an important role in genetic studies of the species.
2. Materials and methods
2.1. Parasites and DNA extraction
Seven P. yoelii lines and one P. berghei line were used in this study. These parasites differed in several phenotypes such as growth rate and disease virulence and can used as parents for genetic crosses. They were P. yoelii 17XLs (a 17X lethal line that has been maintained in the laboratory of Professor Pan Weiqing, Tongji University, Shanghai, China); P. yoelii17XNLs (a nonlethal 17X line, also from Professor Pan Weiqing); four MR4 lines (http://www.mr4.org/) P. yoelii 17XL (a lethal 17X line, MR4 #MRA-426, deposited by W. Peters, London School of Hygiene and Tropical Medicine, London, UK, and R. Killick-Kendrick, Imperial College, London, UK), P. yoelii 17XNL (a nonlethal 17X line, MR4 #MRA-593, deposited by W. Weiss, Naval Medical Research Center, Silver Spring, Maryland, USA), P. yoelii 312 (MR4 #MRA-312, deposited by T.F. McCutchan, NIH), and P. yoelii nigeriensis (MR4 #MRA-427, deposited by W. Peters and B.L. Robinson, London School of Hygiene and Tropical Medicine, London, UK, and R. Killick-Kendrick, Imperial College, London, UK); P. yoelii By265 (a line obtained from the laboratory of Professor Huang Fusheng, the Third Military Medical University, Chongqing, China); and a P. berghei line (PbK173), maintained at T.F. McCutchan’s lab. The 17X related lines were likely derived from a single P. yoelii parasite isolate designated 17X described previously [16]. For simplicity, these lines will be referred to as Py17XLs, Py17XNLs, Py17XL, Py17XNL, Py312, PyNIG, PyBy265 and PbK173, respectively.
Parasite DNA samples were extracted from infected mouse blood 5–7 days after injection of 1×105 parasites into 5- to 7-week-old BALB/c mice using a NIH-approved protocol (LMVR85). Blood samples infected with the parasites were collected from orbital sinus under general anesthesia [17] and centrifuged briefly to pellet the erythrocytes. Genomic DNA was extracted using a commercial blood sample DNA extraction kit (Qiagen).
2.2. Database search for SSRs and polymerase chain reaction (PCR) primer synthesis
We screened the P. yoelii 17XNL clone 1.1 genome (http://www.tigr.org/tdb/e2k1/pya1/) for simple sequence repeats (SSRs) as described previously [8]. Briefly, potential MS were first identified using Tandem Repeat Finder (http://tandem.bu.edu/trf/trf.html) [18] or through simple sequence search using the “Find” function in Microsoft Word after downloading the genome sequence. The criteria for selecting potential MS were 10 repeat units or longer for mono- or di-nucleotide repeats and 6 units or more for tri-nucleotide or longer repeats. DNA sequences flanking the repeats were selected for primer design by visual inspection. Because of the AT richness of the genome and the requirement of specific PCR product sizes, selection of primer sequences using commercial software often failed to pick appropriate primer pairs at specified locations. We therefore followed some general rules for primer selection that would produce primers with an annealing temperature of 50–55°C and expected PCR product size of 100–300 bp. Sequences of 20-mer with five to seven GC were selected, with optimal sequences having 20-mer with six GC. If no such sequences were available, longer sequences with four to five GC or shorter sequences (18-19-mer) with eight to nine GC were also considered. Primer sequences were sent to a commercial company for synthesis (Shanghai Sangon Biological Engineering Technology & Services Co., Ltd, Shanghai, China).
