

SUMMARY
Induced pluripotent stem cells (iPSCs) derived from somatic cells of patients represent a powerful tool for biomedical research and may provide a source for replacement therapies. However, the use of viruses encoding the reprogramming factors represents a major limitation of the current technology since even low vector expression may alter the differentiation potential of the iPSCs or induce malignant transformation. Here, we show that fibroblasts from five patients with idiopathic Parkinson’s disease can be efficiently reprogrammed and subsequently differentiated into dopaminergic neurons. Moreover, we derived hiPSCs free of reprogramming factors using Cre-recombinase excisable viruses. Factor-free hiPSCs maintain a pluripotent state and show a global gene expression profile, more closely related to hESCs than to hiPSCs carrying the transgenes. Our results indicate that residual transgene expression in virus-carrying hiPSCs can affect their molecular characteristics and that factor-free hiPSCs therefore represent a more suitable source of cells for modeling of human disease.
INTRODUCTION
Reprogramming of mouse and human somatic cells to a pluripotent state has been achieved by viral transduction of four transcription factors, OCT4, KLF4, SOX2, and c-MYC (Aasen et al., 2008; Dimos et al., 2008; Hockemeyer et al., 2008; Lowry et al., 2008; Maherali et al., 2008; Nakagawa et al., 2008; Okita et al., 2007; Park et al., 2008a, 2008b; Takahashi et al., 2007; Takahashi and Yamanaka, 2006; Wernig et al., 2007). Generation of such human induced pluripotent stem cells (hiPSCs) with embryonic stem cell (ESC)-like properties opened up the intriguing possibility of generating patient-specific cells (Dimos et al., 2008; Ebert et al., 2009; Park et al., 2008a). hiPSCs, characterized by their ability to self-renew and to differentiate into any cell type of the body, are predicted to become a powerful tool for biomedical research as well as a source for cell-replacement therapies. Although the realization of ESC/induced pluripotent stem cell (iPSC)-based therapies is still at an early stage of development, the possibility of modeling human disease in vitro could make patient-specific hiPSCs immediately valuable. This is particularly relevant for diseases of the central nervous system (CNS) such as Parkinson’s disease (PD), where primary neuronal tissue is not available.
PD is the second most common chronic progressive neurodegenerative disorder and is characterized primarily by major loss of nigrostriatal dopaminergic neurons. The discovery of genes linked to rare familial forms of PD has provided vital clues in understanding the cellular and molecular pathogenesis of the disease (Gasser, 2007; Schulz, 2008). However, the majority of cases are sporadic, not linked to a known genetic mutation, and likely the result of complex interactions between genetic and environmental factors (de Lau and Breteler, 2006). One of the major reasons for the lack of understanding of the underlying pathophysiology of PD is the paucity of reliable experimental models that recapitulate all features of the human disease. The derivation of PD patient-specific hiPSCs and subsequent differentiation into dopaminergic neurons would provide patient-specific in vitro models that are otherwise experimentally not accessible.
A major limitation of current reprogramming strategies for clinical application is the presence of viral vectors used to transduce the reprogramming factors. It has been demonstrated in the mouse system that iPSC-derived chimeras frequently develop tumors resulting from reactivation of the oncogene c-Myc (Markoulaki et al., 2009; Okita et al., 2007). Although reprogramming has been achieved in the absence of c-MYC, though with longer latency and substantially reduced efficiency (Nakagawa et al., 2008; Wernig et al., 2008), the remaining integrated reprogramming factors could also cause tumor formation (Hochedlinger et al., 2005). Furthermore, it has been proposed that residual transgene expression may explain some of the observed differences between ESCs and iPSCs, such as the altered differentiation into functional cell types (Yu et al., 2007). More recently, reprogramming of mouse somatic cells has been achieved without stable integration through the use of transient transfection or adenoviral infection to deliver the reprogramming factors (Okita et al., 2008; Stadtfeld et al., 2008). Because of the substantially lower efficiency of these methods, it remains unclear whether similar approaches would be successful in the human system.
Here, we show that fibroblasts from five patients with sporadic PD could be efficiently reprogrammed and demonstrate that these patient-derived hiPSCs could be subsequently differentiated in vitro into dopaminergic neurons. Moreover, using doxycycline (DOX)-inducible lentiviral vectors that could be excised with Cre-recombinase, we generated hiPSCs that are free of the reprogramming factors. These factor-free hiPSCs maintained all of the characteristics of a pluripotent ESC-like state after removal of the transgenes. Importantly, genome-wide transcription analysis revealed that residual transgene expression from the partially silenced viral vectors did in fact perturb overall gene expression in hiPSCs such that the factor-free hiPSCs more closely resembled embryo-derived human embryonic stem cells (hESCs) than the parental virus-carrying hiPSCs.
RESULTS
Reprogramming of Fibroblasts from PD Patients by DOX-Inducible Lentiviral Vectors
Dermal fibroblasts from five patients with idiopathic PD (age of biopsy between 53 and 85 years) and from two unaffected subjects were obtained from the Coriell Institute for Medical Research (see Table 1). To induce reprogramming, we infected 1 × 106 fibroblasts with a constitutively active lentivirus expressing the reverse tetracycline transactivator (FUW-M2rtTA) together with DOX-inducible lentiviruses transducing either four (OCT4, SOX2, c-MYC, KLF4) or three (OCT4, SOX2, KLF4) reprogramming factors. We will subsequently refer to hiPSC lines derived by transduction of four factors as hiPSC4F and those obtained by three factors as hiPSC3F. Colonies with well-defined hESC-like morphology were selected and manually picked 3 to 5 weeks after DOX-induced transgene expression. All fibroblasts obtained from PD patients and non-PD patients gave rise to stable hiPSCs that were maintained in the absence of DOX for more than 30 passages. At least one cell line from each donor fibroblast line was analyzed in detail (Table 1). All of these hiPSCs uniformly expressed the pluripotency markers Tra-1-60, SSEA4, OCT4, SOX2, and NANOG as determined by immunocytochemistry (Figure 1A). In addition, all hiPSC lines analyzed by quantitative RT-PCR showed reactivation of the endogenous pluripotency-related genes OCT4, SOX2, and NANOG with similar levels of expression as seen in hESCs (Figure 1B). As expected for hiPSCs, the OCT4 promoter region of PD patient-derived hiPSCs was found to be hypomethylated in contrast to its hypermethylated state in the parental fibroblasts (Figure 1C). In order to test for pluripotency, hiPSCs isolated from each donor fibroblast line were injected into SCID mice. All hiPSCs formed teratomas comprised of tissues developing from all embryonic germ layers including cartilage, bone, smooth muscle (mesoderm), neural rosettes, pigmented neural epithelium (ectoderm), and intestinal epithelium with goblet- and Paneth-like cells (endoderm) (Figure 2A).
Table 1.
