

Abstract
Biological systems have powerful inbuilt mechanisms of control intended to maintain homeostasis. Cytokines are no exception to this rule, and imbalance in cytokine activities may lead to inflammation with subsequent tissue and organ damage, altered function, and death. Balance is achieved through multiple, not mutually exclusive, mechanisms including the simultaneous production of agonist and antagonistic cytokines, expression of soluble receptors or membrane-bound nonsignaling receptors, priming and/or reprogramming of signaling, and uncoupling of ligand/receptor pairing from signal transduction. Insight into cytokine balance is leading to novel therapeutic approaches particularly in autoimmune conditions, which are intimately linked to a dysregulated cytokine production.
Introduction
To explore the complex regulation of cytokine activities it may be of help to bear in mind the example of rheumatoid arthritis (RA). A major step forward in RA treatment was achieved when it became possible to control disease manifestations such as joint destruction by blocking TNF. This could indicate that a single cytokine, in this case TNF, drives unopposed a series of events that lead to inflammation and destruction. The situation is less simple inside the joint, however, where proinflammatory cytokines co-exist alongside their endogenous inhibitors. This is a consequence of ongoing processes in which proinflammatory stimuli induce their anti-inflammatory counterparts and the imbalance between the two results in disease.
The cytokine network is a homeostatic system that may be comparable with the acid/base equilibrium. The biological activity of any cytokine in biological fluids can be interpreted correctly only by taking into account the activities of other synergistic or antagonistic cytokines, of their respective inhibitors, and the extent to which each cytokine receptor is expressed. Interactions between intracellular signals modulate further cytokine activities. In addition, cell types with polarized patterns of cytokine production contribute to the balance. Owing to their potent activities in many different processes - including cell growth and differentiation, organ development, inflammation, immune response, and repair processes aiming at homeostasis - cytokine activities have to be tightly controlled. Since one of the main functions of cytokines is to mediate interactions between the immune and inflammatory responses, it is thought that chronic immuno-inflammatory diseases might be caused in part by the uncontrolled production of cytokines. Furthermore, depending on the stage of inflammation or the biological effect under scrutiny, the same cytokine may have proinflammatory or anti-inflammatory activities. Many different mechanisms of regulation have been identified affecting both cells and soluble mediators (Table 1).
Table 1.
Balance in cytokine activities according to biological processes
| Process | Cytokines |
|---|---|
| Inflammation | IL-1/IL-1 receptor antagonist, IL-1 receptor II, soluble IL-1 receptor I, soluble IL-1 receptor II |
| TNF/soluble TNF receptor I, soluble TNF receptor II | |
| IL-6/soluble gp130 | |
| IL-18/IL-18 binding protein | |
| IL-22/IL-22 binding protein | |
| IL-13/IL-13 receptor alpha | |
| CXCLELR+ /CXCLELR- | |
| Several proinflammatory chemokines (CXC and CC)/Duffy antigen receptor for chemokines | |
| Several proinflammatory chemokines (CC not CXC)/D6 | |
| CCL19, CCL21, CCL25, CXCL13/CCX-CKR | |
| Chemerin 9/chemerin 15 | |
| Immune cell responses | Th1 cells/Th2 cells |
| Th17 cells/Th2 cells | |
| Th17 cells/T cells with regulatory function | |
| T cells with regulatory function/Th1, Th2, Th17 cells | |
| Tissue repair and remodeling | Transforming growth factor beta/TNF |
| IL-1/IFNγ | |
| IL-4/IFNγ | |
| CD4 T-cell differentiation | IL-12/IL-4 |
| Transforming growth factor beta/IL-6 + T-cell growth factor beta | |
| Tissue destruction | Osteoprotegerin/RANKL |
| WNT/Dickkopf-1 | |
| Metabolism | Adiponectin/leptin, vistatin, resistin |
In view of the pleiotropic actions of cytokines, the table presents a far from complete view of possible opposing activities of cytokines and their ligands. The back slash (/) separates the opposing molecules in respect of a given biological activity. RANKL, receptor activator of NKκB ligand; WNT, wingless integration site.
The present review describes the key levels of imbalance that have been associated with chronic inflammation and tissue destruction. This has to be integrated in general processes of disease initiation through the innate and adaptive immune responses ending in tissue and organ damage (Figure 1).
Figure 1.
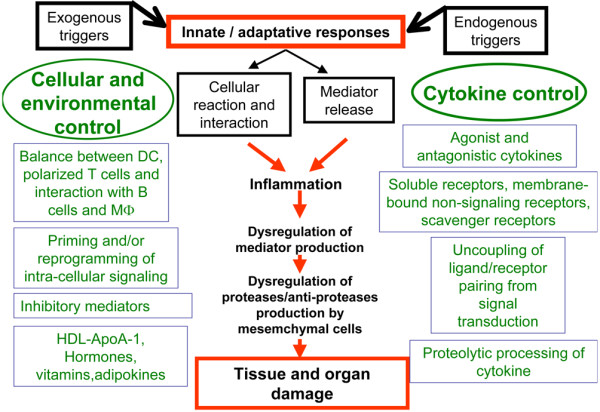
Conceptual framework for the role of cytokine imbalance in the pathogenesis of chronic inflammatory diseases. DC, dendritic cells; HDL-ApoA-1, high-density lipoprotein apolipoprotein A1; MΦ, macrophage.
Balance in cytokines
Balance between IL-1 and IL-1 natural antagonists
Amongst the most powerful proinflammatory cytokines, IL-1 stands out as a paradigmatic example of fine-tuned regulation of biological activities through a complex system of ligands with agonist and antagonist functions, as well as signaling and nonsignaling receptors (Figure 2). First of all, a natural ligand of IL-1 receptors - IL-1 receptor antagonist (IL-1Ra) -prevents recruitment of the accessory protein needed to signal, thus acting as a competitor to IL-1 [1]. Interestingly, IL-1Ra is preferentially produced by monocytes/macrophages stimulated by anti-inflammatory cytokines (see below). Second, two IL-1 receptors (Il-1RI and IL-1RII) are expressed at the surface of many cell types. An important functional difference, however, exists between the two receptors. Indeed, in contrast to IL-1RI, which transduces the signal, IL-1RII does not transduce and acts as a decoy receptor. Furthermore, both receptors may be shed from the cell surface by matrix metalloproteinases, and by binding to IL-1 or IL-1Ra soluble receptors may modulate their bioavailability, ultimately affecting cell responses. One of the many members of the IL-1 family, IL-1F5, also has inhibitory activities [2]. Some patients have autoantibodies to IL-1α and these may also play a role by blocking IL-1 biological activity. Regulation is also provided by single immunoglobulin IL-1-related receptor (SIGIRR), also known as Toll-IL-1 receptor 8, which is a member of the Toll-like receptor/IL-1R family. Its small single extracellular immunoglobulin domain does not support ligand binding. Besides, the intracellular domain of SIGIRR cannot activate NFκB because it lacks two essential amino acids (Ser447 and Tyr536) in its highly conserved Toll-IL-1 receptor domain. SIGIRR rather acts as an endogenous inhibitor of Toll-like receptor and IL-1 signaling, because overexpression of SIGIRR in Jurkat or HepG2 cells substantially reduced lipopolysaccharide-induced or IL-1-induced activation of NFκB. Furthermore, lupusprone mice have an accelerated course of disease when lacking Toll-IL-1 receptor 8 [3,4].