2.3. MS typing
The MS typing procedure using a single fluorescently labeled universal M13-tailed primer has been described [8]. Briefly, for each typing reaction, three PCR primers were included, with a pair of forward and reverse sequence specific primers and a third primer that was tagged with fluorescent dyes. One of the sequence-specific primers also contained the M13-universal sequence (5′-CACGACGTTGTAAAACGAC-3′) that acts as an anchor sequence for the third primer. PCR cycling parameters were: 94°C, 2 min for initial denaturation; and 94°C, 20 s; 50°C, 20 s; and 60°C, 30 s for 40 cycles. PCR products were fluorescently labeled after incorporation of the M13 primer tagged with D4-PA or D3-PA dyes (Sigma-Aldrich) and were detected in a Beckman CEQ 8000 DNA analyzer (Beckman Coulter). The sizes of PCR products were estimated using the size standard kit 400 from the same company. The primer sequences, sizes of PCR products and numbers of polymorphic MSs (alleles) among parasite lines generated using each pair of the forward and reverse primers are shown in Supplementary Table 1.
2.4. Assigning MS markers to chromosomes
MS markers were first assigned to contigs where they were initially identified. The positions of the contigs were “mapped” to their positions on the rodent malaria composite chromosome/contig maps according to Kooij et al. [15]. After the specific positions for the MS markers on a chromosome were identified, a physical chromosome with locations of each MS marker was drawn.
3. Results and Discussion
3.1. Search of MS and marker diversity
Extensive database searches of ~20 Mb P. yoelii genome sequences available when we conducted the search identified 625 SSRs as potential MS markers. PCR primer pairs flanking the 625 SSRs were synthesized and used to amplify the loci from P. yoelii and P. berghei DNAs using the M13-tailed primer method described previously [8]. After testing the SSRs on DNA from eight parasite lines, we identified 591 (or 94.6%) polymorphic MS (Table 1 and Supplemental Table 1), including 77 MS markers reported previously [8]. These MS markers provide a density of approximately one polymorphic MS per 28.4 kb or approximately 2 cM per MS marker assuming that P. yoelii has a similar recombination frequency as that seen in P. falciparum and Plasmodium chaboudi chaboudi [19, 20].
Table 1.
Summary of unique, mixed, and missing alleles of microsatellites from Plasmodium yoelii and Plasmodium bergheia
| Chr | MS typed | Poly MS | Py17XL | Py17XLs | Py17XNL | Py17XNLs | Py312 | PyBy265 | PyNIG | PbK173 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 34 | 34 | 0/1/0 | 1/1/0 | 0/1/0 | 0/1/0 | 0/1/0 | 27/1/0 | 30/1/3 | 19/3/14 |
| 2 | 16 | 14 | 0/0/0 | 0/0/0 | 0/0/0 | 0/0/0 | 0/0/0 | 12/0/0 | 12/0/2 | 5/1/9 |
| 3 | 22 | 21 | 2/0/0 | 2/0/0 | 0/0/0 | 1/0/0 | 0/0/0 | 17/0/3 | 17/1/4 | 7/3/14 |
| 4 | 27 | 27 | 0/1/0 | 0/1/0 | 0/1/0 | 0/1/0 | 0/1/0 | 19/1/1 | 26/1/1 | 15/3/11 |
| 5 | 38 | 35 | 0/0/0 | 0/0/0 | 0/0/0 | 0/0/0 | 0/0/0 | 30/2/0 | 25/0/7 | 16/0/19 |
| 6 | 31 | 30 | 2/0/0 | 1/0/0 | 1/0/0 | 0/0/0 | 0/0/0 | 22/0/1 | 23/0/5 | 10/1/20 |
| 7 | 26 | 25 | 2/0/0 | 1/0/0 | 0/0/0 | 0/0/0 | 0/0/0 | 21/0/0 | 24/0/1 | 10/1/15 |
| 8 | 45 | 43 | 1/0/1 | 1/2/0 | 0/0/0 | 0/2/0 | 1/1/0 | 36/2/0 | 38/1/3 | 22/4/21 |
| 9 | 53 | 51 | 2/1/0 | 1/0/0 | 1/2/0 | 1/1/0 | 1/2/0 | 40/0/3 | 41/2/7 | 28/1/23 |
| 10 | 43 | 41 | 3/1/0 | 2/0/0 | 0/0/0 | 1/0/0 | 1/0/0 | 32/1/1 | 31/0/6 | 14/1/26 |
| 11 | 61 | 56 | 3/1/1 | 1/2/0 | 0/2/0 | 2/2/0 | 1/1/1 | 38/2/2 | 52/1/1 | 34/4/22 |