Summary of hiPSCs Derived from Primary Fibroblasts
| Parental Cell Line | Donora | Age at Onset of PD | Age at Biopsy | Reprogramming Factors | Number of iPSC Clones Characterized | iPSC Clone ID |
|---|---|---|---|---|---|---|
| AG20443 (PDA) | Parkinson’s disease patient, idopathic, male | NA | 71 | FUW-tetO 3 factors (OCT4, SOX2, KLF4) | 2 | PDA3F -1, -5 |
| AG20442 (PDB) | Parkinson’s disease patient, idopathic, male | 51 | 53 | FUW-tetO 3 factors (OCT4, SOX2, KLF4) | 5b | PDB3F-1, -5, -8, -9, PDB3F-d12 |
| AG20442 (PDB) | Parkinson’s disease patient, idopathic, male | 51 | 53 | FUW-tetO 4 factors (OCT4, SOX2, KLF4, c-MYC) | 5c | PDB4F-1, -2, -3, -4, -5 |
| AG20446 (PDC) | Parkinson’s disease patient, idopathic, male | 50 | 57 | FUW-tetO 3 factors (OCT4, SOX2, KLF4) | 1 | PDC3F-1 |
| AG20445 (PDD) | Parkinson’s disease patient, idopathic, male | 44 | 60 | FUW-tetO 3 factors (OCT4, SOX2, KLF4) | 3 | PDD3F-1, -4, -7 |
| AG20445 (PDD) | Parkinson’s disease patient, idopathic, male | 44 | 60 | FUW-tetO 4 factors (OCT4, SOX2, KLF4, c-MYC) | 5 | PDD4F-1, -4, -5, -8, -9 |
| AG08395 (PDE) | Parkinson’s disease patient, idopathic, female | 83 | 85 | FUW-tetO 3 factors (OCT4, SOX2, KLF4) | 2 | PDE3F-3, -4 |
| GM01786 | Dyskeratosis congenital carrier, female | - | 30 | FUW-tetO 3 factors (OCT4, SOX2, KLF4) | 2 | M3F-1, -2 |
| GM01660 | Lesh-Nyhan carrier, female | - | 11 | FUW-tetO 3 factors (OCT4, SOX2, KLF4) | 2d | A1, A6 |
| MRC-5 | male, embryonic fibroblasts | FUW-tetO 4 factors (OCT4, SOX2, KLF4), c-MYC | 2d | D1, D4 |
NA, data not available.
Additional information about these fibroblast cell lines can be obtained from the Coriell Institute.
PDB3F-12d was isolated in experiments to determine the temporal requirements of transgene expression. PDB3F-12d was isolated from cultures exposed for 12 days to doxycycline.
These cells were derived in experiments to determine the temporal requirements of transgene expression. PDB4F-1 to -3 were isolated from cultures exposed for 8 days to doxycyline, whereas PDB4F-4 and -5 were exposed to doxycycline for 10 and 12 days, respectively.
These hiPSCs cells have been previously characterized in Hockemeyer et al., 2008.
Figure 1. Characterization of DOX-Inducible hiPSCs Derived from Fibroblasts from PD Patients.

(A) Phase contrast picture and immunofluorescence staining of hiPSC lines M3F-1 (non-PD hiPSCs), PDA3F-1, PDB3F-5, PDC3F-1, PDD3F-1, and PDE3F-3 for pluripotency markers SSEA4, Tra-1-60, OCT4, SOX2, and NANOG.
(B) Quantitative RT-PCR for the reactivation of the endogenous pluripotency-related genes NANOG, OCT4, and SOX2 in indicated hiPSC lines, hESCs, and primary fibroblasts. Relative expression levels were normalized to expression of these genes in fibroblasts.
(C) Methylation analysis of the OCT4 promoter region. Light gray squares indicate unmethylated and black squares methylated CpGs in the OCT4 promoter of hiPSCs and parental primary fibroblasts cells.
Figure 2. PD Patient-Derived hiPSCs Carry Low Copy Numbers of Viral Integrations.
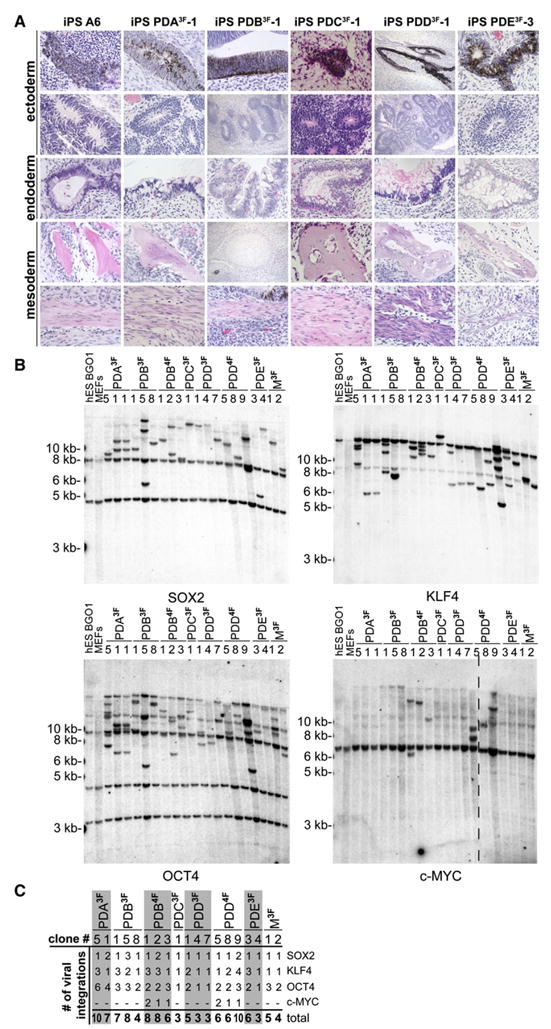
(A) Hematoxylin and eosin staining of teratoma sections generated from hiPSC lines A6 (non-PD hiPSCs), PDA3F-1, PDB3F-1, PDC3F-1, PDD3F-1, and PDE3F-3 showing: top row panels, pigmented neural epithelium; second row panels, neural rosettes; third row panels, intestinal epithelium; fourth row panels, bone/cartilage; and bottom row panels, smooth muscle.
(B) Southern blot analysis of hESC line BG01, mouse embryonic fibroblast (MEF) feeder cells, and the indicated PD patient-derived hiPSCs (and non-PD hiPSC line M3F-1) for proviral integrations of XbaI-digested genomic DNA with 32P-labeled DNA probes against OCT4, KLF4, SOX2, and c-MYC.
(C) Table summarizing the approximate number of proviral integrations for the four reprogramming factors in hiPSCs based on Southern blot analysis shown in (B).
Cytogenetic analysis of PD-specific hiPSC lines revealed a normal karyotype in 11 out of 12 lines (Figure S1 available online). Only one out of three clones derived from the fibroblast line PDD that had been transduced with four factors (iPS PDD4F-5) showed an unbalanced translocation between the long arm of chromosome 18 and the long arm of chromosome 22, resulting in a derivative chromosome 18 and a single copy of chromosome 22. Two independent hiPSCs derived from a non-PD patient fibroblast line (iPS M3F-1 and iPS M3F-2) showed a balanced translocation between the short and long arms of chromosomes 4 and 7, suggesting that the 4;7 translocation was already present in the donor fibroblasts (Figure S1). DNA fingerprinting of the PD patient-derived hiPSCs and the parental fibroblasts was performed to confirm the origin of the hiPSCs and to rule out crosscontaminations with existing pluripotent cell lines (data not shown). Southern blot analysis probing for lentiviral integrations showed distinct patterns for each of the hiPSC lines, confirming that each line analyzed was derived from independently infected fibroblasts carrying a total of three to ten proviral copies (Figures 2B and 2C).
