

Abstract
Rheumatoid arthritis (RA) is recognized to be an autoimmune disease that causes preclinical systemic abnormalities and eventually leads to synovial inflammation and destruction of the joint architecture. Recently identified genetic risk factors and novel insights from animal models of spontaneous arthritis have lent support to the concept that thymic selection of an autoreactive T-cell repertoire is an important risk factor for this disease. With advancing age, defects in the homeostatic control of the T-cell pool and in the setting of signaling thresholds lead to the accumulation of pro-inflammatory T-effector cell populations and loss of tolerance to neo-antigens, such as citrullinated peptides. As the breakdown of tolerance to modified self-antigens can precede synovitis by decades, repair of homeostatic defects may open a unique window of opportunity for preventive interventions in RA. The end result of RA, destruction of cartilage and bone, appears to be driven by cytokine- and cell contact-induced activation of synoviocytes and monocytic cells, some of which differentiate into tissue-destructive osteoclasts. Targeting mediators involved in this process has greatly improved the management of this chronic inflammatory syndrome.
Introduction
Understanding of the chronic inflammatory disease rheumatoid arthritis (RA) has evolved considerably during the past decade. Introduction of novel therapeutic strategies has had a major impact not only on how we treat affected patients but also on how we conceptualize the disease process [1]. RA has served as a model to enhance our knowledge of the pivotal role played by cytokines during the effector stages of human disease; has been instrumental in clarifying the place of cytokines in the maintenance and chronicity of inflammation; and has been instrumental in deciphering the involvement of cytokine networks in tissue damage [2,3].
This enormous progress was made possible by the introduction of cytokine-directed therapies, the prototype of which is the neutralization of tumor necrosis factor (TNF)-α activity [4]. Inhibition of IL-6, another apparently effective treatment, is entering clinical application [5], and additional cytokine inhibitors are currently in clinical studies [6]. The availability of this therapeutic armamentarium has fundamentally changed the management of RA and has re-emphasized the primarily inflammatory character of this autoimmune syndrome. In support of the concept that cytokine-driven inflammation and not uncontrolled proliferation of synoviocytes is the primary disease process, inflammatory markers have emerged as the best predictors of clinical outcome [1].
As much as we have learned about the cytokines that are involved in the disease process and can be therapeutically targeted, our understanding of the upstream mechanisms that eventually lead to a destructive inflammatory reaction has received less attention. However, there is agreement within the scientific community that changing RA from a manageable into a curable disease entity will eventually require identification of etiologic factors and initiating pathways. RA is not a prototypic autoimmune disease such as type 1 diabetes mellitus or autoimmune thyroid disease, in which a failure in tolerance to a tissue-specific antigen results in selective and organ-destructive immune responses. Although the synovial inflammation is clinically prominent, the disease is systemic at all stages. The two most characteristic auto-antibodies, rheumatoid factor and antibodies to citrullinated peptides, are directed at common antigens widely expressed outside of the joint; their presence can precede synovial inflammation by decades [7,8]. Systemic complications manifest themselves as rheumatoid nodules, rheumatoid vasculitis, Felty's syndrome, or interstitial lung disease.
Interestingly, major organ manifestations of RA have become less frequent in clinical practice [9]. This decline in incidence started in the 1980s, before aggressive treatment of RA was introduced and the advent of biologics, suggesting that not only treatment but also changes in lifestyle and environment influence the clinical pattern of RA. As we move from successful palliative management to the goal of curative and preventive interventions, it is important to understand the mechanisms that initiate the disease and to identify the endogenous and environmental determinants that cause pathology upstream of synovial inflammation.
Clues to RA pathogenesis
Genetic risk factors in humans
Genetic factors have a substantial influence on determining the susceptibility to develop RA. Twin studies have demonstrated a fourfold higher concordance rate in monozygotic (15%) than in dizygotic (3.6%) twins [10]. The risk in siblings of patients compared with that in a 'normal' population has been estimated at between two- and 17-fold greater [11]. It is now clear that the relative risk for each genetic polymorphism is rather minor, making it unlikely that individual genetic polymorphisms will gain value in RA diagnosis or in identifying healthy individuals at risk. Also, preliminary studies, mostly of anti-TNF-treated patients, have indicated that large cohorts will be necessary to identify genetic polymorphisms that correlate with treatment response and that predictive power in individual cases will be small [12]. The primary promise of identifying disease-associated genes lies in the potential to define pathways that are important in disease pathogenesis. Recent advances made in linkage and in genome-wide association studies and the availability of large RA cohorts have allowed several novel risk genes to be identified. Although none of them was an obvious candidate gene, it is of interest to note that all of the confirmed disease-associated genes represent genes that are involved in immune responses, again emphasizing the immune pathogenesis of the disease.