| 12 | 52 | 49 | 3/0/0 | 2/0/0 | 0/0/0 | 0/0/0 | 0/0/0 | 39/1/0 | 40/1/3 | 21/1/28 |
| 13 | 78 | 71 | 2/1/1 | 1/2/0 | 1/1/0 | 0/1/0 | 0/0/0 | 52/3/2 | 61/2/5 | 39/6/32 |
| 14 | 61 | 58 | 0/0/1 | 2/1/0 | 0/0/0 | 1/1/0 | 1/0/0 | 43/2/3 | 51/0/3 | 32/2/26 |
| UA | 38 | 36 | 1/4/2 | 1/5/0 | 0/5/0 | 0/6/0 | 0/6/0 | 27/4/2 | 30/2/5 | 17/4/19 |
| Total | 25 | 591 | 21/10/6 | 16/14/0 | 3/12/0 | 6/15/0 | 5/12/1 | 455/19/18 | 501/12/56 | 289/35/299 |
Abbreviations: Chr, chromosome; MS typed, number of primer pairs used to amplify parasite DNA; Poly MS, number of microsatellites that were polymorphic among the parasites; Py17XL and the rest of parasite names, unique alleles different from the common Py17X alleles/numbers of markers with mixed alleles/numbers of markers with missing alleles (no calls) for each parasite. For example, 0/1/0 under Py17XL on chromosome 1 indicates that 0 markers were different from those of other Py17X-derived lines (no unique alleles), 1 marker had more than one alleles (mixed alleles), and 0 markers had no PCR products (all produced products); UA, number of unassigned markers.
The estimated genome size of P. yoelii was ~23 Mb [14], but only approximately 20 Mb sequences were searched. Assuming that we could identity MS at a similar rate for the whole genome, we could have obtained ~680 potential MS, or one MS per ~34 kb. This frequency is lower than that of P. falciparum, with an estimated one or more polymorphic MS per kb [10, 19, 21], although the sequences not present in the current P. yoelii genome sequences could have higher AT content and therefore more AT-related repeats. The lower AT content of the P. yoelii genome (~78% vs. 82% for P. falciparum) may contribute to the lower frequency of AT-related MS in the genome. Low MS frequency was also observed in Plasmodium vivax, a human parasite that has a much higher GC content than those of P. falciparum and P. yoelii [22, 23].
Of the 591 markers, 557 (94.2%) were polymorphic among the P. yoelii parasites and 34 were polymorphic between the P. yoelii isolates and P. berghei only (no differences among P. yoelii parasites). As expected, the P. yoelii 17X-derived lines (Py17XLs, Py17XNLs, Py17XL, Py17NXL, and Py312) had almost identical genotypes with only a few different MS alleles, reflecting recent mutations accumulated during propagation in different laboratories from a single parasite isolate designated as P. yoelii 17X (Table 1 and Supplemental Table 1). These differences, ranging from three unique MS for Py17XNL to 21 unique MS for Py17XL, can be used to distinguish these closely related parasites. There are many laboratory-derived lines of Py17X in different laboratories. A recent study using AFLP markers classified the lines into two distinct genotypes (genotype 1 and 2) [24]; different lines within each genotype exhibited either a fast or slow growth phenotype. Judging by the low levels of polymorphism (maximum 3.6%) among the Py17X parasites in our study compared with hundreds of differences (279) in AFLP markers between parasites with genotype 1 and 2 [24], the Py17X lines in our study likely belong to a single genome type. These results are in agreement with the findings of Pattaradilokrat and colleagues [24] showing that P. yoelii 17X related lines, except P. yoelii 17A, were derived from a single parasite with almost identical genomes. In contrast, MS from PyNIG, PyBy265, and Py17X-related lines (as a group) were very different from each other, with the majority of the 591 MS (77.0% for PyBy265 and 84.8% for PyNIG) having unique alleles.