In order to further characterize the usefulness of this system, we determined the reprogramming efficiencies for one fibroblast line (PDB) in detail. Reprogramming efficiencies were calculated after 20 days on the basis of immunocytochemistry for the pluripotency markers Tra-1-60 and NANOG. hiPSCs arose with an efficiency of approximately 0.005% after transduction with three factors and approximately 0.01% after transduction with four factors. This is comparable to previously reported efficiencies with either Moloney-based retroviral vectors or constitutively active lentiviral vectors (Nakagawa et al., 2008; Takahashi et al., 2007; Yu et al., 2007). Immunocytochemistry for NANOG and Tra-1-60 at different time points after DOX addition revealed that small pluripotent colonies could be detected in four-factor-transduced fibroblasts as early as 8 days after transgene induction (Figure S2A). We also determined the temporal requirement for the expression of the reprogramming factors by varying the time of DOX-induced transgene expression in fibroblasts transduced with either three or four reprogramming factors. After 24 days, we were able to isolate hiPSC colonies from four-factor-transduced fibroblasts exposed to DOX for only 8 days (PDB4F-1, -2, -3), whereas hiPSCs from three-factor-transduced cells could be isolated only after exposure to DOX for at least 12 days (PDB3F-d12). Although the reprogramming factors were only expressed for a limited period, all of the picked cells gave rise to fully reprogrammed hiPSCs that stained for pluripotency markers (Figure S2B), reactivated the endogenous OCT4, NANOG, and SOX2 genes (Figure S2C), and formed teratomas comprised of cells derived from the three developmental germ layers (Figure S2D). Our results suggest that reprogramming by three factors is less efficient and takes longer than reprogramming by four factors, in agreement with previous observations (Nakagawa et al., 2008; Wernig et al., 2008). However, we find that derivation of hiPSCs with three factors is more practical, since the infected fibroblast cultures are not overgrown by granulated, fast growing non-hiPSC colonies, as has been described previously for cultures infected with four factors (Nakagawa et al., 2008; Takahashi et al., 2007).
The results described so far show that DOX-inducible delivery of the reprogramming factors can efficiently generate hiPSCs from skin biopsies obtained from PD patients in the absence of c-MYC with similar kinetics and efficiencies, as previously reported with other approaches. Importantly, eight of 13 three-factor hiPSCs carried a total of only three to five proviral integrations (Figures 2B and 2C), which is significantly less than observed in previous studies (Wernig et al., 2007).
Generation of Dopaminergic Neurons from PD Patient-Derived hiPSCs
Several protocols for the directed differentiation of hESCs into dopaminergic neurons have been established (Elkabetz et al., 2008; Kim et al., 2006; Perrier et al., 2004; Roy et al., 2006; Sonntag et al., 2007). To test whether PD-specific hiPSCs were able to generate dopaminergic neurons, we induced neural differentiation by embryoid body (EB) formation in hiPSC lines derived from each PD fibroblast line, as well as from a non-PD fibroblast line (iPS M3F-1) and embryo-derived hESCs. Small clusters of hESCs or hiPSCs were cultured in EB medium on nonadherent culture plates for 8 days to form simple EBs. Subsequently, neural precursor cells were selected in ITS medium containing fibronectin, dissociated, and expanded in the presence of the growth factors FGF2, FGF8, and sonic hedgehog (SHH). Terminal differentiation was induced by growth factor withdrawal for 8 days. At the end of this differentiation protocol, cultures were stained for tyrosine hydroxylase (TH) and neuron-specific class III-β-tubulin (TUJ1) to label dopaminergic neurons. TH-and TUJ1-positive neurons were generated from non-PD hiPSCs as well as from all PD patient-derived hiPSC lines regardless of whether the cells had been reprogrammed with three or four factors (Figure 3A). No obvious differences in the ability to generate dopaminergic neurons were observed between PD-and non-PD-derived hiPSCs or hESCs. In order to confirm these results, we employed a different protocol based on the combination of MS5 stromal cell coculture with the BMP antagonist Noggin to induce neural differentiation (Perrier et al., 2004; Sonntag et al., 2007). All PD-derived hiPSCs tested with this protocol generated TH- and TUJ1-positive dopaminergic neurons with approximately the same efficiency as did non-PD hiPSCs and hESCs (Figures 3B and 3C).
Figure 3. Generation of Dopaminergic Neurons from PD Patient-Specific hiPSCs.

(A) Immunofluorescence staining of neuronal cultures derived from hESC line BG01, non-PD hiPSCs M3F-1, and the indicated PD patient-specific hiPSCs for neuron-specific class III β-tubulin (TUJ1; green) and the dopaminergic neuron-specific marker tyrosine hydroxylase (TH; red). Neuronal cultures were derived with an embryoid body (EB)-based differentiation protocol.
(B) Immunofluorescence staining of neuronal cultures derived from hESC line H9, non-PD hiPSCs A6, and the indicated PD patient-specific hiPSCs for neuron specific class III β-tubulin (TUJ1; red) and the dopaminergic neuron-specific marker tyrosine hydroxylase (TH; green). Neuronal cultures were derived with a stromal cell coculture-based protocol.
(C) Quantification of TUJ1-positive neurons (left graph) and TH-positive dopaminergic neurons (right graph) after neuronal differentiation of hESCs (BG02, H9), non-PD hiPSCs (iPS A6, M3F-2) and indicated PD patient-specific hiPSCs using a stromal cell coculture-based protocol (as shown in [B]). Graphs indicate the percentage of cells that stain positive relative to nuclear Hoechst staining. Error bars indicate the SEM generated from triplicates of the same experiment.
Generation PD Patient-Derived hiPSCs Free of Viral Reprogramming Factors
In order to derive hiPSCs that were free of proviruses, we generated lentiviral vectors that could be excised after integration using Cre-recombinase. The human ubiquitin promoter of the FUGW-loxP lentivirus, which contains a loxP site in the 3′ long terminal repeat (LTR) (Hanna et al., 2007), was replaced with a DOX-inducible, minimal cytomegalovirus (CMV) promoter followed by the human cDNAs for OCT4, KLF4, or SOX2. Upon proviral replication, the loxP site in the 3′ LTR is duplicated into the 5′ LTR, resulting in an integrated transgene flanked by loxP sites in both LTRs (Figure 4A). One million fibroblasts (PDB) were transduced simultaneously with these three viruses, as well as a constitutively active lentivirus expressing the reverse tetracycline transactivator (FUW-M2rtTA). Twenty-four hiPSC lines (PDB2lox-1 to -24) were isolated 3 to 4 weeks after DOX addition with similar kinetics and efficiency as described above. Southern blot analysis for 12 cell lines showed that four PDB2lox lines (PDB2lox-5, PDB2lox-17, PDB2lox-21, PDB2lox-22) contained only five to seven integrations of the reprogramming factors (Figure S3). These PDB2lox cell lines were maintained in the absence of DOX for more than 20 passages and displayed all of the characteristics of hiPSCs, such as expression of pluripotency-related marker proteins Tra-1-60, SSEA4, OCT4, SOX2, and NANOG (Figure 4B) and the reactivation of the endogenous pluripotency-related genes OCT4, NANOG, and SOX2 (as shown below). Furthermore, all tested PDB2lox clones (PDB2lox-5, PDB2lox-17, PDB2lox-21, PDB2lox-22) demonstrated in vitro multilineage differentiation in EBs (Figure S4) and formed teratomas with contributions to all three embryonic germ layers after subcutaneous injection into SCID mice (Figure 4C).