The only genetic region that has emerged in linkage and in genome-wide association studies in all ethnic groups is the major histocompatibility complex (MHC) region [13]. The strength of the association varies significantly, depending upon the ethnic group [14], but the shared epitope hypothesis - first formulated during the 1980s [15] - has held up. Human leukocyte antigen (HLA)-DRB1 alleles expressing the amino acid sequence stretch Q/R-K/R-R-A-A at positions 70 to 74 are the major risk factor within the MHC region in individuals of diverse ethnic origin; for example, HLA-DRB1*0101, *0401, and *0404 in individuals of European ancestry or *0405 and *0901 in Asians. In addition to disease-associated alleles, a disease protective HLA-DRB1 polymorphism (DERAA) may exist; however, this notion of an active protective mechanism versus the absence of a disease risk gene is difficult to ascertain. HLA alleles appear to be more closely associated with the presence of antibodies to IgG Fc or to citrullinated peptides than with RA itself [16,17], suggesting that the polymorphisms primarily predispose to autoantibody production and that seronegative RA is fundamentally different from seropositive RA. Only DRB1*0401 and *0405 carry relative risks greater than 3; all other epitope-positive alleles contribute only a minor risk. Overall, it has been estimated that HLA polymorphisms account for 30% to 50% of the genetic load [18].
All other disease risk genes identified so far confer relative risks of about 1.3 to 1.5. Although these disease risk genes have been confirmed in independent studies, their association is not universal but occurs only within the context of particular ethnic backgrounds. A polymorphism within the PTPN22 gene has been unequivocally associated with RA in several studies in Canada, Europe, and the USA [19-21]. The polymorphism is responsible for an amino acid exchange from an arginine to a tryptophan within the coding region of the gene. This polymorphism represents a minor allele that is infrequent in healthy control individuals as well as in the RA population (8.7% versus 14.4%) [22]. A disease association in the Japanese population has not been found [23]; in fact, the polymorphism does not exist in Asians [24]. The PTPN22 protein is a tyrosine phosphatase that exerts negative feedback regulation in T-cell receptor (TCR) signaling [25]. The phosphatase binds to the regulatory kinase Csk; the complex of PTPN22 and Csk is responsible for terminating TCR signaling by phosphorylating Lck at position 505 and dephosphorylating Lck at position 394. The genetic polymorphism acts by directly modifying the phosphatase activity of PTPN22 and/or controlling its binding to Csk [26].
Surprisingly, studies have shown that the polymorphism is a gain-of-function mutation [27] (the carriers of the polymorphism are more likely to terminate TCR signaling), which is counterintuitive as a risk factor for an autoimmune disease. It has therefore been proposed that the underlying mechanism does not involve signaling of peripheral T cells, but that the signaling defect impairs negative thymic selection, resulting in the selection of an autoreactive repertoire. In this model, a defect in central tolerance sets the stage for the eventual development of a chronic inflammatory disease. This model not only applies to RA but also to a number of autoimmune syndromes, including type 1 diabetes mellitus, systemic lupus erythematosus, juvenile idiopathic arthritis, Graves' disease and vitiligo, each of which has been found to be associated with the PTPN22 polymorphism [28].
A genetic polymorphism of peptidylarginine deiminase 4 (PADI4) is important in the Asian population [29-31]. This polymorphism could very well play a role in the citrullination of proteins and therefore influence the development of antibodies to citrullinated antigens, which are among the auto-immune hallmarks of RA. Although this polymorphism also exists in Caucasian populations, an association with RA could not be demonstrated [32-35]. Because antibodies to citrullinated antigens are a general phenomenon in RA, independent of ethnicity, the meaning of this discrepancy is currently unclear.
Three additional risk regions have been identified during the past year. All three of these genetic regions have in common that they confer a 50% risk increase and represent a single nucleotide polymorphism (SNP) close to an immune response gene. The functional implications of these disease risk regions are unclear, and it is therefore premature to develop pathogenetic models. Linkage studies and subsequent SNP mapping identified a region on chromosome 1q in the third intron of the STAT4 gene [36]. The association originally identified in a North American study was confirmed in a Swedish and in a Korean cohort [37]. An influence of the polymorphism on STAT4 transcription or function could have implications for signal calibration of a number of cytokine receptors, including type I IFN, IL-12, and IL-23. Whole-genome association studies identified two additional regions, one on chromosome 6q23 and one on chromosome 9q33-34. One SNP on chromosome 6q23 is between the genes encoding oligodendrocyte lineage transcription factor 3 and TNF-α-induced protein 3 [38,39]. TNF-α-induced protein 3, if confirmed to be the relevant variant, would be of interest because it functions as a negative regulator of nuclear factor-κB activation in response to Toll-like receptors, and mice deficient for TNF-α-induced protein 3 develop an auto-inflammatory syndrome [40-42]. The second region, on chromosome 9q33-34, was confirmed in independent candidate gene studies and maps between the complement 5 gene and the TNF receptor-associated factor 1 [43-45]. The latter functions as a signaling molecule of receptors of the TNF receptor superfamily, including type 2 TNF receptor and CD40 ligand. Again, it remains to be determined whether functional polymorphisms can be identified. CD40 has also been identified as a disease-associated gene [46].