3.2. MS repeat types and frequencies
Similar to the P. falciparum genome, the majority of the repeats in the P. yoelii genome are AT-rich repeats. Among the polymorphic MS, 124 were A or T mononucleotide repeats (21.0%), 193 were AT dinucleotide repeats (32.7%), 43 were AAT repeats (7.3%), 26 were ATT repeats (4.4%), and 111 (18.8%) were a mixture of different repeats (Table 2). Different types of repeats are distributed among the 14 chromosomes, although some chromosomes might have slightly higher proportions of some particular repeat types.
Table 2.
Repeat types and frequencies of microsatellites identified from the Plasmodium yoelii chromosomes
| Chra | Markers | T | A | AT | AAT | ATT | CAT | ATG | CATA | TATG | ATTT | AAAT | Mixedb | Othersc |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 34 | 4 | 5 | 9 | 2 | 3 | 0 | 2 | 1 | 1 | 0 | 0 | 6 | 1 |
| 2 | 14 | 1 | 2 | 2 | 1 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 4 | 1 |
| 3 | 21 | 7 | 2 | 5 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 4 | 0 |
| 4 | 27 | 6 | 2 | 7 | 4 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 3 |
| 5 | 35 | 9 | 5 | 7 | 1 | 1 | 0 | 0 | 2 | 2 | 1 | 0 | 6 | 1 |
| 6 | 30 | 4 | 2 | 8 | 3 | 0 | 0 | 0 | 1 | 2 | 1 | 0 | 9 | 0 |
| 7 | 25 | 1 | 1 | 9 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 5 | 2 |
| 8 | 43 | 2 | 6 | 11 | 2 | 3 | 3 | 0 | 0 | 0 | 2 | 2 | 9 | 3 |
| 9 | 51 | 8 | 6 | 17 | 3 | 4 | 1 | 0 | 1 | 2 | 0 | 1 | 5 | 3 |
| 10 | 41 | 4 | 5 | 15 | 1 | 2 | 1 | 0 | 0 | 1 | 1 | 1 | 9 | 1 |
| 11 | 56 | 8 | 5 | 22 | 4 | 3 | 0 | 0 | 0 | 1 | 3 | 0 | 7 | 3 |
| 12 | 49 | 6 | 5 | 12 | 4 | 1 | 1 | 0 | 4 | 2 | 0 | 0 | 13 | 1 |
| 13 | 71 | 5 | 2 | 27 | 7 | 3 | 2 | 1 | 2 | 2 | 0 | 1 | 15 | 4 |
| 14 | 58 | 5 | 2 | 24 | 3 | 3 | 2 | 0 | 1 | 0 | 0 | 1 | 13 | 4 |
| UA | 36 | 2 | 2 | 18 | 7 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 2 | 2 |
| Total | 591 | 72 | 52 | 193 | 43 | 26 | 11 | 6 | 16 | 15 | 10 | 7 | 111 | 29 |
Chr, chromosome; Markers, numbers of markers on each chromosome; T, A, and AT, units of simple sequence repeats.
Mixed, microsatellites that have more than one repeat type.
Others, rare repeat units not listed.