Figure 4. Generation of PD Patient-Derived hiPSCs with loxP-Excisable Reprogramming Factors.

(A) Schematic drawing of the DOX-inducible lentiviral construct FUW-tetO-loxP and the genomic locus after proviral integration (2lox) and Cre-recombinase-mediated excision (1lox). The FUW-TetO-loxP vector contains a tetracycline response element (TRE) located 5′ of a minimal CMV promoter and a unique MfeI site used for diagnostic Southern blot digests. The reprogramming factors are flanked by EcoRI restriction sites. The 3′ LTR of this lentiviral vector contains a single loxP site, which is duplicated during proviral replication into the 5′ LTR. This duplication results in a transgene flanked by two loxP sites after genomic integration of the provirus (2lox). This allows the excision of the transgene in combination with the complete promoter sequences with Cre-recombinase (1lox). (WRE, woodchuck response element.)
(B) Phase contrast picture and immunofluorescence staining of hiPSC lines PDB2lox-17 and PDB2lox-21 for pluripotency markers SSEA4, Tra-1-60, OCT4, SOX2, and NANOG. PDB2lox-17 and PDB2lox-21 were derived by expression of the three reprogramming factors OCT4, SOX2, and KLF4 from the FUW-tetO-loxP virus shown in (A). In these cells, all three reprogramming factors are flanked by loxP sites at their genomic integration site.
(C) Hematoxylin and eosin staining of teratoma sections generated from PDB2lox-17 and PDB2lox-21 cells carrying excisable reprogramming factors.
We focused on two clones, with either five (PDB2lox-21) or seven (PDB2lox-17) total integrations of the reprogramming factors to test whether the excision of the loxP site-flanked lentiviral vectors would generate transgene-free cells. Two different strategies for Cre-mediated vector excision were used (Figure 5A): (1) HiPSCs were transiently transfected with an expression vector encoding Cre-recombinase and the puromycin resistance gene (pCre-PAC). After electroporation, cells were selected with puromycin for 48 hr to enrich for cells that transiently expressed Cre-recombinase and puromycin. (2) HiPSCs were cotransfected with plasmids for Cre-recombinase and EGFP and subsequently sorted for EGFP-positive and Cre-expressing cells 60 hr after transfection using fluorescence-activated cell sorting (FACS). Using these two methods, we isolated a total of 180 clones 10 to 14 days after electroporation (Figure 5A). Initial Southern blot analysis to screen for the excision of KLF4 (highest number of integrations) with an internal EcoRI digest showed that 48 clones were negative for KLF4 lentiviral integrations (data not shown). Subsequent Southern blot analysis for KLF4, OCT4, and SOX2 proviral integrations with an external XbaI restriction digest revealed that seven clones derived from PDB2lox-17 and nine clones derived from PDB2lox-21 had no integration of any of the reprogramming factors (Figure 5B, referred to as PDB1lox clones and summarized in Figure 5D). Excision of all reprogramming factors was confirmed by an additional Southern blot analysis with a different restriction digest (Figure S5). Furthermore, PCR of genomic DNA with primers specific for Cre-recombinase confirmed that none of the PDB1lox clones had stably integrated the electroporated plasmids (data not shown). Southern blot analysis for the integration of the reverse tetracycline transactivator M2rtTA showed one integration for line PDB2lox-17 and two integrations for line PDB2lox-21 (Figure S6). This means that the overall number of proviral integrations including the transactivator in line PDB2lox-21 is the same as the number of excised transgenes from PDB2lox-17, suggesting that the excision of all transgenes including the transactivator should be possible. Cytogenetic analysis demonstrated that 14 out of 14 analyzed clones showed a normal karyotype after Cre-mediated transgene excision (Figure 5C and data not shown).
Figure 5. Generation and Characterization of Reprogramming Factor-free hiPSCs.

(A) Schematic overview of Cre-mediated excision of the transgenes to generate reprogramming factor-free hiPSCs. IPS PDB2lox cells were derived with FUW-tetO-loxP lentiviral vectors transducing three reprogramming factors, OCT4, KLF4, and SOX2.
(B) Southern blot analysis for proviral integrations of parental fibroblasts (PDB), provirus-carrying PDB2lox clones (PDB2lox-17 and PDB2lox-21), and the indicated PDB1lox clones after Cre-recombinase-mediated excision of the transgenes. Puro indicates PDB1lox clones, which were isolated by puromycin selection; GFP indicates PDB1lox clones isolated by FACS sorting for EGFP (as shown in [A]). Genomic DNA was digested with XbaI and probed for proviral integrations using 32P-labeled DNA probes against OCT4, KLF4, and SOX2. PDB1lox clones indicated in blue were disregarded because of remaining transgene integrations based on the MfeI digest shown in Figure S5.
(C) Cytogenetic analysis of hiPSC lines PDB1lox-17Puro-5 and PDB1lox-21Puro-12 shows normal karyotype after Cre-mediated excision of the transgenes.
(D) Summary of the generation of factor-free hiPSCs.
All factor-free clones retained a stable hESC-like morphology upon prolonged culture for more than 15 passages and maintained all the characteristics of hiPSCs, such as expression of the hESC-related marker proteins Tra-1-60, SSEA4, OCT4, SOX2, and NANOG as shown by immunocytochemistry (Figure 6A), and the expression of the endogenous pluripotency-related genes OCT4, SOX2, and NANOG (Figure 6B) at levels comparable to hESCs and to the parental hiPSCs before excision of the transgenes. In order to demonstrate that the reprogramming factor-free PDB1lox clones maintain pluripotency after the excision of the reprogramming factors, we differentiated independent PDB1lox clones by in vitro EB formation or subcutaneous injection into SCID mice. All tested PDB1lox clones showed multilineage differentiation in vitro (Figure S4) and developed into teratomas with contributions to all three embryonic germ layers (Figure 6C). Furthermore, using the above-described EB-based protocol to induce neural differentiation, we were able to derive dopaminergic neurons from all PDB1lox-puro clones (Figure 6D and data not shown).
Figure 6. Characterization of Reprogramming Factor-free hiPSCs.

(A) Phase contrast picture and immunofluorescence staining of reprogramming factor-free hiPSC lines PDB1lox-17Puro-5 and PDB1lox-21Puro-12 for pluripotency markers SSEA4, Tra-1-60, OCT4, SOX2, and NANOG.