The common theme that emerges from these genetic linkage and association studies is the possible involvement of signaling pathways transmitting activation signals into cells of the immune system (Figure 1). The major genetic risk factor continues to be shared epitope-expressing HLA-DRB1 alleles, which function in triggering the TCR. The minor genetic risk factors identified thus far are mostly related to signal calibrations, either to antigen recognition by TCRs or B-cell receptors, or in response to certain cytokines. The genetic polymorphisms are neither necessary nor sufficient for disease development because they are too infrequent and the associated risk is low; however, they indicate that these pathways are of importance in rendering an individual susceptible to RA development.
Figure 1.
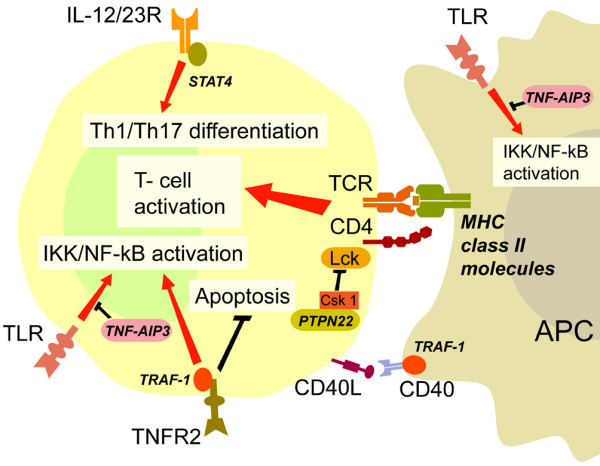
Shown is a T cell-APC interaction to illustrate biologic pathways involving rheumatoid arthritis-associated genes (indicated in italics). APC, antigen-presenting cell; IKK, IκB kinase; MHC, major histocompatibility complex; NF-κB, nuclear factor-κB; TCR, T-cell receptor; Th, T-helper; TLR, Toll-like receptor; TNFR2, type 2 TNF receptor.
Mouse models of arthritis
Several mouse models with arthritis of spontaneous onset have become available during the past decade. Earlier animal models were based on the premise that RA results from an adaptive immune response to a joint-specific antigen. Models such as collagen-induced or proteoglycan-induced arthritis have been very helpful in providing evidence for the paradigm that autoimmunity to joint-specific antigens can lead to arthritis [47,48]; these models have allowed investigators to study effector mechanisms in the arthritic process and to test therapeutic interventions. In contrast to spontaneously occurring arthritis models, models of induced arthritis are already built upon the notion that synovial inflammation is mediated by a response to a particular autoantigen, and therefore they do not permit study of upstream mechanisms. One of the first models that exhibited spontaneous onset of arthritis was the TNF-α transgenic mouse [49]. The finding that the over-production of TNF-α alone is sufficient to induce erosive arthritis emphasizes the sensitivity and response of synoviocytes to circulating cytokines, a concept that was first introduced by Feldman and Maini [4] and is now the basis for the treatment of human disease with anti-TNF inhibitors.
Four recently discovered mouse strains provide opportunities to decipher mechanisms upstream of synoviocyte activation. The spontaneous occurrence of arthritis in these models was unexpected, but all four models point toward T-cell repertoire selection as a critical determinant in initiating and sustaining arthritis (Figure 2). In the first model, Mathis and colleagues [50] crossed a TCR transgene onto the NOD background. This TCR transgene happened to recognize a ubiquitously expressed protein, namely glucose-6 phosphate isomerase, but thymic negative selection failed to purge this autoreactive receptor from the T-cell repertoire [51]. The mice, known as K/B × N mice, develop an early onset, rapidly progressive arthritis that is mediated by autoantibodies that bind glucose-6 phosphate isomerase. Arthritis can be transferred by antibodies, clearly demonstrating that the generation of a particular autoantibody due to defective thymic selection can induce disease. Unfortunately, autoantibodies to glucose-6 phosphate isomerase do not appear to play a role in RA, therefore limiting the applicability of this model beyond the notion that thymic selection may be important.
Figure 2.

Central and peripheral T-cell selection and differentiation as risk factors for synovial inflammation. HPC, hematopoietic progenitor cells; MHC, major histocompatibility complex; TCR, T-cell receptor.
Similar conclusions can be drawn from a second TCR transgene model. Caton and colleagues [52] engineered mice that expressed an influenza hemagglutinin antigen in combination with a transgene for hemagglutinin-reactive TCR. Different strains were constructed carrying TCR with varying affinity for the antigen [52,53]. Mice that expressed the low-affinity TCR failed negative selection and developed erosive arthritis, again illustrating the notion that inclusion of autoreactive TCR in the T-cell repertoire can eventually lead to synovial inflammation, mimicking the conditions in RA.