3.3. Assignment of MS markers to chromosomes
The MS markers were assigned to the rodent malaria composite chromosomal groups described by Kooij et al. [15]. Among the 591 polymorphic MS markers, 555 were successfully assigned to one of the 14 chromosomes (Fig. 1 and Supplemental Table 1). Chromosome 13 had the largest number, with 71 MS markers, while chromosome 2 had only 14 markers. The MS markers appeared to be distributed randomly across the 14 chromosomes, although there were some regions with no MS markers and regions having clusters of MS. The regions without MS markers were mostly located toward the chromosome ends, with the largest segment having no MS markers located at one end of chromosome 10 (Fig. 1). Because our searches for MS were conducted randomly, it is difficult to explain the absence of MS markers in some chromosomal regions. One possibility for the absence of MS markers at a specific chromosome region could be the lack of P. yoelii sequences in the region, because the composite maps consist of sequences from P. c. chabaudi, P. yoelii, and P. berghei; and we only searched P. yoelii sequences for MS. To address this possibility, we compared the numbers of total P. yoelii nucleotides and MS identified in segments of similar sizes (400 kb) on chromosomes 10 and 13. The first 400-kb segment of chromosome 10 has one MS marker, and the next 400-kb sequence (400 kb to 800 kb) has 13 MS markers; however, the first 400 kb has 364,186 bp non-overlapping P. yoelii sequences, whereas the second 400 kb has 308,959 non-overlapping P. yoelii sequences. Similar results of lack of correlation between the numbers of P. yoelii nucleotides and MS identified were obtained from other segments on chromosomes 10 and 13 (data not shown). Clearly, the differences in the total P. yoelii sequences could not explain the absence of MS at the first 400 kb sequence of chromosome 10.
Fig. 1.

Distribution of Plasmodium yoelii polymorphic microsatellite markers on the rodent malaria composite chromosome maps. The markers were assigned to composite chromosome maps of rodent malaria parasites described by Kooij et al. [15]. The chromosomes are as marked from 1 to 14; the vertical ticks on top of the chromosome lines indicate the position of microsatellites.
3.4. Marker distribution among chromosomes
The numbers of MS markers on each chromosome generally correlated with the chromosome sizes. A plot of total chromosome length (bp) against the number of MS markers on each chromosome showed that the numbers of MS on the chromosomes generally correlated with the chromosome sizes (R2 = 0.86), although the numbers of MS on some chromosomes deviated from those expected based on chromosome sizes (Fig. 2). Chromosome 1 had significantly (P<0.001) more MS markers than the expected numbers of MS based on its chromosome size (34 observed vs. 17 expected), while chromosome 14 had significant fewer (P=0.05) MS than expected. The deviations of MS numbers on chromosomes 2, 5, 8, 10 and 11 were not statistically significant. Because the SSRs were identified from the whole genome sequences before assigning them to chromosomes, it was unlikely that the observed number of MS markers on chromosome 1 was due to bias during database search or marker selection. One possibility for the observation of biased MS distribution was the differences in GC content among the chromosomes. Because the majority of the MS were AT-rich repeats, higher GC content might lead to a drop in the AT-related repeats. We therefore investigated the relationship of GC content and the numbers of SSRs detected. There appeared to be no correlation between the numbers of MS identified and the GC contents of each chromosome. All of the chromosomes (nucleotides from contigs assigned to each chromosome only) had an AT content of between 77% and 78%, with chromosome 2 having the lowest AT content (77.2%) and chromosome 8 having the highest AT content (78.1%). Chromosome 1, which had twice the expected number of MS, had an AT content of 77.6%, approximately the genome average of 77.5%. The small variation in AT content among the chromosomes could not explain the observed variations in MS numbers in some chromosomes. Clearly, the larger chromosomes had more DNA sequences and therefore more MS. The reason for the deviation from the expected numbers of MS seen in some chromosomes is still unknown.
Fig. 2.