Figure 2.
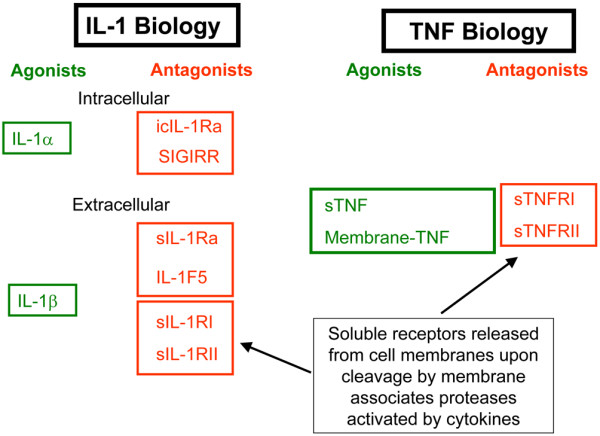
Schematic representation of agonists and antagonists determining the biological activities of IL-1 and TNF. icIL-1Ra, intracellular IL-1 receptor antagonist; SIGIRR, single immunoglobulin IL-1-related receptor; sIL-1Ra, soluble IL-1 receptor antagonist; sIL-1R, soluble IL-1 receptor; sTNF, soluble TNF; sTNFR, soluble TNF receptor.
The production by monocytes-macrophages of IL-1 and IL-1Ra is dependent on many distinct stimuli, including T-cell contact. Of interest, apolipoprotein A1, a negative acute-phase reactant, may act as negative feedback regulator by reducing IL-1 but not IL-1Ra production induced by T-cell contact. IFNβ favors the production of IL-1Ra while simultaneously inhibiting IL-1. Similar activities are shared by IL-4, IL-13 and transforming growth factor beta (TGFβ), which in this context are generally considered anti-inflammatory in that they increase IL-1Ra and, to a lesser extent, decrease IL-1 production (Table 2). A similar type of regulation is provided by leptin, which can modulate the expression of IL-1Ra and the release of IL-1β by beta cells in human islets [5].
Table 2.
Cytokine roles categorized according to their contribution to inflammation in rheumatoid arthritis
| Proinflammatory | Ambivalent | Anti-inflammatory |
|---|---|---|
| TNF | IFNγ | IL-1 receptor antagonist |
| IL-1 | Transforming growth factor beta | IL-4a |
| IL-12 | IL-6b | IL-13 |
| IL-15 | IL-10c | |
| IL-17A/IL-17F | IL-25 | |
| IL-18 | IL-27 | |
| CXCL8 | IL-35 | |
| CCL3 | ||
| CCL2 | 7ND |
7ND, N-terminal natural deletion variant of monocytes chemotactic protein-1/CCL2. aIL-4 is anti-inflammatory in the context of rheumatoid arthritis synovial inflammation. By impacting on IgE production, however, IL-4 is a key cytokine in IgE-mediated inflammation. Similar considerations apply to IL-13. bIL-6 may be proinflammatory or anti-inflammatory according to the circumstances. IL-6 blockade has been shown to be clinically useful to control rheumatoid arthritis in randomized trials. cIL-10 is usually anti-inflammatory, but upon priming of monocytes with IFNα it induces proinflammatory responses.
Phosphatidylinositide 3 kinase is among the most important signaling pathways involved in the control of the IL-1/IL-1Ra balance in human monocytes, in so far as inhibition of phosphatidylinositide 3 kinase delta markedly decreases IL-1 while increasing IL-1Ra [6,7]. A further example of the plasticity of the IL-1/IL-1Ra balance in human monocytes is the increase in IL-1Ra but decrease in T-cell-induced IL-1β in the presence of glatiramer acetate, a therapeutic agent used in multiple sclerosis [8].
Balance in TNF and IL-6 activities
TNF and IL-6 have become successful targets of biological therapies in a variety of inflammatory conditions starting with RA, thus underling their pivotal role in inflammation. Several excellent reviews have been devoted to these two cytokines and their relevance in human diseases [9-13]. Therefore we shall here overview only the basic mechanisms involved in the regulation of their biological activities, in particular stressing differences in the activity of their respective soluble receptors. Trimeric TNF, mostly produced by activated macrophages and T cells, acts by binding to two distinct TNF receptors: TNF-RI (p55), which is widely expressed; and TNF-RII (p75), mostly present on cells of the immune system (Figure 2). Both receptors can be enzymatically shed from the surface of the cells and, once in the body fluids, both can bind TNF and neutralize its biological activity [14]. The receptors therefore act as natural inhibitors of TNF, and their production is regulated by several stimuli including TNF itself.
At variance with TNF, IL-6 acts by binding to a heterodimeric receptor composed of the common gp130 chain, shared with oncostatin M, IL-11, ciliary neurotrophic factor-1, cardiotropin-1, and leukemia inhibitor factor, and to its specific IL-6 receptor alpha (IL-6Rα). The signaling chain is gp130, affinity of which for IL-6 is increased in the presence of IL-6Rα. Of interest, IL-6Rα exists as a cell-bound form expressed on few cell types - particularly hepatocytes, phagocytes, and some lymphocytes - but also in a soluble form abundantly present in body fluids. Soluble IL-6Rα (sIL-6Rα) has the capacity of binding to IL-6 and to increase its affinity for gp130. Since gp130 is ubiquitously expressed, sIL-6Rα offers the opportunity to cells that do not express IL-6Rα to become responsive to IL-6, a phenomenon called trans-signaling. In transgenic mice sIL-6Rα functions as a carrier protein for its ligand, thereby markedly prolonging the plasma half-life of IL-6, indicating that IL-6 signaling is increased by sIL-6Rα [15]. The agonistic properties of sIL-6Rα by enhancing IL-6 signaling are well documented. There are results indicating also antagonistic properties of sIL-6Rα, however, which may explain why IL-6 may in some circumstances acts as an anti-inflammatory mediator [16].
Besides a soluble form of IL-6Rα, a soluble form of gp130 (sgp130) has been detected in healthy human sera with antagonistic properties. Of interest, the antagonistic activity of sgp130 is markedly enhanced in the presence of sIL-6Rα [17]. Cell responses to IL-6 are therefore finely tuned by the ratios between-cell bound gp130 and IL-6Rα on the one side, and on the other by available IL-6, sIL-6Rα and sgp130.