Whereas the investigator teams led by Mathis and Caton used TCR transgenic mice to study central tolerance mechanisms and unexpectedly observed RA-like disease, investigators at the laboratory of Hirano [54] engineered mice that lacked a negative feedback loop in gp130 signaling, creating conditions of unopposed cytokine signaling. gp130 is a necessary constituent of a class of cytokine receptors that bind IL-6, leukemia-inhibiting factor, oncostatin M, and IL-11. A single point mutation at position 759 of gp130 prevents recruitment of negative regulatory molecules, such as SHP-2 and SOCS-3, thus causing sustained signaling. Mice transgenic for this gp130 variant develop an erosive arthritis. Defective cytokine signal calibration as a risk factor for arthritis would be consistent with synovial fibroblasts being highly sensitive to cytokine action, similar to the TNF-hyper-producing mice. However, subsequent studies have shown that the pathogenesis in the gp130 mutant transgenic mice is dependent on T cells, because arthritis does not occur in RAG-deficient mice and includes polyclonal T-cell and B-cell stimulation with the production of rheumatoid factor and antinuclear antibodies. Subsequent studies of TCR transgenic mice expressing the gp130 mutant again described a defect in negative thymic selection.
A defect in thymic function has been postulated also to cause the arthritis in the SKG mouse model. SKG mice have a spontaneously incurred loss-of-function mutation in the Zap70 gene [55]. TCR signaling is therefore attenuated. Using appropriate TCR transgenic mouse systems, positive, as well as negative, selection in the thymus was found to be impaired. Both defects may contribute to the emergence of peripheral autoimmunity [56]. Defective negative selection would bias the TCR repertoire toward autoreactivity. Defective positive selection may cause lymphopenia, which has been shown to be a risk factor for autoimmunity [57,58]. Peripheral T cells in the SKG mouse continue to be hypo-responsive, but adoptive transfer of these T cells into T/B-cell-deficient mice reproduces joint inflammation, clearly demonstrating that the T cells are sufficient to transfer disease. Given their low responsiveness, there must be a strong peripheral stimulus to overcome peripheral tolerance. In support of this notion, mice maintained in germ-free conditions do not develop disease. In fact, fungal infection and the IL-6-mediated development of T-helper-17 response appear to be instrumental in disease development [56].
None of the genetic polymorphisms that cause disease in mice has been associated with RA. However, it is striking that all of these disease models involve TCR threshold calibration and thymic selection. Of the genes associated with RA, HLA-DRB1 and PTPN22 are also directly involved in TCR stimulation. In particular, the PTPN22 polymorphism attenuates TCR signaling and may be associated with defective negative selection.
The mouse models have the potential to improve our understanding of how misguided T-cell responses translate into synovial inflammation and the other organ manifestations in RA patients. In the K/B × N model, this transition is made by the induction of autoantibodies to a joint nonspecific antigen; the disease can be rapidly transferred by glucose-6 phosphate-specific autoantibodies. For the SKG and the gp130 mutant models, specific autoantigens have not been identified. Instead, these mice have a broadly autoreactive repertoire. Although the TCR signaling capacity is low, T cells develop into polyclonal effector T cells that mediate arthritis. Based on these animal models, Cope and colleagues [59,60] have postulated that a similar mechanism is functional in RA and that autoreactive T cells that are generally low reactive, but can be activated to develop into very potent effector cells, hold the pathogenetic key to RA. One factor that calibrates the TCR threshold in these T cells and enables their differentiation into effector T cells may be lymphopenia and compensatory homeostatic proliferation [61].
T-cell abnormalities in RA patients
In the majority of patients, RA occurs at an age when formation of the TCR repertoire has been concluded for many decades and thymic function is already severely reduced or has even completely ceased. Although possibly a predisposing factor, it is difficult to conceptualize how the process of central tolerance established early in life would only fail after many decades of disease-free survival. Rather, peripheral tolerance appears to be much more important in determining self/nonself distinction in a host older than 50 years (Figure 2).
The most remarkable finding in the T-cell compartment of RA patients is that the T cells exhibit a signature that is reminiscent of accelerated immune aging [62]. Of particular interest, this fingerprint of premature immune senescence is not limited to memory T cells but mostly affects antigen-inexperienced naïve T cells. One hallmark of immune aging is the loss of telomeric sequences. Telomeres are repeat sequences at the end of linear chromosomes that are being continuously shortened with each cycle of cellular division unless telomeric ends are replenished by telomerase. Telomeric sequences of proliferating cell populations decline with age; T cells, which are under explicit proliferative demand, are no exception to this rule. During adulthood telomeres in T cells shorten by 50 to 100 base pairs per year [63]. In patients with RA, telomeric erosion in T cells is premature; with a loss of about 1,500 kilobases, RA T cells resemble control T cells that are 20 years older [64]. Possible mechanisms include an increased replicative history and accumulated DNA damage arising from a defective DNA repair response in RA. Of interest, age-inappropriate loss of telomeric ends in RA is not limited to T cells, but also involves the myeloid lineage and hematopoietic precursor cells, suggesting a defect in the homeostasis of bone marrow-derived progenitor cells [65,66].