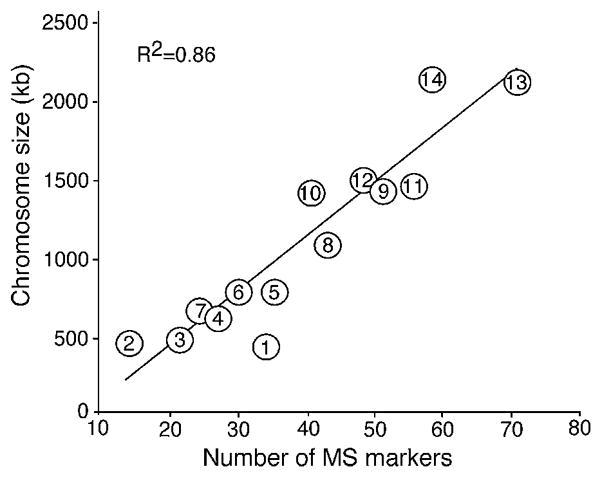
Relationship of the number of microsatellite (MS) on each chromosome and the chromosome length. Total numbers of nucleotide numbers (base pairs) from all the contigs assigned to a chromosome were plotted against the numbers of MS markers identified. The numbers inside each circle indicate each of the 14 chromosomes. R2 is the correlation coefficiency for the regression line.
3.5. Amplification of P. berghei sequences with primers having multiple substitutions
Interestingly, approximately 47% of the primer sets also produced PCR products from P. berghei DNA, with the majority (99.0%) of the P. berghei alleles different in size and/or sequences from those of the P. yoelii lines. The primers that did not generate any PCR products for P. berghei were likely due to nucleotide substitutions within the primer sequences. To investigate this possibility, we blasted the primer sequences of the 34 MS markers on chromosome 1 against the P. berghei genome sequence [25] (www.PlasmoDB.org). Primer sequences from 26 of the 34 MS markers had matches in the P. berghei genome, including 23 that had sequences matching both primers (Supplemental Table 2). Visual inspection of the P. berghei sequences flanked by the primers and the estimated sizes of PCR products suggested that the primers amplified the correct homologous sequences from the P. berghei genome (Supplemental Table 2). Surprisingly, only one MS marker (Py60) had perfect matches in both primer sequences, and many of the primers with one or more nucleotide substitutions could still produce PCR products from P. berghei genomic DNA. Despite mismatches in primer sequences, 19 primer pairs produced products from the P. berghei DNA, some of which had up to five substitutions in one of the primers (Supplemental Table 2). Additionally, five of the primer pairs that produced products from P. berghei DNA (Py277, Py349, Py619, Py1346, and Py2056) had substitutions in both primers. Among the 14 primer pairs that did not produce any products, 9 had matching primers identified from the P. berghei genome; all of the 9 primer pairs had substitutions in both primers, suggesting that substitutions in the primers prevented the amplification of P. berghei DNA (Supplemental Table 2). For the eight MS that did not have primer matches in the P. berghei genome, three produced a PCR product (Py1618-2, Py821 and Py451). It was likely that the P. berghei sequences were so different (or absent) for these eight MS that BLAST search could not identify them. These data showed that some mismatches in primer sequences could still produce good PCR products in MS typing and that the rodent malaria parasites were highly diverse, even among independent P. yoelii isolates.
A large number of AFLP markers has been developed for genotyping the rodent malaria parasite P. c. chabaudi, and more than 800 AFLP bands have been identified between two P. c. chabaudi clones, AS and AJ. Of the 819 markers, 403 fragments were specific to AS and 416 to AJ [5]. A genetic linkage map with 614 AFLP markers was subsequently developed for P. c. chabaudi using 28 independent recombinant progeny from a genetic cross between AS and AJ clones with a mean map unit size of 13.7 kb/cM [20]. The AFLP method was also applied to characterize in a quantitative manner the proportion of parasites in a mixture based on the intensities of the AFLP bands [6]. The quantitative AFLP and other methods eventually led to the development of a mapping strategy called linkage group selection (LGS) that greatly simplified genetic mapping in rodent malaria parasites [12]. A similar strategy can be applied to study P. yoelii traits. Unfortunately, genetic markers and genetic crosses for P. yoelii are limited compared with those available for P. c. chabaudi. It would be interesting and valuable to investigate the possibility of using the MS markers to accurately estimate the proportion of alleles in a mixture, because not all MS can accurately predict allelic proportions of mixed DNA samples [26]. MS markers that can accurately estimate the proportion of DNA in a mixture could be applied to LGS mapping strategy to map selectable traits in P. yoelii.