Balance generated by soluble osteoprotegerin
Another cytokine whose biological activities are modulated by soluble receptors or natural antagonists is osteoprotegerin, which is a secreted member of the TNF receptor family that binds OPGL and blocks its activity. Genetic (including gene-targeting) studies and functional studies in vitro and in vivo indicate that osteoprotegerin is a pure, soluble decoy receptor [18]. Osteoprotegerin also binds and neutralizes TNF-α-related apoptosis-inducing ligand [19].
Additional cytokines whose biological activities are regulated by the balance of agonist and soluble nonsignaling receptors include IL-18/IL-18 binding protein, IL-22/IL-22 binding protein, and IL-13/IL-13 receptor alpha. These will not be discussed in the present review, however, owing to the shortage of space.
Balance in chemokine responses
A balance in chemokine responses is generated via several distinct, but not mutually exclusive, operational mechanisms. As previously shown for other cytokines, distinct chemokines may fulfill opposing functions for a given task. A classical example is the propensity of CXC chemokines sharing the ELR motif (CXCL1, CXCL3, CXCL5, CXCL6, and CXCL8) to exert angiogenic properties, while CXC chemokines lacking the ELR motif (CXCL9, CXCL10, CXCL11) are more angiostatic [20]. Similarly, chemokines may play opposing roles in proliferation and apoptosis susceptibility. In addition, a peculiarity of some chemokine receptors is that they bind chemokines but fail to signal [21]. Chemokines signal through seven-transmembrane domain, G-protein-coupled receptors, of which 19 have been molecularly defined. These receptor families reflect the two major (CC and CXC) chemokine families and two minor (C and CX3C) chemokine families [22]. In addition, chemokine receptors whose structural features are inconsistent with signaling functions have been described. By binding to chemokines, non-signaling receptors act as a decoy, scavenge receptors, and regulate inflammatory and immune responses. The family of silent chemokine receptors comprises Duffy antigen receptor for chemokines (DARC), D6 (also known as CC chemokine binding protein 2), and CCX-CKR (also known as CCRL1). It is noteworthy that the silent chemokine receptors, which lack the key residues needed for coupling with G-proteins, have unusual expression patterns and a wide range of chemokine-binding properties.
DARC is expressed on erythrocytes and endothelial cells of postcapillary veins in many organs - including, amongst others, high endothelial venules in lymphoid organs [23]. DARC binds 11 proinflammatory (both CC and CXC) but not homeostatic chemokines, and preferentially angiogenic but not angiostatic chemokines [24]. Chemokines injected in DARC-/- mice rapidly disappear from circulation, indicating a role of erythrocyte DARC as a sink or reservoir. Endothelial DARC, however, appears to have a downregulating effect on inflammation. Overexpression of endothelial DARC in animal models is therefore associated with both decreased angiogenesis and tumor growth, while a lack in DARC is associated with increased tumor growth, metastasis formation and increased concentrations of CXCL1 and CXCL3 [25,26].
D6 binds most inflammatory CC chemokines, but not CXC and homeostatic CC chemokines. D6 is expressed at high concentrations on lymphatic and venular endothelium, particularly in the skin, gut, lung, and placenta [27]. D6 mediates chemokine degradation, being constitutively internalized through clathrin-coated pits. D6-/- mice are prone to exaggerated inflammatory responses induced by phorbol ester myristate acetate application to the skin or subcutaneous injections of complete Freund's adjuvant [28,29]. Lack of D6 expression in syncytiotrophoblast increases the susceptibility to inflammation-induced fetal loss [30]. In contrast, transgenic expression of D6 in keratinocytes dampens cutaneous inflammation and reduces tumor growth [31].
CCX-CKR appears to have a more limited chemokine-binding repertoire that includes CCL19, CCL21, CCL25, and CXCL13, and it is expressed exclusively by stromal cells in the thymus and lymph nodes, by lymph vessels in the intestine and by the epidermis [32]. In CCX-CKR-/- mice, trafficking of dendritic cells to lymph nodes under steady-state conditions appears to be decreased, as well as the recruitment of hematopoietic precursors to the thymus.
Pathogen-encoded decoys also affect chemokine activities. Indeed, molecular mimicry of chemokines and their receptor is an important immune-evasion strategy used by pathogens, of which numerous examples are known. Viral chemokine binding protein and Schistosoma mansonii chemokine binding protein have been described.
The receptor functions of some chemokines appear to vary according to the context in which they operate. For instance, IL-10 uncouples CCR2 binding from signaling, and therefore CCR2 functionally becomes a decoy receptor [33]. An additional example is the high level of CCR5 expressed in response to lipoxin A4 on apoptotic neutrophils and T cells. Lipoxin A4 is produced late during the inflammatory response when significant tissue damage has already occurred. By increasing the expression of CCR5 on dying cells, lipoxin A4 contributes to scavenging CCR5 ligands, which therefore are no longer available for recruiting new cells, which in turn reduces inflammation.
An additional mechanism regulating chemokine activities is related to modifications of their primary structure. For instance, the N-terminal natural deletion variant of monocytes chemotactic protein-1/CCL2 (called 7ND) inhibits chemotaxis mediated by monocytes chemotactic protein-1, and the extension of RANTES/CCL5 by a single methionine (met-RANTES) creates a potent and selective RANTES antagonist.
The particular example of chemerin
Chemerin is a plasma protein known for its proinflammatory properties exerted upon binding to the G-protein coupled receptor ChemR23/CMKLR1 - expressed on macrophages and plasmacytoid dendritic cells - where it induces cell migration. Chemerin is secreted as an inactive precursor and is processed by proteases before becoming an active mediator. As for conventional chemokines, the biologically active chemerin binds to ChemR23 with its COOH-terminal portion.
Of interest, different proteases generate different chemerin peptides, which possess opposite functions. Serine proteases mainly produced by activated neutrophils - early mediators in inflammation - therefore generate chemerin 9 (9 AA peptide), which is an agonist in the nanomolar range. Cysteine proteases - mainly produced by macrophages - which arrive later at the inflammatory site, however, generate chemerin 15 (15 AA peptide). This peptide in the picomolar range acts as an antagonist, expressing potent anti-inflammatory activities and contributing to reduce inflammation [34].
A further layer of complexity has been added recently with the description of an additional chemerin receptor named CCRL2, selectively expressed on mouse mast cells. Upon binding to this receptor, chemerin induces neither cell migration nor calcium flux. CCRL2 is therefore supposed to scavenge chemerin. The experimental test of this hypothesis led to the opposite result, however, indicating enhanced inflammation in a rodent model of IgE-mediated passive cutaneous anaphylaxis. A possible explanation could be that mast cells bind the N-terminal portion of chemerin with CCRL2 and present the COOH-terminal portion to cells expressing ChemR23, which are thus potently activated [35].