Recent studies have uncovered a defect in telomeric repair in RA T cells. Specifically, naïve T cells undergoing priming typically upregulate telomerase to repair the chromosomal ends. This induction of telomerase is blunted in RA T cells because of transcriptional repression of the human telomerase reverse transcriptase (hTERT) component of the enzyme telomerase [67]. hTERT deficiency renders T cells from RA patients more susceptible to apoptosis, identifying a broader role for this enzyme in regulating T-cell fate. Knockdown of hTERT in healthy T cells impaired survival rates. Restoring telomerase activity in RA T cells rescued such cells from excessive apoptosis. In essence, telomeres and the telomeric surveillance machinery emerge as critical regulators of T-cell death and life. Inappropriate culling of T cells during the priming process potentially aggravates a vicious cycle of increased cell death, lymphopenia, compensatory homeostatic cell proliferation, and cellular senescence. Controlling nuclear integrity now emerges as a novel theme in assessing cell fate decisions in T cells, cells that are basically programmed to undergo cycles of expansion and contraction, with some of them living for extended time periods.
A recent study has shed light on defects in DNA repair mechanisms in RA T cells, linking the accumulation of damaged DNA to deficiency in the ataxia telangiectasia mutated (ATM) surveillance and repair pathway. Again, the inability of RA T cells to effectively repair DNA breaks was associated with increased cell death, straining T-cell regenerative mechanisms [68]. In support of this interpretation, TCR excision circles (TRECs) containing T cells are reduced in RA patients [64]. TRECs are DNA episomes generated during TCR rearrangement [69]. High numbers of TREC-positive T cells therefore reflect thymic activity, whereas decreased numbers are indicative of T-cell loss that is not compensated for by thymic production of new T cells [70]. Telomeric erosion, increased susceptibility to cell death due to defective telomerase activity and DNA repair mechanisms, as well as peripheral loss of TREC-positive cells, are all consistent with a model in which RA patients have a history of lymphopenia and accelerated homeostatic proliferation [61].
Homeostatic proliferation of naïve CD4+ and CD8+ T cells is dependent on recognition of MHC class II and class I molecules, respectively, and will therefore eventually be associated with peripheral selection of a T-cell repertoire with high affinity to self [71]. In support of this interpretation, the diversity of the naïve TCR repertoire in patients with RA is contracted by a factor of about 10 [72]. Thus, in addition to defective central thymic selection, peripheral selection over the years could set the stage for an autoimmune disposition. This model would also fit with the observation that the best characterized autoimmune responses in patients with RA are directed at neoantigens. A pathognomonic autoantibody in RA patients is that directed against citrullinated peptides, which are generated mostly in matrix molecules by converting an arginine to a citrullin [73]. Even the second hallmark of RA, namely the antibody response to the constant region of IgG measured as rheumatoid factor, may be directed to neoantigens because glycosylation differences of the Fc fragment have been shown to be important in autoantibody recognition [74].
Peripheral repertoire selection is only one of the mechanisms by which lymphopenia and compensatory homeostatic proliferation increase the risk of autoimmunity. In many spontaneous animal models of autoimmunity, a transient, often minute state of lymphopenia is a prerequisite for developing autoimmune disease. This was first described in the NOD mouse model of immune-mediated diabetes [57]. Development of autoimmune phenomena in NOD mice, which are slightly lymphopenic at a young age, is dependent on IL-21-driven homeostatic proliferation. Similarly, Calzascia and coworkers [58] demonstrated that homeostatic proliferation, in this case in response to IL-7, released autoreactive CD4+ cells from inhibitory networks. Lymphocyte depletion largely enhanced the activity of CD4+ T cells to license dendritic cells and to initiate a cascade of CD4+ and CD8+auto-reactive responses, eventually leading to disease. As one possible mechanism, homeostatic proliferation lowers the TCR threshold that antigen recognition must surpass to deliver an activating signal. Recent studies have provided direct evidence supporting a model in which TCR calibration is altered in RA patients. RA T cells have a spontaneously hyper-responsive Ras/Raf-MEK-ERK (Ras/Raf-mitogen-activated protein kinase kinase/extracellular signal-regulated kinase) module. As originally proposed by Germain and colleagues [75,76], increased extracellular signal-regulated kinase activity inhibits a negative feedback loop in response to TCR stimulation and therefore lowers the TCR activation threshold, eventually breaking tolerance. Hyperactivity of this pathway in healthy T cells can be induced by exposure to homeostatic cytokines [77]. Within the panel of homeostatic cytokines, IL-7 appears to be reduced in RA [78]; however, IL-15 and IL-21 are increased [79,80], and this increase appears to precede disease development.