This study identifies and characterizes hundreds of polymorphic MS markers that will be useful for genetic mapping of P. yoelii traits. Typing progeny from P. yoelii crosses using these MS markers will lead to a high-resolution genetic map that may guide the assembly of the parasite chromosomes and genome. These MS markers may also be used in LGS if a reliable quantitative typing method can be developed.
Supplementary Material
Acknowledgments
We thank Professors Weiqing Pan and Fusheng Huang for making the parasite lines available to us, Dr. S. Pattaradilokrat for constructive comments, and NIAID intramural editor Brenda Rae Marshall for assistance. This study was support by the National Basic Research Program of China, 973 Program 2007CB513103; by a graduate student fund from Xiamen University, China; and by the Intramural Research Program of the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Because T.F.M. and X-z.S. are US government employees and this is a government work, the work is in the public domain in the United States. Notwithstanding any other agreements, the NIH reserves the right to provide the work to PubMed Central for display and use by the public, and PubMedCentral may tag or modify the work consistent with its customary practices. You can establish rights outside of the U.S. subject to a government use license.
Abbreviations
- AFLP
amplified fragment-length polymorphism
- LGS
linkage group selection
- MS
microsatellites
- PCR
polymerase chain reaction
- SNP
single nucleotide polymorphism
- SSR
simple sequence repeat
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Culleton R, Martinelli A, Hunt P, Carter R. Linkage group selection: rapid gene discovery in malaria parasites. Genome Res. 2005;15(1):92–7. doi: 10.1101/gr.2866205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter R, Voller A. The distribution of enzyme variation in populations of Plasmodium falciparum in Africa. Trans R Soc Trop Med Hyg. 1975;69(4):371–6. doi: 10.1016/0035-9203(75)90191-1. [DOI] [PubMed] [Google Scholar]
- 3.Walliker D. Genetic factors in malaria parasites and their effect on host-parasite relationships. In: Taylor AE, Muller R, editors. Genetic aspects of host-parasite relationships. Oxford: Blackwell; 1976. pp. 25–44. [Google Scholar]
- 4.Wellems TE, Panton LJ, Gluzman IY, do Rosario VE, Gwadz RW, Walker-Jonah A, et al. Chloroquine resistance not linked to mdr-like genes in a Plasmodium falciparum cross. Nature. 1990;345(6272):253–5. doi: 10.1038/345253a0. [DOI] [PubMed] [Google Scholar]
- 5.Grech K, Martinelli A, Pathirana S, Walliker D, Hunt P, Carter R. Numerous, robust genetic markers for Plasmodium chabaudi by the method of amplified fragment length polymorphism. Mol Biochem Parasitol. 2002;123(2):95–104. doi: 10.1016/s0166-6851(02)00142-1. [DOI] [PubMed] [Google Scholar]
- 6.Martinelli A, Hunt P, Cheesman SJ, Carter R. Amplified fragment length polymorphism measures proportions of malaria parasites carrying specific alleles in complex genetic mixtures. Mol Biochem Parasitol. 2004;136(2):117–22. doi: 10.1016/j.molbiopara.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 7.Kidgell C, Volkman SK, Daily J, Borevitz JO, Plouffe D, Zhou Y, et al. A systematic map of genetic variation in Plasmodium falciparum. PLoS Pathog. 2006;2(6):e57. doi: 10.1371/journal.ppat.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Zhang Y, Sullivan M, Hong L, Huang L, Lu F, et al. Typing Plasmodium yoelii microsatellites using a simple and affordable fluorescent labeling method. Mol Biochem Parasitol. 2007;155(2):94–102. doi: 10.1016/j.molbiopara.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeffares DC, Pain A, Berry A, Cox AV, Stalker J, Ingle CE, et al. Genome variation and evolution of the malaria parasite Plasmodium falciparum. Nat Genet. 2007;39(1):120–5. doi: 10.1038/ng1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mu J, Awadalla P, Duan J, McGee KM, Keebler J, Seydel K, et al. Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat Genet. 2007;39(1):126–30. doi: 10.1038/ng1924. [DOI] [PubMed] [Google Scholar]