The Th1/Th2 balance
In the late 1980s Mosmann and colleagues described the Th1/Th2 balance when studying a large series of mouse CD4+ T-cell clones [36]. They observed that some clones would produce IFNγ but not IL-4, while others would do the opposite. Therefore, based on the dicotomic production of two key cytokines, it was possible to classify T-cell clones into two groups, which were named Th1 and Th2. The same concepts were verified by studying human T-cell clones [37]. Naïve T cells could be induced to become Th1 or Th2 simply by modifying the cytokine present in the milieu during priming, although the dose of antigen, the amount of co-stimulation, and the age of antigen-presenting cells could also affect polarization.
Of major importance, Th1 cytokines were shown to inhibit Th2 cytokine production and function, and vice versa. This observation included cytokines important for priming: IL-12 and IFNγ for Th1 cells, and IL-4 for Th2 cells. Starting investigations with mouse models of human diseases, it was found that models of multiple sclerosis - such as the antigen-induced experimental acute encephalomyelitis (EAE) - or of RA - such as type II collagen arthritis - were associated with the overexpression of IFNγ but not of IL-4. In sharp contrast, models of allergic diseases such as asthma were associated with IL-4 without IFNγ expression. In these models, forced expression of counteracting T-helper cytokines could in many instances abrogate disease expression [38,39].
Addition of the Th17 pattern
In 2005 the above classification was amended when it was shown in the mouse that IL-17 was produced by a particular T-helper cell, named Th17 [40,41] (Figure 3). As early as 1999, however, it was shown that some T-cell clones obtained from the synovium of RA patients were producing IL-17 and differed from the classical Th1/Th2 clones [42]. Indeed, they did not produce IL-4 and produced little, if any, IFNγ.
Figure 3.

Cytokines, hormones, and other soluble mediators controlling biology of Th17 cells leading to tissue destruction. Summary of some of the many mediators involved in Th17 differentiation, expansion, acquisition of effector function and their relationship with macrophages, which may then mediate tissue destruction. Orange arrows, enhancement; blunted black heads, inhibition; black arrows, production. AHR, aryl-hydrocarbon receptor; APO-A-1, apolipoprotein A1; MMP, matrix metalloproteinase; MΦ, macrophage; PGE2, prostaglandin E2; RORγt, retinoic acid-related orphan receptor γt; STAT, signal transducer and activator of transcription; TGFβ, T-cell growth factor beta; Treg = T cell with regulatory function.
The Th1/Th2 paradigm was then revisited; key observations were made based on the murine EAE model [43]. This model was previously associated with Th1 responses. Th1 cells are induced by IL-12 produced by monocytes and dendritic cells. IL-12 is a heterodimer composed of p35 and p40 subunits. Protection from EAE was afforded when IL-12 was blocked with anti-IL-12p40. IL-23 is also a heterodimer, however, composed of the IL-12/IL-23 common p40 subunit and the specific p19 subunit. When inhibitors specific to IL-23 or p19-deficient mice were used, it was recognized that IL-23 and not IL-12 was responsible for EAE induction by assisting the expansion of Th17 cells. Many chronic inflammatory diseases previously thought to be associated with Th1 have therefore been reclassified as Th17 diseases [44]. The opposing roles of Th2 and Th17 responses are now clear, since IL-4 strongly inhibits IL-17 differentiation. For Th1 and Th17 cells, a more balanced view is now accepted [45]. In both human and murine conditions, a large proportion of T cells can express simultaneously IFNγ and IL-17. This is clearly seen with T-cell clones from peripheral blood. The simultaneous production of the two cytokines appears uncommon, however, in inflammatory tissues where T cells producing cytokines take on a plasma cell-like appearance, possibly indicating full differentiation with a fixed phenotype [46].
In addition to the production of IL-17 (now referred to as IL-17A), Th17 cells can produce other cytokines - including IL-17F (a close member of the IL-17 family), IL-21, and IL-22. IL-21 acts as an endogenous amplifier of the Th17 lineage [41]. IL-22 appears more specifically associated with skin defense [47]. IL-17A and IL-17F share a large number of functions, with a strong correlation between the genes induced in RA synoviocytes by the two cytokines, IL-17F being less potent [48]. In addition, synergistic activities are seen when combining TNF with IL-17A or IL-17F. IL-17A and IL-17F may, however, have different roles in mouse models of inflammation and host defense [49].
IL-17E (also termed IL-25) is a very different member of the IL-17 family. IL-17E is more a Th2 cytokine, involved in allergic reactions and inhibiting the Th17 pathway [50]. Consequently, there is another balance between the effects of IL-17A and IL-17F and those of IL-17E/IL-25.
Balance between Th17 and T cells with regulatory function
Th1, Th2, and Th17 cells are effector cells contributing to key functions of the immune response. An additional heterogeneous subset of T cells with regulatory function (Tregs) has recently been identified. Some Tregs occur naturally, whereas others are induced in response to antigens. Characteristically, Tregs express the transcription factor Foxp3, as well as CD4 and CD25. The immunomodulating effects of Tregs are mediated by membrane molecules (for example, cytotoxic T-lymphocyte-associated protein 4, glucocorticoid-induced TNF receptor, and OX40) and by cytokines including IL-10 and TGFβ.
TGFβ is key to the induction of Foxp3-positive regulatory T cells. Indeed, mice defective in TFGβ die quickly from a massive uncontrolled inflammatory disease [51]. Contrasting with the effect of TGFβ alone, the simultaneous presence of TGFβ and IL-6 favors the emergence of Th17 cells alongside the inhibition of the Tregs [52]. IL-6 - a cytokine with pleiotropic inflammatory effects - therefore plays a pivotal part, at least in the mouse, in directing the differentiation of T cells toward the Th17 or Treg pathways. TNF, IL-1, and IL-17 interact together to induce massive amounts of IL-6. Increased inflammation therefore has a positive effect on the Th17 pathway and a negative effect on its regulation.
The inhibitory functions of IL-27 and IL-35
Some recently identified cytokines such as IL-27 and IL-35 appear to be more involved in dampening the immune response. IL-27 belongs to the IL-12 cytokine family that also comprises IL-23 and IL-35, all involved in the regulation of T-helper cell differentiation. IL-27 is unique in that it induces Th1 differentiation while simultaneously suppressing immune responses. The immunosuppressive effects of IL-27 depend on inhibition of the development of Th17 cells and induction of IL-10 production [53]. IL-27 exerts potent anti-inflammatory effects in several infectious and experimental autoimmune models. In particular, suppressive effects on helper T cells - which are implicated in the pathogenesis of multiple sclerosis - suggest that IL-27 may be therapeutically relevant in multiple sclerosis. While exciting discoveries have been made, however, these are still at an early stage and further studies are required to understand the pathophysiological roles of IL-27 and its therapeutic potential in humans [54].