Excessive proliferative turnover and premature senescence not only change the phenotype and function of naïve peripheral CD4+ cells, but also have consequences for the memory subpopulations. Again, these appear to be global phenomena and not limited to a small fraction of expanded antigen-specific T cells. Telomeres in the RA memory population are shortened, and dominant oligoclonal T-cell populations are more frequently detected [64,81-83]. These populations have a phenotype of effector memory or even terminally differentiated effector cells. CD28 and CD27 are lost [84], expression of lymphocyte function-associated antigen-1 (LFA-1) is increased [85], and the chemokine receptor profile is consistent with the differentiation state of effector cells [86]. End-differentiated memory T cells in RA frequently acquire expression of the fractalkine receptor CX3CR1 (chemokine [C-X3-C motif] receptor 1) [87], as well as regulatory receptors that are usually found on natural killer cells, such as natural-killer group 2, member D (NKG2D) and killer immunoglobulin-like receptors [88-90]. In the periphery, these cells are high producers of effector cytokines and are capable of perforin-mediated cytotoxicity [91,92]. Their frequency in peripheral blood correlates with disease severity and the presence of extra-articular manifestations including co-morbidities such as cardiovascular disease [93-95]. Because of their phenotype and functional properties, these cells are prone to be tissue invasive and to be regulated by environmental cues (cytokines; stress-induced ligands binding to NKG2D; MHC class I molecules engaging killer immunoglobulin-like receptors) rather than the classical costimulatory signals.
It is conceivable and even likely that the forces that drive remodeling of the T-cell compartment also affect the frequency and function of regulatory T cells. Depletion or functional degeneration of regulatory T cells could cause a tolerance defect and favor inflammatory responses. The data on regulatory T cells in RA thus far are conflicting. The frequencies of these cells appear to be increased, but their function is compromised, possibly secondary to the effects of TNF-α [75-77,96].
In the synovial tissue, most T cells exhibit features of lymphocyte exhaustion. Characteristic is a loss of the CD3 ζ-chain [97]. Over-expression of PD1, which has been implicated in lymphocyte exhaustion with chronic viral infections [98], has not yet been described. Several factors probably contribute to the exhausted state of synovial T cells, including chronic TCR stimulation and the redox state in the synovial tissue [99,100]. It is also possible that synovial T cells are not truly exhausted but activated by cytokines. Cytokine activation generates an effector function profile that may in part be responsible for the synovial inflammation [101]. In fact, some of these features are reversible upon TNF withdrawal [102]. Importantly, T-cell exhaustion should not be mistaken for T-cell anergy; the two states have different transcriptional profiles [103].
Characterization of novel autoantigens
Production of autoantibodies to the Fc portion of IgG, known as rheumatoid factors, has been the serologic hallmark of RA for the past five decades. Despite considerable effort, attempts to identify autoantibodies to joint-related antigens have yielded inconsistent results. Antigens that are now recognized as relatively specific targets for autoantibodies include the perinuclear factor and keratin. In 1998, van Venrooij and colleagues [104] first reported that these antibodies were directed against deiminated peptides. Subsequent studies showed that the epitopes preferentially recognized in RA are citrullinated peptides of a number of different matrix proteins including fillaggrin, keratin, fibrinogen, and vimentin [73,105]. These antibodies can be measured through their recognition of cyclic citrullinated peptides, now commonly used in clinical practice. Based on these autoantibody profiles, RA patients fail to maintain or induce tolerance to post-translational modifications of common cellular proteins.
Of note, another post-translational modification, glycosylation of IgG Fc, has been implicated in the generation of rheumatoid factors. IgG Fc glycosylation defects are not specific to RA but occur in a number of inflammatory conditions [106]. Similarly, citrullination is not specific for RA or for the synovium, but occurs in most individuals with aging to a varying degree and in numerous tissues. Quantitative difference in the degree of citrullination may play a role in the initiation of an immune response. The finding that Asian RA patients are more likely to have inherited an enzyme variant of PADI-4 (peptidylarginine deiminase 4), the enzyme that is responsible for arginine deimination and citrullination, is consistent with this notion. In addition, smoking, which has been proposed to represent an environmental risk factor for RA, has been correlated with increased citrullination in lung tissue and generation of citrullinated peptide-specific antibodies [107]. Smoking induced an anti-cyclic citrullinated peptide response only in individuals carrying a shared epitope allele, which fits with the immune response gene hypothesis of the HLA-DRB1 association of RA [108]. For reasons unclear, an impact of smoking was observed in Europe but not in the USA [107,109,110].
However, the primary defect in patients with RA appears not to be a defect in post-translational modification but a defect in inducing or maintaining peripheral tolerance, which is very much in line with the global changes in the T-cell compartment observed in patients with RA described above. If RA patients have a broad tolerance defect, then autoantibody responses to an increasing array of self-antigens must be expected. Indeed, Auger and colleagues [111] identified antibodies to PADI-4 and several signaling molecules, including BRAF (v raf murine sarcoma viral oncogene homologue B1 catalytic domain), PKCβ1 (protein kinase Cβ1), and PIP4K2C (phosphatylinositol 4 phosphate 5 kinase type II γ), using protein arrays. Goeb and colleagues [112] used mass spectrometry to identify antibodies to glycolytic enzymes and to chaperones. Confirmatory studies and epitope mapping are needed, but preliminary data indicate that some but not all of these immune responses are once again directed against citrulline modifications.