(B) Quantitative RT-PCR for the reactivation of the endogenous pluripotency-related genes NANOG, OCT4, and SOX2 in hESCs, fibroblasts (PDB), provirus-carrying PDB2lox clones (PDB2lox-17 and PDB2lox-21), and indicated PDB1lox clones after Cre-recombinase-mediated excision of the transgenes. Relative expression levels were normalized to the expression of these genes in fibroblasts.
(C) Hematoxylin and eosin staining of teratoma sections generated from factor-free PDB1lox-17puro-5 and PDB1lox-21puro-26 cells.
(D) Immunofluorescence staining of neuronal cultures derived from factor-free PDB1lox-17puro-31 and PDB1lox-21puro-13 hiPSCs for neuron-specific class III β-tubulin (TUJ1; green) and the dopaminergic neuron-specific marker tyrosine hydroxylase (TH; red). Neuronal cultures were derived with an embryoid body (EB)-based differentiation protocol.
(E) Quantitative RT-PCR for residual transgene expression of OCT4, KLF4, and SOX2 in hESCs (BG01), primary fibroblasts (PDB), primary infected fibroblasts (PDD3F ± DOX), hiPSCs (M3F3-1), PD-derived hiPSCs (PDA3F-1, PDB3F-5, PDC3F-1, PDD3F-1, PDE3F-3), provirus-carrying PDB2lox clones (PDB2lox-17 and PDB2lox-21), and the reprogramming factor-free PDB1lox clones (PDB1lox-17Puro-5, PDB1lox-17Puro-31, PDB1lox-21Puro-12, PDB1lox-21Puro-20). Relative expression levels are normalized to DOX-induced expression in primary infected fibroblasts.
(F) Comparison of gene expression profile of hESCs (BG01, H9), factor-carrying PDB2lox lines (PDB2lox-5, PDB2lox-17, PDB2lox-21, PDB2lox-22), and factor-free PDB1lox lines (PDB1lox-17Puro-5, PDB1lox-17Puro-10, PDB1lox-21Puro-20, PDB1lox-21Puro-26). All genes (n = 1434) showing at least 2-fold differential expression between factor-free hiPSCs and factor-carrying hiPSCs cells were ordered by this ratio and hierarchically clustered by sample (using uncentered correlation and average linkage). All log2 ratios are relative to expression in fibroblasts. Numbers shown in dendrogram indicate Pearson correlation between clusters. The expression profile of factor-free hiPSCs is higher correlated to hESCs than the expression profile of factor-carrying hiPSCs as tested by Fisher’s Z transformation (p < 1e-7). Confidence of the hierarchical clustering was computed with multiscale bootstrap resampling, generating in more than 99% a cluster between the hESCs and all factor-free PDB1lox lines.
(G) Venn diagram displaying the number of differentially expressed genes (p < 0.05 determined by moderated t test, corrected for false discovery rate) between provirus-carrying PDB2lox lines (PDB2lox-5, PDB2lox-17, PDB2lox-21, PDB2lox-22) compared to hESCs (H9, BG01), or reprogramming factor-free PDB1ox lines (PDB1lox-17Puro-5, PDB1lox-17Puro-10, PDB1lox-21Puro-20, PDB1lox-21Puro-26) compared to hESCs (H9, BG01), respectively.
In order to compare residual transgene expression between distinct hiPSCs with integrated transgenes and factor-free hiPSCs, we performed quantitative RT-PCR using transgene-specific PCR primers. As reported previously with either lentiviral or Moloney-based retroviral vectors (Dimos et al., 2008; Ebert et al., 2009; Hockemeyer et al., 2008; Park et al., 2008a; Yu et al., 2007), we detected residual expression of the reprogramming factors for most of the transgenes in all cell lines with integrated viruses but not in uninfected fibroblasts, hESCs, or PDB1lox lines (Figure 6E). Our results indicate that the use of loxP-flanked vectors for reprogramming followed by Cre-mediated excision can efficiently generate reprogramming factor-free hiPSCs.
To address whether residual transgene expression could affect the overall gene expression profile of the reprogrammed cells, we compared hESCs, the parental fibroblasts, and hiPSCs before and after transgene excision by genome-wide gene expression analysis. Initial correlation analysis based on all genes that show at least a 4-fold expression difference between fibroblasts and hESCs confirmed that all hiPSCs are closely related to hESCs regardless of whether the transgenes were removed (Figure S7). Despite the similarity of hESCs and hiPSCs, statistical analysis comparing PDB1lox and PDB2lox cells in correlation to hESCs demonstrated that PDB1lox cells are more similar to hESCs than the parental PDB2lox cells (Figure S7). Notably, correlation analysis based on all genes showing at least a 2-fold expression difference between hiPSCs either with or without transgenes confirmed that the gene expression profile of each individual PDB1lox line was more closely related to hESCs than to PDB2lox lines (Figure 6F). In hiPSCs with viral integrations, 271 genes showed statistically significant differential expression as compared to hESCs (p < 0.05) (Figure 6G). Similar differences have been reported previously (Takahashi et al., 2007). In contrast, only 48 genes were differentially expressed between factor-free hiPSCs and hESCs (Figure 6G). This represents a reduction of more than 80% of deregulated genes upon removal of the reprogramming factors. The remaining differentially expressed genes in factor-free hiPSCs are most likely due to either the diverse genetic background of hESCs and hiPSCs or the expression of the transactivator or a genetic memory of the reprogrammed somatic cell of origin. A detailed list of the differentially regulated genes is shown in Table S1.
DISCUSSION
In this work, we derived hiPSCs from skin biopsies obtained from patients with idiopathic PD. We developed a robust reprogramming protocol that allows the reproducible generation of patient-specific hiPSCs carrying a low number of proviral vector integrations. The use of modified lentiviruses carrying a loxP site flanking the integrated proviruses allowed the efficient removal of transgene sequences and generated reprogramming factor-free hiPSCs. The factor-free hiPSCs were pluripotent, and, as judged by molecular criteria, were more similar to embryo-derived hESCs than to the conventional vector-carrying parental hiPSCs.
Generation of Dopaminergic Neurons from hiPSCs Isolated from Somatic PD Patient Cells
Efforts to understand the underlying pathophysiology of many neurodegenerative diseases such as PD are hampered by the lack of genuine in vitro models. Using hiPSC technology, we established hiPSC lines from five patients with idiopathic PD using DOX-inducible lentiviral vectors transducing either three or four reprogramming factors. These cells were shown to have all of the features of pluripotent hESCs, including the ability to differentiate into cell types of all embryonic lineages.