The inhibitory cytokine IL-35 contributes to regulatory T-cell function, being specifically produced by Tregs and required for maximal suppressive activity [55]. Ectopic expression of IL-35 confers regulatory activity on naive T cells, whereas recombinant IL-35 suppresses T-cell proliferation. The role of Tregs in RA has been established in both patients and animal models. The Tregs increase in patients who are responding to anti-TNFα therapy. Of the current hypotheses, Treg expansion or transfer may hold promise for the treatment of RA [56].
Cytokines, hormones, vitamins, arachidonic acid metabolites and lipoproteins
A further layer of control at the level of expression of cytokines, cytokine inhibitors and acute-phase proteins is provided by hormones. Estrogens as well as androgens inhibit the production of IL-1β and TNFα by monocytes-macrophages. Androgens antagonize stimulatory effects of estrogens. Some studies suggest that estradiol is more inhibitory to Thl cytokines (for example, IFNγ, IL-2) while testosterone is inhibitory to Th2 cytokines (for example, IL-4). On the other hand, cytokines control the hypothalamic-hypophyseal-adrenal gland axis as well as the sex hormones [57]. Vitamins may also affect cytokine production by influencing the polarization of effector CD4+ T cells. For instance, retinoic acid enhances Treg expansion while simultaneously inhibiting Th17 cells [58]. Conversely, vitamin D favors Th2 polarization and diverts Tregs from their regulatory function [59,60]. Finally, prostaglandin E2 - a metabolite of arachidonic acid - may also affect cytokine production by favoring the expansion of Th17 cells [61].
Destruction/repair balance
Chronic inflammatory diseases such as RA are so severe because the disease process affects matrix metabolism. Although RA is seen as a destructive disease, it is not well appreciated that the main problem is in fact the inhibition of repair activity. Any type of chronic joint inflammation, whether infectious, inflammatory, or autoimmune, will result in joint destruction within months or, at best, within a few years, but it will take decades to observe some kind of joint repair - even in conditions like osteoarthritis where repair activity is maintained. In a model of cell interaction between synoviocytes and T-cell clones, it was found that Th1 and Th17 clones induced defects in collagen synthesis in vitro, indicating an inhibition of their repair activity (Figure 1). In sharp contrast, Th2 cells induce collagen synthesis, indicating their beneficial role in repair activity [62]. Very similar conclusions were obtained when monocytes were incubated with Th1 or Th2 clones. The interaction with a Th1 clone led to the production of IL-1, a key marker of destructive inflammation, whereas the use of a Th2 clone led to production of IL-1Ra along with its anti-inflammatory and anti-destructive properties [63].
Wingless integration site (Wnt) proteins make up a family of secreted growth factors, identified in virtually every organism; they regulate key aspects of cellular functions such as growth, differentiation, and death. Several members of the Wnt pathway play an important part in bone remodeling. Dickkopf-1, a soluble inhibitor of the Wnt pathway, controls bone remodeling. Increased Dickkopf-1 levels are linked to bone resorption, and decreased levels are linked to new bone formation. Low-density lipoprotein receptor-related protein 5, the main receptor that mediates Wnt signaling, plays a critical role in bone mass regulation. Gain-of-function mutations of lipoprotein receptor-related protein 5 cause high bone mass phenotypes, whereas loss-of-function mutations are linked to severe osteoporosis [64].
Adipose tissue in inflammation: a protective role via IL-1 receptor antagonist?
Adipokines are beginning to emerge as mediators of inflammation. Knowledge of their precise activities remains in its infancy, however, and is still controversial [65]. Many of the adipokines appear to have proinflammatory properties. In general, adiponectin is considered anti-inflammatory, and leptin, vistatin and resistin are considered proinflammatory. The formation of adipose tissue could be due to abnormal metabolic processes and, at the local level, due to chronic inflammatory processes such as those occurring in the synovium in RA or osteoarthritis, or in the peritoneal cavity in various inflammatory processes of the digestive system.
Adipocytes are said to produce many hormones and pro-inflammatory mediators. White adipose tissue in humans, however, is assumed to be the main source of IL-1Ra, and also contains IL-10. Furthermore, IFNβ was found to be the principal cytokine inducing IL-1Ra in various white adipose tissues, such as that present in the synovium. It is possible that, in addition to other functions, adipose tissue may be part of a mechanism limiting local inflammation and that fibroblasts in the vicinity may further induce IL-1Ra in adipocytes via the production of IFNβ [66].
Influence of signal transduction in cytokine balance
Cytokines may have opposing effects on the same cell depending on the circumstances in which they hit their target. The timing and the previous activation status are major determinants of responses that cytokines elicit (Figure 4). Differential outcomes could be sensitization or amplification of proinflammatory signals (that is, priming), reprogramming of signaling resulting in proinflammatory activity of pleiotropic or anti-inflammatory cytokines, and attenuation of anti-inflammatory signals and homeostatic mechanisms. Signal transducer and activator of transcription (STAT) 1 has been shown in vitro and in vivo to be involved in some of these effects. For instance, transient exposure to subactivating concentrations of IFNα or IL-6 primes primary human monocytes for subsequent exposure to IFNγ, resulting in enhanced interferon regulatory factor 1 and indoleamine-2,3-dioxygenase gene expression in a STAT-1-dependent manner [67,68]. This may explain robust IFN signatures in RA synovium, notwithstanding very low amounts of IFNγ. Enhanced expression of STAT-1-dependent genes upon IFNγ priming of monocytes is a finely tuned process involving Fcγ receptor/DNAX activation protein 12, as demonstrated in Fcγ receptor/DNAX activation protein 12-/- mice in which the priming effect is lost.
Figure 4.

Schematic examples of cytokine signal modulation. (a) Priming: upon exposure to suboptimal levels of type I interferon or IL-6, no signal is generated; but if later the cell (macrophage) sees suboptimal levels of IFNγ, then gene transcription initiates and a signal is generated [67,68]. IDO, indoleamine-2,3-dioxygenase; IFNAR, interferon alpha receptor IL-6Ra, IL-6 receptor alpha; IRF1, interferon regulatory factor 1; STAT, signal transducer and activator of transcription. (b) Uncoupling of signaling: monocytes chemotactic protein-1 (MCP-1)/CCL2 signal upon CCR2 binding. In the presence of IL-10, binding of MCP-1/CCL2 to CCR2 is preserved but signal is abolished [33]. IL-10R, IL-10 receptor. (c) Reprogramming of signaling: in macrophages, Toll-like receptor (TLR) 2 activation induces TNF, production of which is reduced by simultaneously induced homeostatic IL-10 (negative feedback). If the cell has been primed with type I interferon, however, then IL-10 fails to negatively regulate TLR signaling. In turn, IL-10 becomes a proinflammatory cytokine favoring the production of TNF and other cytokines. The signaling cascade induced by IL-10 shifts form anti-inflammatory STAT 3 to proinflammatory STAT 1 [70]. Figures in circles indicate sequences of events. AP-1, activator protein 1.