Translating systemic autoreactivity into synovitis
Most of the abnormalities in the adaptive immune system in RA are systemic in nature, but in patients with established disease synovial manifestations clearly dominate. The question of how systemic abnormalities are translated into inflammation of the synovium is one of the major challenges in elucidating RA pathogenesis. Antibodies to citrullinated peptides and rheumatoid factors can predate the onset of joint manifestations by more than a decade [7,8,113], clearly demonstrating that they are not a consequence of disease and alone are not sufficient to induce disease. This prodromal stage appears to be longer among those patients who develop disease later in life [114], again emphasizing the role played by time and aging in pathogenesis. Similar to auto-antibodies, a case-control study from the Women's Health Study and the Nurses' Health Study [115] found that elevated serum levels of soluble TNF receptor II (as a proxy for TNF-α) and of IL-6 predated disease by up to 12 years. Similar conclusions apply to other cytokines, such as IL-15. In essence, autoimmunity and inflammation exist long before inflammatory lesions are established in the synovial membrane. Epidemiologic data currently do not support the notion of identifiable precipitating events, such as a trauma or an infection, which would turn systemic immune abnormalities into localized tissue inflammation. Rather, it appears that either cumulative changes or stochastically occurring instabilities precipitate the onset of symptoms, suggesting that there is a window of opportunity for preventive interventions.
What is the role played by antigen-specific responses in synovitis? Citrullinated antigens exist in the synovial tissues, but they are hardly specific. An immune response to citrullinated antigens can induce arthritis, as demonstrated in HLA-DR4-IE transgenic mice with citrullinated fibrinogen [116]. In contrast to RA, this arthritis was nonerosive. In the animal model of collagen-induced arthritis, the immune response to citrullinated antigens emerged as an important co-factor to amplify disease manifestations, but by itself it was not sufficient to induce disease [117]. Adoptive transfer of antibodies to citrullinated collagen frequently induced arthritis in naïve mice, however, only when co-administered with antibodies to unmodified collagen [118].
The best evidence for antigen-specific responses in synovial tissue comes from the synovial pathology. The synovial tissue is rich in dendritic cells, which can present antigen and support the activation of T cells [119,120]. About a quarter of the patients have lymphoid follicles with germinal centers, sophisticated structures that facilitate antigen recognition by B and T cells presented by follicular and myeloid dendritic cells [121]. Developing these structures may be a decisive step in sustaining an autoimmune response in the tissue [122]. Important mediators associated with synovial germinal center formation are lymphotoxin-α1β2, IL-7, a proliferation inducing-ligand (APRIL), and CXCL13 (chemokine [C-X-C motif] ligand 13) - cytokines that have also been implicated in the generation of secondary lymphoid structures [123]. Somatic hypermutation of immunoglobulin genes demonstrate the full functionality of these follicles [124]. The antigen recognized by T cells on myeloid dendritic cells and presented by follicular dendritic cells to B cells does not need to be locally produced, but can be taken up by follicular dendritic cells from the bloodstream and can be brought into the synovial tissue by migrating dendritic cells.
Most RA patients do not have germinal centers and do not exhibit unequivocal evidence of antigen recognition in the synovial tissue, although subdued antigen-specific stimulation, as is often seen with exhausted lymphocytes, remains possible. Lymphocytes are scattered in the synovial sublining layer, and T cell-derived cytokines, with the exception of TNF-α and IL-17, are not abundant. IL-17 was originally detected in human synovium from RA patients [125]. Its pathogenetic importance in chronic inflammation has been suggested in a variety of murine model systems. It is attractive to speculate that T cell-derived IL-17 drives the synovial fibroblast activation and cytokine secretion that are characteristic of the rheumatoid synovium [126]. The role played by IFN-γ as a T cell-derived cytokine is less clear in RA. Many T cells isolated out of the environment of the rheumatoid synovitis are able to produce IFN-γ, and studies have shown that the survival of macrophage-like synoviocytes is dependent on IFN-γ production [127]. Furthermore, in humans, contrary to mice, IL-17 and IFN-γ are not mutually exclusive, and IFN-γ/IL-17 double-producing T cells are not infrequent. However, production of IFN-γ in situ is difficult to demonstrate, and treatment of RA patients with IFN-γ has at least not led to disease exacerbation. Synoviocytes are extremely sensitive to cytokine action. Given the multitude of pro- and anti-inflammatory cytokine activities in the synovial tissue, it is difficult to predict hierarchical organization. As recently reviewed, many different cytokines are or will soon be targeted in clinical studies, which will provide insights into the relative contributions made by individual cytokines to the disease process [2,3,6].