In order to create an in vitro culture system for dopaminergic neurons from PD patient-specific hiPSCs, we used two different established protocols for the directed differentiation of hESCs based on either the coculture of hiPSCs with stromal feeder cells or feeder-free embryoid body formation (Elkabetz et al., 2008; Kim et al., 2006; Perrier et al., 2004; Roy et al., 2006; Sonntag et al., 2007). Using both protocols, we were able to generate dopaminergic neurons from all PD patient-derived hiPSCs with efficiencies similar to non-PD patient-derived hiPSCs or hESCs. Such patient-specific cells will provide a system for investigating the proposed molecular and cellular mechanisms of sporadic PD, such as protein aggregation, mitochondrial dysfunction, oxidative stress, and altered kinase activity (Gasser, 2007; Schulz, 2008). Using a similar approach, it was recently shown for spinal muscular atrophy (SMA) that modeling of CNS diseases in vitro with hiPSC technology can recapitulate the underlying pathological mechanism (Ebert et al., 2009). However, because of the relatively short time span of cultured neurons (weeks) compared to the age of onset of PD (approximately >50 years), it may be necessary to accelerate PD-pathology related phenotypes in vitro with exogenous challenges such as the exposure to oxidative stress, neurotoxins such as MPTP, or the overexpression of PD-related genes such as alpha-synuclein or LRKK2. Such in vitro models could be utilized for large-scale genetic or drug-based screens since large numbers of hiPSCs can be generated and robustly differentiated into dopaminergic neurons. Furthermore, our finding that dopaminergic neurons from PD patients can be derived regardless of the underlying disease or the age of the donor substantiates the idea that hiPSC-based cell replacement could become a feasible therapeutic option for PD in the future.
Generation of Reprogramming Factor-free hiPSCs
As discussed previously, the key obstacle to the eventual use of hiPSCs for cell replacement therapies is the use of viral vectors to deliver the factors necessary to initiate reprogramming (Trounson, 2009). So far, two different strategies have been pursued to generate genetically nonmodified iPSCs: (1) Reprogramming of mouse somatic cells has been achieved through transient expression of the reprogramming factors using either adenoviral infection or transient transfection (Okita et al., 2008; Stadtfeld et al., 2008). Because of the substantially lower efficiency of reprogramming, it remains unclear whether similar approaches would be feasible in the human system. (2) Replacement of the reprogramming factors by small molecules or protein delivery is being pursued in many laboratories. Screens to replace the reprogramming factors in the mouse and in the human system have led to the replacement of single factors (Huangfu et al., 2008; Shi et al., 2008a, 2008b). However, it is not yet clear whether the replacement of all reprogramming factors by small molecules will be possible.
Our results indicate that removal of the integrated transgenes by Cre-/lox-mediated recombination can lead to vector-free hiPSCs. A previous report failed to excise transgenes flanked by loxP sites (Takahashi and Yamanaka, 2006). This is probably due to the high number of retroviral integrations (more than 20) that made complete removal of all proviruses impossible or caused catastrophic genomic instability. Our results, based upon DOX-inducible lentiviral transduction, show that hiPSCs carrying as few as three or four viral integrations can be generated. Using DOX-inducible lentiviral vectors with a loxP site within the 3′ LTR, we derived PD patient-specific reprogramming factor-free hiPSCs after Cre-recombinase-mediated excision of the transgenes. Removal of the promoter and transgene sequences in self-inactivating (SIN) lentiviral vectors is expected to considerably reduce the risk of oncogenic transformation due to virus-mediated oncogene activation and/or re-expression of the transduced transcription factors (Allen and Berns, 1996; von Kalle et al., 2004). The remaining risk of gene disruption could in the future be eliminated by targeting of the reprogramming factors as a polycistronic single expression vector flanked by loxP sites into a genomic safe-harbor locus (Carey et al., 2009).
Factor-free hiPSCs Maintain a Pluripotent ESC-like State
Although silencing of transgene expression has been reported for several hiPSCs, all hiPSCs generated to date (including the lines described in this paper prior to removal of the reprogramming factors) sustain a low but detectable residual transgene expression (Dimos et al., 2008; Ebert et al., 2009; Hockemeyer et al., 2008; Park et al., 2008a; Yu et al., 2007). The question of whether hiPSCs depend on the expression of the reprogramming factors to maintain a pluripotent ESC-like state has therefore not been conclusively resolved. The observation that factor-free hiPSCs were morphologically and biological indistinguishable from the parental hiPSCs and maintained all the characteristics of hESCs demonstrates that human somatic cells can be reprogrammed to a self-sustaining pluripotent state that can be maintained in the complete absence of the exogenous reprogramming factors. These results provide additional proof that hiPSCs reestablish a pluripotency-related autoregulatory loop that has been proposed to rely on the activation of the four endogenous transcription factors OCT4, NANOG, SOX2, and TCF3 (Jaenisch and Young, 2008).
Residual Transgene Expression from Partially Silenced Viral Vectors Perturbs the Transcriptional Profile of hiPSCs
Because the genomic integration site of a particular provirus influences proviral silencing as well as its risk of being reactivated, hiPSCs withidentical and predictable properties cannot be generated by approaches relying on stochastic silencing. This is particularly disconcerting because residual transgene expression might affect the differentiation properties of iPSCs. Indeed, significant differences between mouse ES cells and iPSCs in their ability to differentiate into cardiomyocytes (K. Hochedlinger, personal communication) and partially blocked EB-induced differentiation along with incomplete OCT4 and NANOG downregulation of distinct hiPSC clones (Yu et al., 2007) have been observed. These observations are consistent with the possibility that the variable basal transcription of only partially silenced vectors might influence the generation of functional differentiated cells.
In an effort to assess whether the removal of the vectors would affect the properties of the hiPSCs, we compared overall gene expression patterns in parental provirus-carrying hiPSCs, factor-free hiPSCs, and embryo-derived hESCs. As reported previously (Park et al., 2008b; Takahashi et al., 2007; Yu et al., 2007), the provirus-carrying hiPSCs and factor-free hiPSCs clustered closely with the hESCs when compared to the donor fibroblasts. However, a more detailed analysis of the most divergent genes between the different hiPSCs cell types revealed that embryo-derived hESCs and factor-free hiPSCs were more closely related to each other than to the provirus-carrying parental hiPSCs. It is possible that the remaining small difference in gene expression between the factor-free hiPSCs and hESCs may be due to expression of the transactivator that had not been excised in our experiments. These results presented here provide clear evidence that the basal expression of proviruses carried in conventional iPS cells can affect the molecular characteristics of the cells. The system described in this paper provides the basis for further elucidation of the effect of residual transgene expression, especially in the context of in vitro and in vivo differentiation paradigms. Furthermore, these results demonstrate that the derivation of reprogramming factor-free hiPSCs is essential not only for potential future therapeutic applications, but also for biomedical research in order to develop reliable and reproducible in vitro models of diseases. To this end, one of the main advantages of generating transgene-free hiPSCs by Cre-mediated excision is its high efficiency and experimental simplicity. The system described here has the potential to become a routine technology for the derivation of hiPSCs that will allow the generation of standardized hiPSCs from different sources with different combinations of reprogramming factors.