IL-10 contributes to homeostatic responses in proinflammatory conditions. For instance, in human monocytes, Toll-like receptor 2 ligation results in NFκB-dependent TNF production and simultaneously in activator protein-1-dependent IL-10 production [69]. Upon binding to its receptor, IL-10 decreases TNF production in a STAT-3-dependent manner, thus exerting a negative feedback. Pre-exposure of monocytes to IFNα, however, results in IL-10 gaining pro-inflammatory functions. Of interest, this process is STAT 1 dependent. It has therefore been shown in human monocytes primed with IFNα that IL-10 not only fails to reduce the subsequent production of TNF in response to lipopolysaccharide, which may simply indicate a loss of function of the anti-inflammatory activity of IL-10, but in addition primes monocytes to transcribe genes in response to IL-10 usually induced by IFN. It appears that, due to the effect of type I interferons, the balance of IL-10 signaling shifts from STAT 3 (anti-inflammatory) to STAT 1 (proinflammatory) signals. Furthermore, IL-10 induces chemokine production in IFNα-primed macrophages, resulting in recruitment of activated T cells; aberrant IL-10 signaling may therefore contribute to inflammation in conditions with high interferon levels (systemic lupus erythematosus) [70].
The suppressors of cytokine signaling (SOCS) family of intracellular proteins - which encompasses eight members, sharing a central Src homology domain 2 and a C-terminus SOCS box - act as negative regulators of intracellular signaling of the Jak-STAT pathway used by several cytokines. They act by inhibiting the kinase activity, by competing with substrates needed for signal transduction, and by targeting associated proteins to proteasome degradation. Beside negative regulation, SOCS proteins can also affect the quality of signaling. For instance, in the absence of SOCS 3, IL-6 induces a wider transcriptional response, which includes interferon-like gene expression owing to increased STAT 1 phosphorylation. SOCS proteins therefore impact on a number of important mechanisms regulating inflammation and the immune response [71].
Conclusion
Cytokine activities affect most, if not all, biological processes involved in homeostasis as well as in host defense and auto-aggression. A continuous, finely tuned, crosstalk between cytokines, receptors, agonist and antagonist ligands, as well as with mediators belonging to other families of molecules, regulates cytokine biological activities. Furthermore, the context in which cytokines are available, including the temporal sequence of events preceding the availability of a given cytokine, very much impact on their capacity to favor or inhibit inflammation and other biological processes. During the past three decades we have learned that an imbalance in cytokine activities is associated with autoimmune and autoinflammatory disorders. More important, our knowledge of the many levels of cytokine balance has led to the generation of important tools to control inflammatory and destructive diseases. The future will no doubt witness additional major achievements in this area of medicine.
Abbreviations
CCR: CC-family chemokine receptor; DARC: Duffy antigen receptor for chemokines; EAE: experimental allergic encephalomyelitis; Foxp3: forkhead box p3; IFN: interferon; IL: interleukin; IL-1R: IL-1 receptor; IL-6Rα: IL-6 receptor alpha; IL-1Ra: IL-1 receptor antagonist; NF: nuclear factor; RA: rheumatoid arthritis; RANTES: regulated on activation, normal T-cell expressed and secreted; SIGIRR: single immunoglobulin IL-1-related receptor; sIL-6Rα: soluble IL-6Rα; SOCS: suppressors of cytokine signaling; STAT: signal transducer and activator of transcription; TGFβ: transforming growth factor beta; Th: T-helper type; TNF: tumor necrosis factor; Treg: T cell with regulatory function; Wnt: wingless integration site.
Competing interests
The authors declare that they have no competing interests.
Note
The Scientific Basis of Rheumatology: A Decade of Progress
This article is part of a special collection of reviews, The Scientific Basis of Rheumatology: A Decade of Progress, published to mark Arthritis Research & Therapy's 10th anniversary.
Other articles in this series can be found at: http://arthritis-research.com/sbr
Contributor Information
Carlo Chizzolini, Email: carlo.chizzolini@unige.ch.
Jean-Michel Dayer, Email: jean-michel.dayer@unige.ch.
Pierre Miossec, Email: miossec@univ-lyon1.fr.
Acknowledgements
The field of cytokine balance is very large and imprecisely defined. The authors would like to apologize to the many authors having contributed to this fascinating field whose work has not been quoted in the present review. CC was supported in part by grant No 31003A_124941/1 from the Swiss National Science Foundation.