- 11.Volkman SK, Sabeti PC, DeCaprio D, Neafsey DE, Schaffner SF, Milner DA, Jr, et al. A genome-wide map of diversity in Plasmodium falciparum. Nat Genet. 2007;39(1):113–9. doi: 10.1038/ng1930. [DOI] [PubMed] [Google Scholar]
- 12.Martinelli A, Cheesman S, Hunt P, Culleton R, Raza A, Mackinnon M, et al. A genetic approach to the de novo identification of targets of strain-specific immunity in malaria parasites. Proc Natl Acad Sci U S A. 2005;102(3):814–9. doi: 10.1073/pnas.0405097102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pattaradilokrat S, Cheesman SJ, Carter R. Linkage group selection: towards identifying genes controlling strain specific protective immunity in malaria. PLoS ONE. 2007;2(9):e857. doi: 10.1371/journal.pone.0000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, et al. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature. 2002;419(6906):512–9. doi: 10.1038/nature01099. [DOI] [PubMed] [Google Scholar]
- 15.Kooij TW, Carlton JM, Bidwell SL, Hall N, Ramesar J, Janse CJ, et al. A Plasmodium whole-genome synteny map: indels and synteny breakpoints as foci for species-specific genes. PLoS Pathog. 2005;1(4):e44. doi: 10.1371/journal.ppat.0010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landau I, Killick-Kendrick R. Rodent plasmodia of the Republique Centrafricaine: the sporogony and tissue stages of Plasmodium chabaudi and P. berghei yoelii. Trans R Soc Trop Med Hyg. 1966;60(5):633–49. doi: 10.1016/0035-9203(66)90010-1. [DOI] [PubMed] [Google Scholar]
- 17.Hoff J. Methods of blood collection in the mouse. Lab Animal. 2000;29(10):47–53. [Google Scholar]
- 18.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su X-z, Ferdig MT, Huang Y, Huynh CQ, Liu A, You J, et al. A genetic map and recombination parameters of the human malaria parasite Plasmodium falciparum. Science. 1999;286(5443):1351–3. doi: 10.1126/science.286.5443.1351. [DOI] [PubMed] [Google Scholar]
- 20.Martinelli A, Hunt P, Fawcett R, Cravo PV, Walliker D, Carter R. An AFLP-based genetic linkage map of Plasmodium chabaudi chabaudi. Malar J. 2005;4(1):11. doi: 10.1186/1475-2875-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su X-z, Wellems TE. Toward a high-resolution Plasmodium falciparum linkage map: polymorphic markers from hundreds of simple sequence repeats. Genomics. 1996;33(3):430–44. doi: 10.1006/geno.1996.0218. [DOI] [PubMed] [Google Scholar]
- 22.Feng X, Carlton JM, Joy DA, Mu J, Furuya T, Suh BB, et al. Single-nucleotide polymorphisms and genome diversity in Plasmodium vivax. Proc Natl Acad Sci U S A. 2003;100(14):8502–7. doi: 10.1073/pnas.1232502100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, et al. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455(7214):757–63. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pattaradilokrat S, Cheesman SJ, Carter R. Congenicity and genetic polymorphism in cloned lines derived from a single isolate of a rodent malaria parasite. Mol Biochem Parasitol. 2008;157(2):244–7. doi: 10.1016/j.molbiopara.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Hall N, Karras M, Raine JD, Carlton JM, Kooij TW, Berriman M, et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science. 2005;307(5706):82–6. doi: 10.1126/science.1103717. [DOI] [PubMed] [Google Scholar]
- 26.Liu S, Mu J, Jiang H, Su X-z. Effects of Plasmodium falciparum mixed infections on in vitro antimalarial drug tests and genotyping. Am J Trop Med Hyg. 2008;79(2):178–84. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.