EXPERIMENTAL PROCEDURES
Cell Culture
All primary fibroblast cell lines described in this paper were purchased from the Coriell Cell Repository. Fibroblasts were cultured in fibroblast medium (Dulbecco’s modified Eagle’s medium [DMEM] supplemented with 15% fetal bovine serum [FBS; Hyclone], 1 mM glutamine [Invitrogen], 1% nonessential amino acids [Invitrogen], and penicillin/streptomycin [Invitrogen]).
hiPSCs and the hESC lines BG01 and BG02 (National Institutes of Health [NIH] code: BG01 and BG02; BresaGen, Inc., Athens, GA) were maintained on mitomycin C (MMC)-inactivated mouse embryonic fibroblast (MEF) feeder layers in hESC medium (DMEM/F12 [Invitrogen] supplemented with 15% FBS [Hyclone], 5% KnockOut Serum Replacement [Invitrogen], 1 mM glutamine [Invitrogen], 1% nonessential amino acids [Invitrogen], 0.1 mM β-mercaptoethanol [Sigma], and 4 ng/ml FGF2 [R&D Systems]). Cultures were passaged every 5 to 7 days either manually or enzymatically with collagenase type IV (Invitrogen; 1.5 mg/ml). HESC line H9 (NIH code: WA09; Wisconsin Alumni Research Foundation, Madison, WI) was maintained on MMC-inactivated MEFs or on MMC-inactivated human fibroblasts (D551; American Type Culture Collection, Manassas, VA) according to the manufacturer’s protocol. For EB-induced differentiation, hESC/hiPSC colonies were harvested with 1.5 mg/ml collagenase type IV (Invitrogen), separated from the MEF feeder cells by gravity, gently triturated, and cultured for 10 days in nonadherent suspension culture dishes (Corning) in DMEM supplemented with 15% FBS. Differentiation of dopaminergic neurons was performed as described previously with either an EB-based feeder-free protocol (Kim et al., 2006) or a protocol based on the combination of MS5 stromal cell coculture with the BMP antagonist Noggin (Perrier et al., 2004; Sonntag et al., 2007). Detailed description of both methods can be found in Supplemental Experimental Procedures.
For Cre-recombinase-mediated vector excision, hiPSC lines were cultured in Rho Kinase (ROCK) inhibitor (Calbiochem; Y-27632) 24 hr prior to electroporation. Cells were harvested with 0.05% trypsin/ethylenediaminetetraacetic acid (EDTA) solution (Invitrogen), and 1 × 107 cells resuspended in PBS were either transfected with pCre-PAC (50 μg) (Taniguchi et al., 1998) or cotransfected with pTurbo-Cre (40 μg; GenBank accession number AF334827) and pEGFP-N1 (10 μg; Clontech) by electroporation as described previously (Costa et al., 2007) (Gene Pulser Xcell System, Bio-Rad: 250 V, 500 μF, 0.4 cm cuvettes). Cells were subsequently plated on MEF feeder layers (DR4 MEFs for puromycin selection) in hESC medium supplemented with ROCK inhibitor for the first 24 hr. Cre-recombinase-expressing cells were selected with one of the following methods: (1) addition of puromycin (2 μg/ml) 2 days after electroporation for a period of 48 hr or (2) FACS sorting (FACS-Aria; BD Biosciences) of a single cell suspension for EGFP-expressing cells 60 hr after electroporation followed by replating at a low density in ROCK inhibitor containing hESC medium. Individual colonies were picked 10 to 14 days after electroporation.
Viral Constructs
The FUW-M2rtTA lentiviral vector and lentiviral vectors containing the human cDNAs for KLF4 (FUW-tetO-hKLF4), OCT4 (FUW-tetO-hOCT4), SOX2 (FUW-tetO-hSOX2), and c-MYC (FUW-tetO-hMYC) under the control of the tetracycline operator and a minimal CMV promoter have been described previously (Hockemeyer et al., 2008). For generation of the Cre-recombinase-excisable DOX-inducible lentiviral vectors, a NotI/BSU36I fragment containing the tetracycline operator/minimal CMV promoter and the human cDNAs for KLF4, OCT4, or SOX2 were subcloned from each FUW-tetO vector into the NotI/BSU36I sites of the FUGW-loxP, which contains a loxP site in the 3′ LTR (Hanna et al., 2007).
Lentiviral Infection and hiPSC Derivation
Vesicular stomatitis virus G protein (VSVG)-coated lentiviruses were generated in 293 cells as described previously (Brambrink et al., 2008). In brief, culture medium was changed 12 hr after transfection, and virus-containing supernatant was collected 60–72 hr after transfection. Viral supernatant was filtered through a 0.45 μm filter. Virus-containing supernatants were pooled for three-and four-factor infections and supplemented with FUW-M2rtTA virus and an equal volume of fresh culture medium. One million human fibroblasts were seeded 24 hr before transduction in T75 flasks. Four consecutive infections in the presence of 2 μg/ml of polybrene were performed over a period of 48 hr. Culture medium was changed 12 hr after the last infection. Five days after transduction, fibroblasts were passaged with trypsin and replated at different densities between 5 × 104 and 2 × 105 cells per 10 cm gelatin-coated dish. For induction of reprogramming, culture medium was replaced 48 hr later by hESC medium supplemented with DOX (Sigma-Aldrich; 2 μg/ml). hiPSCs colonies were picked manually on the basis of morphology between 3 and 5 weeks after DOX induction and manually maintained and passaged according hESC protocols in the absence of DOX. For determination of reprogramming efficiencies, 1 × 105 human fibroblasts were seeded onto 10 cm gelatin-coated dishes. Reprogramming efficiencies were calculated after 20 days on the basis of immunocytochemistry for the pluripotency markers Tra-1-60 and NANOG.
Microarray Gene Expression Analysis
RNA was isolated from hESCs and iPSCs, which were mechanically separated from feeder cells, with the RNeasy Mini Kit (QIAGEN). Two micrograms of total RNA was used to prepare biotinylated cRNA according to the manufacturer’s protocol (Affymetrix One Cycle cDNA Synthesis Kit). In brief, this method involves SuperScript II-directed reverse transcription using a T7-Oligo(dT) Promoter Primer to create first strand cDNA. RNase H-mediated second-strand cDNA synthesis is followed by T7 RNA Polymerase-directed in vitro transcription, which incorporates a biotinylated nucleotide analog during cRNA amplification. Samples were prepared for hybridization with 15 μg biotinylated cRNA in a 1× hybridization cocktail according the Affymetrix hybridization manual. GeneChip arrays (Human U133 2.0) were hybridized in a GeneChip Hybridization Oven at 45°C for 16 hr at 60 RPM. Washing was done with a GeneChip Fluidics Station 450 according to the manufacturer’s instructions, with the buffers provided in the Affymetrix GeneChip Hybridization, Wash, and Stain Kit. Arrays were scanned on a GeneChip Scanner 3000, and images were extracted and analyzed with GeneChip Operating Software v1.4.
U133 Plus 2.0 microarrays (Affymetrix) were processed with the MAS5 algorithm, and absent/present calls for each probeset were determined with the standard Affymetrix algorithm, both as implemented in Bioconductor. Probe sets that were absent in all samples were removed for subsequent analysis. Differential expression was determined by a moderated t test with the “limma” package in R (corrected for false discovery rate) or by fold change. Where a gene was represented by multiple probe sets (based on annotation from Affymetrix), gene expression log ratios and p values were calculated as the mean and minimum of these probesets, respectively. Hierarchical clustering was performed on log-transformed gene expression ratios with uncentered Pearson correlation and pairwise average linkage. Correlations were compared with Fisher’s Z transformation. Confidence of the hierarchical clustering was computed with multiscale bootstrap resampling with the R package “pvclust.”