References
- Arend WP, Dayer JM. Cytokines and cytokine inhibitors or antagonists in rheumatoid arthritis. Arthritis Rheum. 1990;33:305–315. doi: 10.1002/art.1780330302. [DOI] [PubMed] [Google Scholar]
- O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–18. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- Garlanda C, Riva F, Polentarutti N, Buracchi C, Sironi M, De Bortoli M, Muzio M, Bergottini R, Scanziani E, Vecchi A, Hirsch E, Mantovani A. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc Natl Acad Sci USA. 2004;101:3522–3526. doi: 10.1073/pnas.0308680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lech M, Kulkarni OP, Pfeiffer S, Savarese E, Krug A, Garlanda C, Mantovani A, Anders HJ. Tir8/Sigirr prevents murine lupus by suppressing the immunostimulatory effects of lupus autoantigens. J Exp Med. 2008;205:1879–1888. doi: 10.1084/jem.20072646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Sergeev P, Ehses JA, Mathe Z, Bosco D, Berney T, Dayer JM, Reinecke M, Halban PA, Donath MY. Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1β in human islets. Proc Natl Acad Sci USA. 2004;101:8138–8143. doi: 10.1073/pnas.0305683101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnarfi N, Gruaz L, Dayer JM, Burger D. Opposite regulation of IL-1β and secreted IL-1 receptor antagonist production by phosphatidylinositide-3 kinases in human monocytes activated by lipopolysaccharides or contact with T cells. J Immunol. 2007;178:446–454. doi: 10.4049/jimmunol.178.1.446. [DOI] [PubMed] [Google Scholar]
- Molnarfi N, Brandt KJ, Gruaz L, Dayer JM, Burger D. Differential regulation of cytokine production by PI3Kδ in human monocytes upon acute and chronic inflammatory conditions. Mol Immunol. 2008;45:3419–3427. doi: 10.1016/j.molimm.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Burger D, Molnarfi N, Weber MS, Brandt KJ, Benkhoucha M, Gruaz L, Chofflon M, Zamvil SS, Lalive PH. Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1β in human monocytes and multiple sclerosis. Proc Natl Acad Sci USA. 2009;106:4355–4359. doi: 10.1073/pnas.0812183106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–196. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Maini SR. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol Rev. 2008;223:7–19. doi: 10.1111/j.1600-065X.2008.00626.x. [DOI] [PubMed] [Google Scholar]
- Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–3545. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6: from basic science to medicine - 40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Aletaha D, Koeller M, Weisman MH, Emery P. New therapies for treatment of rheumatoid arthritis. Lancet. 2007;370:1861–1874. doi: 10.1016/S0140-6736(07)60784-3. [DOI] [PubMed] [Google Scholar]
- Seckinger P, Isaaz S, Dayer JM. Purification and biologic characterization of a specific tumor necrosis factor alpha inhibitor. J Biol Chem. 1989;264:11966–11973. [PubMed] [Google Scholar]
- Peters M, Jacobs S, Ehlers M, Vollmer P, Mullberg J, Wolf E, Brem G, Meyer zum Buschenfelde KH, Rose-John S. The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J Exp Med. 1996;183:1399–1406. doi: 10.1084/jem.183.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knupfer H, Preiss R. sIL-6R: more than an agonist? Immunol Cell Biol. 2008;86:87–91. doi: 10.1038/sj.icb.7100113. [DOI] [PubMed] [Google Scholar]
- Muller-Newen G, Kuster A, Hemmann U, Keul R, Horsten U, Martens A, Graeve L, Wijdenes J, Heinrich PC. Soluble IL-6 receptor potentiates the antagonistic activity of soluble gp130 on IL-6 responses. J Immunol. 1998;161:6347–6355. [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/S0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, Goddard AD, Godowski P, Ashkenazi A. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–821. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- Mehrad B, Keane MP, Strieter RM. Chemokines as mediators of angiogenesis. Thromb Haemost. 2007;97:755–762. [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Bonecchi R, Locati M. Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat Rev Immunol. 2006;6:907–918. doi: 10.1038/nri1964. [DOI] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Peiper SC, Wang ZX, Neote K, Martin AW, Showell HJ, Conklyn MJ, Ogborne K, Hadley TJ, Lu ZH, Hesselgesser J, Horuk R. The Duffy antigen/receptor for chemokines (DARC) is expressed in endothelial cells of Duffy negative individuals who lack the erythrocyte receptor. J Exp Med. 1995;181:1311–1317. doi: 10.1084/jem.181.4.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner L, Patterson AM, Ashton BA, Stone MA, Middleton J. The human Duffy antigen binds selected inflammatory but not homeostatic chemokines. Biochem Biophys Res Commun. 2004;321:306–312. doi: 10.1016/j.bbrc.2004.06.146. [DOI] [PubMed] [Google Scholar]
- Du J, Luan J, Liu H, Daniel TO, Peiper S, Chen TS, Yu Y, Horton LW, Nanney LB, Strieter RM, Richmond A. Potential role for Duffy antigen chemokine-binding protein in angiogenesis and maintenance of homeostasis in response to stress. J Leukoc Biol. 2002;71:141–153. [PMC free article] [PubMed] [Google Scholar]
- Addison CL, Belperio JA, Burdick MD, Strieter RM. Overexpression of the duffy antigen receptor for chemokines (DARC) by NSCLC tumor cells results in increased tumor necrosis. BMC Cancer. 2004;4:28. doi: 10.1186/1471-2407-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibbs RJ, Kriehuber E, Ponath PD, Parent D, Qin S, Campbell JD, Henderson A, Kerjaschki D, Maurer D, Graham GJ, Rot A. The beta-chemokine receptor D6 is expressed by lymphatic endothelium and a subset of vascular tumors. Am J Pathol. 2001;158:867–877. doi: 10.1016/s0002-9440(10)64035-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson T, Cook DN, Nibbs RJ, Rot A, Nixon C, McLean P, Alcami A, Lira SA, Wiekowski M, Graham GJ. The chemokine receptor D6 limits the inflammatory response in vivo. Nat Immunol. 2005;6:403–411. doi: 10.1038/ni1182. [DOI] [PubMed] [Google Scholar]
- Martinez de la Torre Y, Locati M, Buracchi C, Dupor J, Cook DN, Bonecchi R, Nebuloni M, Rukavina D, Vago L, Vecchi A, Lira SA, Mantovani A. Increased inflammation in mice deficient for the chemokine decoy receptor D6. Eur J Immunol. 2005;35:1342–1346. doi: 10.1002/eji.200526114. [DOI] [PubMed] [Google Scholar]
- Martinez de la Torre Y, Buracchi C, Borroni EM, Dupor J, Bonecchi R, Nebuloni M, Pasqualini F, Doni A, Lauri E, Agostinis C, Bulla R, Cook DN, Haribabu B, Meroni P, Rukavina D, Vago L, Tedesco F, Vecchi A, Lira SA, Locati M, Mantovani A. Protection against inflammation- and autoantibody-caused fetal loss by the chemokine decoy receptor D6. Proc Natl Acad Sci USA. 2007;104:2319–2324. doi: 10.1073/pnas.0607514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibbs RJ, Gilchrist DS, King V, Ferra A, Forrow S, Hunter KD, Graham GJ. The atypical chemokine receptor D6 suppresses the development of chemically induced skin tumors. J Clin Invest. 2007;117:1884–1892. doi: 10.1172/JCI30068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel K, Benz C, Bleul CC. A silent chemokine receptor regulates steady-state leukocyte homing in vivo. Proc Natl Acad Sci USA. 2007;104:8421–8426. doi: 10.1073/pnas.0608274104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amico G, Frascaroli G, Bianchi G, Transidico P, Doni A, Vecchi A, Sozzani S, Allavena P, Mantovani A. Uncoupling of inflammatory chemokine receptors by IL-10: generation of functional decoys. Nat Immunol. 2000;1:387–391. doi: 10.1038/80819. [DOI] [PubMed] [Google Scholar]