Supplementary Material
Acknowledgments
We thank Tobias Brambrink for providing the FUGW-loxP plasmid. We thank Raaji Alagappan, Ping Xu, Kristen Lee, and Elizabeth Marlow for technical support and Jessica Daussman, Ruth Flannery, and Dongdong Fu for their help with animal husbandry and processing of teratomas. We would like to thank the members of the Whitehead Genome Technology Core for their help with the microarray expression analysis. We thank all the members of the Jaenisch lab for helpful discussions and comments on the manuscript. D.H. is a Merck Fellow of the Life Science Research Foundation. R.J. was supported by NIH grants R37-CA084198, RO1-CA087869, and RO1-HD045022. This research was supported in part by a Collaborative Innovation Award from the Howard Hughes Medical Institute. O.I. is supported by Udall Parkinson’s Disease Center of Excellence grant P50NS39793 and the Michael Stern Foundation. R.J. is an advisor to Stemgent, which has obtained a license from the Massachusetts Institute of Technology (MIT) to distribute some of the reagents used in this paper. F.S., D.H., and R.J. designed the experiments and wrote the paper. C.B. designed and performed Southern blot analysis. Q.G. analyzed all teratomas. G.B. analyzed the gene expression profile. E.G.C. assisted with the methylation analysis. G.H., A.B., O.C., and O.I. performed and analyzed differentiation experiments of Figure 3B, and C. M.M. provided human ES cell controls. F.S. and D.H. performed all other experiments.
Footnotes
ACCESSION NUMBERS
Microarray data are available at the NCBI Gene Expression Omnibus database under the series accession number GSE14711.
Supplemental Data include Supplemental Experimental Procedures, seven figures, and one table and can be found with this article online at http://www.cell.com/supplemental/S0092-8674(09)00151-2.
References
- Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Vassena R, Bilic J, Pekarik V, Tiscornia G, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- Allen JD, Berns A. Complementation tagging of cooperating oncogenes in knockout mice. Semin Cancer Biol. 1996;7:299–306. doi: 10.1006/scbi.1996.0038. [DOI] [PubMed] [Google Scholar]
- Brambrink T, Foreman R, Welstead GG, Lengner CJ, Wernig M, Suh H, Jaenisch R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell. 2008;2:151–159. doi: 10.1016/j.stem.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Markoulaki S, Hanna J, Saha K, Gao Q, Mitalipova M, Jaenisch R. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc Natl Acad Sci USA. 2009;106:157–162. doi: 10.1073/pnas.0811426106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Dottori M, Sourris K, Jamshidi P, Hatzistavrou T, Davis R, Azzola L, Jackson S, Lim SM, Pera M, et al. A method for genetic modification of human embryonic stem cells using electroporation. Nat Protocols. 2007;2:792–796. doi: 10.1038/nprot.2007.105. [DOI] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Ebert AD, Yu J, Rose FF, Jr, Mattis VB, Lorson CL, Thomson JA, Svendsen CN. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22:152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser T. Update on the genetics of Parkinson’s disease. Mov Disord. 2007;22(Suppl 17):S343–S350. doi: 10.1002/mds.21676. [DOI] [PubMed] [Google Scholar]
- Hanna J, Wernig M, Markoulaki S, Sun CW, Meissner A, Cassady JP, Beard C, Brambrink T, Wu LC, Townes TM, et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–477. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Cook EG, Gao Q, Mitalipova M, Jaenisch R. A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell Stem Cell. 2008;3:346–353. doi: 10.1016/j.stem.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, Muhlestein W, Melton DA. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26:1269–1275. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BK, Kim SE, Shim JH, Woo DH, Gil JE, Kim SK, Kim JH. Neurogenic effect of vascular endothelial growth factor during germ layer formation of human embryonic stem cells. FEBS Lett. 2006;580:5869–5874. doi: 10.1016/j.febslet.2006.09.053. [DOI] [PubMed] [Google Scholar]
- Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R, Clark AT, Plath K. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci USA. 2008;105:2883–2888. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N, Ahfeldt T, Rigamonti A, Utikal J, Cowan C, Hochedlinger K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell. 2008;3:340–345. doi: 10.1016/j.stem.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markoulaki S, Hanna J, Beard C, Carey BW, Cheng AW, Lengner CJ, Dausman JA, Fu D, Gao Q, Wu S, et al. Transgenic mice with defined combinations of drug-inducible reprogramming factors. Nat Biotechnol. 2009;27:169–171. doi: 10.1038/nbt.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949–953. doi: 10.1126/science.1164270. [DOI] [PubMed] [Google Scholar]
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008a;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008b;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Perrier AL, Tabar V, Barberi T, Rubio ME, Bruses J, Topf N, Harrison NL, Studer L. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci USA. 2004;101:12543–12548. doi: 10.1073/pnas.0404700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy NS, Cleren C, Singh SK, Yang L, Beal MF, Goldman SA. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat Med. 2006;12:1259–1268. doi: 10.1038/nm1495. [DOI] [PubMed] [Google Scholar]
- Schulz JB. Update on the pathogenesis of Parkinson’s disease. J Neurol. 2008;255(Suppl 5):3–7. doi: 10.1007/s00415-008-5011-4. [DOI] [PubMed] [Google Scholar]
- Shi Y, Desponts C, Do JT, Hahm HS, Scholer HR, Ding S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. 2008a;3:568–574. doi: 10.1016/j.stem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Shi Y, Do JT, Desponts C, Hahm HS, Scholer HR, Ding S. A combined chemical and genetic approach for the generation of induced pluripotent stem cells. Cell Stem Cell. 2008b;2:525–528. doi: 10.1016/j.stem.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Sonntag KC, Pruszak J, Yoshizaki T, van Arensbergen J, Sanchez-Pernaute R, Isacson O. Enhanced yield of neuroepithelial precursors and midbrain-like dopaminergic neurons from human embryonic stem cells using the bone morphogenic protein antagonist noggin. Stem Cells. 2007;25:411–418. doi: 10.1634/stemcells.2006-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Sanbo M, Watanabe S, Naruse I, Mishina M, Yagi T. Efficient production of Cre-mediated site-directed recombinants through the utilization of the puromycin resistance gene, pac: a transient gene-integration marker for ES cells. Nucleic Acids Res. 1998;26:679–680. doi: 10.1093/nar/26.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trounson A. Rats, cats, and elephants, but still no unicorn: induced pluripotent stem cells from new species. Cell Stem Cell. 2009;4:3–4. doi: 10.1016/j.stem.2008.12.002. [DOI] [PubMed] [Google Scholar]
- von Kalle C, Fehse B, Layh-Schmitt G, Schmidt M, Kelly P, Baum C. Stem cell clonality and genotoxicity in hematopoietic cells: gene activation side effects should be avoidable. Semin Hematol. 2004;41:303–318. doi: 10.1053/j.seminhematol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Cassady JP, Jaenisch R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell. 2008;2:10–12. doi: 10.1016/j.stem.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.