- Cash JL, Hart R, Russ A, Dixon JP, Colledge WH, Doran J, Hendrick AG, Carlton MB, Greaves DR. Synthetic chemerin-derived peptides suppress inflammation through ChemR23. J Exp Med. 2008;205:767–775. doi: 10.1084/jem.20071601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel BA, Nakae S, Zuniga L, Kim JY, Ohyama T, Alt C, Pan J, Suto H, Soler D, Allen SJ, Handel TM, Song CH, Galli SJ, Butcher EC. Mast cell-expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J Exp Med. 2008;205:2207–2220. doi: 10.1084/jem.20080300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann TR, Schumacher JH, Street NF, Budd R, O'Garra A, Fong TA, Bond MW, Moore KW, Sher A, Fiorentino DF. Diversity of cytokine synthesis and function of mouse CD4+ T cells. Immunol Rev. 1991;123:209–229. doi: 10.1111/j.1600-065X.1991.tb00612.x. [DOI] [PubMed] [Google Scholar]
- Romagnani S. Human TH1 and TH2 subsets: doubt no more. Immunol Today. 1991;12:256–257. doi: 10.1016/0167-5699(91)90120-I. [DOI] [PubMed] [Google Scholar]
- Miossec P, Berg W van den. Th1/Th2 cytokine balance in arthritis. Arthritis Rheum. 1997;40:2105–2115. doi: 10.1002/art.1780401203. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Joosten LA, Chabaud M, Bersselaar L van Den, Oppers B, Coenen-De Roo CJ, Richards CD, Miossec P, Berg WB van Den. IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J Clin Invest. 2000;105:1697–1710. doi: 10.1172/JCI7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol. 2007;19:652–657. doi: 10.1016/j.coi.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarvak T, Chabaud M, Miossec P, Natvig JB. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. 1999;162:1246–1251. [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Miossec P. Diseases that may benefit from manipulating the Th17 pathway. Eur J Immunol. 2009;39:667–669. doi: 10.1002/eji.200839088. [DOI] [PubMed] [Google Scholar]
- Steinman L. A rush to judgment on Th17. J Exp Med. 2008;205:1517–1522. doi: 10.1084/jem.20072066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page G, Sattler A, Kersten S, Thiel A, Radbruch A, Miossec P. Plasma cell-like morphology of Th1-cytokine-producing cells associated with the loss of CD3 expression. Am J Pathol. 2004;164:409–417. doi: 10.1016/S0002-9440(10)63131-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- Zrioual S, Ecochard R, Tournadre A, Lenief V, Cazalis MA, Miossec P. Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. J Immunol. 2009;182:3112–3120. doi: 10.4049/jimmunol.0801967. [DOI] [PubMed] [Google Scholar]
- Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, Hippe A, Corrigan CJ, Dong C, Homey B, Yao Z, Ying S, Huston DP, Liu YJ. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med. 2007;204:1837–1847. doi: 10.1084/jem.20070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Yoshiyuki M. Regulation of immune responses by interleukin-27. Immunol Rev. 2008;226:234–247. doi: 10.1111/j.1600-065X.2008.00710.x. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DC, Rostami A. Therapeutic potential of IL-27 in multiple sclerosis? Expert Opin Biol Ther. 2009;9:149–160. doi: 10.1517/14712590802646936. [DOI] [PubMed] [Google Scholar]
- Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- Boissier MC, Assier E, Biton J, Denys A, Falgarone G, Bessis N. Regulatory T cells (Treg) in rheumatoid arthritis. Joint Bone Spine. 2009;76:10–14. doi: 10.1016/j.jbspin.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Burger D, Dayer JM. Cytokines, acute-phase proteins, and hormones: IL-1 and TNF-alpha production in contact-mediated activation of monocytes by T lymphocytes. Ann N Y Acad Sci. 2002;966:464–473. doi: 10.1111/j.1749-6632.2002.tb04248.x. [DOI] [PubMed] [Google Scholar]
- Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-β-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol. 2008;181:2277–2284. doi: 10.4049/jimmunol.181.4.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O'Garra A. 1α,25-Dihydroxyvitamin has a direct effect on naive D3 CD4(+) T cells to enhance the development of Th2 cells. J Immunol. 2001;167:4974–4980. doi: 10.4049/jimmunol.167.9.4974. [DOI] [PubMed] [Google Scholar]
- Urry Z, Xystrakis E, Richards DF, McDonald J, Sattar Z, Cousins DJ, Corrigan CJ, Hickman E, Brown Z, Hawrylowicz CM. Ligation of TLR9 induced on human IL-10-secreting Tregs by 1α,25-dihydroxyvitamin D3 abrogates regulatory function. J Clin Invest. 2009;119:387–398. doi: 10.1172/JCI32354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizzolini C, Chicheportiche R, Alvarez M, de Rham C, Roux-Lombard P, Ferrari-Lacraz S, Dayer JM. Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood. 2008;112:3696–3703. doi: 10.1182/blood-2008-05-155408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabaud M, Aarvak T, Garnero P, Natvig JB, Miossec P. Potential contribution of IL-17-producing Th(1)cells to defective repair activity in joint inflammation: partial correction with Th(2)-promoting conditions. Cytokine. 2001;13:113–118. doi: 10.1006/cyto.2000.0811. [DOI] [PubMed] [Google Scholar]
- Chizzolini C, Chicheportiche R, Burger D, Dayer JM. Human Th1 cells preferentially induce interleukin (IL)-1α while Th2 cells induce IL-1 receptor antagonist production upon cell/cell contact with monocytes. Eur J Immunol. 1997;27:171–177. doi: 10.1002/eji.1830270125. [DOI] [PubMed] [Google Scholar]
- Goldring SR, Goldring MB. Eating bone or adding it: the Wnt pathway decides. Nat Med. 2007;13:133–134. doi: 10.1038/nm0207-133. [DOI] [PubMed] [Google Scholar]
- Fantuzzi G. Adiponectin and inflammation: consensus and controversy. J Allergy Clin Immunol. 2008;121:326–330. doi: 10.1016/j.jaci.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Dayer JM, Chicheportiche R, Juge-Aubry C, Meier C. Adipose tissue has anti-inflammatory properties: focus on IL-1 receptor antagonist (IL-1Ra) Ann N Y Acad Sci. 2006;1069:444–453. doi: 10.1196/annals.1351.043. [DOI] [PubMed] [Google Scholar]
- Hu X, Herrero C, Li WP, Antoniv TT, Falck-Pedersen E, Koch AE, Woods JM, Haines GK, Ivashkiv LB. Sensitization of IFN-γ Jak-STAT signaling during macrophage activation. Nat Immunol. 2002;3:859–866. doi: 10.1038/ni828. [DOI] [PubMed] [Google Scholar]
- Tassiulas I, Hu X, Ho H, Kashyap Y, Paik P, Hu Y, Lowell CA, Ivashkiv LB. Amplification of IFN-α-induced STAT1 activation and inflammatory function by Syk and ITAM-containing adaptors. Nat Immunol. 2004;5:1181–1189. doi: 10.1038/ni1126. [DOI] [PubMed] [Google Scholar]
- Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Sharif MN, Tassiulas I, Hu Y, Mecklenbrauker I, Tarakhovsky A, Ivashkiv LB. IFN-α priming results in a gain of proinflammatory function by IL-10: implications for systemic lupus erythematosus pathogenesis. J Immunol. 2004;172:6476–6481. doi: 10.4049/jimmunol.172.10.6476. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]