In addition to cytokines, the inflammatory infiltrate influences resident synoviocytes through contact-dependent mechanisms (Figure 3). Dayer and colleagues [128] first reported that T cells regulate the production of inflammatory cytokines and metalloproteinases by fibroblasts through cell-to-cell contact. In parallel, direct T cell-synoviocyte interaction inhibits the production of matrix proteins. A number of receptor-ligand interactions in the inflamed synovium have been identified [79,129]. Some of these receptors are constitutively expressed on tissue-infiltrating inflammatory cells, and the mere presence of a cellular infiltrate is sufficient to elicit the responses. Others are activation dependent; however, even for T cells, the activation may not require antigen recognition, but merely cytokine exposure.
Figure 3.

Major tissue destructive pathways in the rheumatoid joint. (a) Osteoclast differentiation and (b) fibroblast-like synoviocyte (FLS) proliferation. CX3CR1, chemokine [C-X3-C motif] receptor 1; FLS, fibroblast-like synoviocyte; HPC, hematopoietic progenitor cells; ICAM, intercellular adhesion molecule; LFA, lymphocyte function-associated antigen; LT, lymphotoxin; M, macrophage; MHC, major histocompatibility complex; RANKL, receptor activator of nuclear factor-κB ligand; SCF, stem cell factor; TCR, T-cell receptor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
NKG2D and its ligands MIC-A and MIC-B contribute to the persistence of the inflammatory infiltrate [88]. Interaction of lymphocyte function-associated antigen-1 with intercellular adhesion molecule-2 influences synoviocyte fibroblastic activation and survival [85]. The fractalkine receptor expressed on cytotoxic effector and terminally differentiated CD4+ T cells binds to cell-bound fractalkine on synovial fibroblasts [87]. The interaction provides a reciprocal activation signal on T cells and synoviocytes, and the subsequent production of soluble fractalkine is a major growth factor for synovial fibroblasts [130]. Cytokine-activated T cells can also directly interact with synovial fibroblasts through membrane-integrated TNF-α expressed on the T cells [131]. Most important is the expression of receptor activator of nuclear factor-κB (RANK) ligand on CD4+ T cells and other infiltrating cells that promote bone erosion through differentiation of monocytic cells into osteoclasts [132]. This list of receptor-ligand interactions is far from inclusive, but it illustrates how the interaction between inflammatory and resident cells develops an architecture that has the ability to be self-perpetuating and tissue damaging.
How does synovitis cause joint destruction?
If not appropriately treated, RA progressively leads to articular destruction and functional disability. In contrast to many tissue-specific autoimmune diseases, the tissue injury is not directly immune-mediated by antigen-specific antibodies or T cells, but is an active remodeling process of the synovium in response to the inflammatory attack.
At least three components contribute to joint destruction: transformation of the synovium into a proliferative, tissue-invasive pannus; generation of osteoclasts that lead to local resorption of bone; and effects of cytokines on cartilage cell function and survival (Figure 3). The normal synovium is a thin layer of macrophage-like and fibroblast-like synoviocytes without an endothelial or epithelial layer and without a true basement membrane. Synovium produces extracellular matrix, ensures a low-resistance surface at joint interface, and possibly has a role in clearing debris. Cadherin-11 has been identified as a critical organizer in forming the synovial lining [133]. Cadherins mediate homotypic cell-to-cell adhesion and are expressed in fibroblast-like synoviocytes. Absence of cadherin in mice results in a hypoplastic synovium [134], whereas forced expression in fibroblasts in vitro produces synovial lining-like structures [135]. Of particular interest, targeting cadherin-11 suppresses arthritis [134]. Cadherin-11-deficient mice do not develop erosive disease; blocking of cadherin-11 by monoclonal antibodies or fusion protein constructs prevents or treats arthritis in the appropriate animal models.
Synovial fibroblasts are very responsive to a large number of stimuli, including cytokines and growth factors produced by the inflammatory infiltrate, and are also responsive to direct receptor-ligand interactions [133]. In addition, the chemokine milieu in the synovial inflammation allows for the recruitment of fibroblast-like synoviocytes, as was recently demonstrated in mice chimeric for green fluorescent protein expression in the bone marrow [136]. The synovium in these mice contained a large proportion of bone marrow-derived fibroblasts when arthritis was induced. The precise chemokines that control this recruitment are not known. Recruitment and local proliferation eventually form a hyperplastic membrane of syoviocytes that exhibits tissue-invasive character, targeting bone and cartilage. This neo-tissue has been termed 'pannus'. Several growth factors, including fibroblast growth factor, platelet-derived growth factor, transforming growth factor-β, and fibronectin, promote synoviocyte proliferation. Studies in mouse models have shown that the tyrosine kinase inhibitor imatinib suppresses arthritis, presumably by inhibiting the platelet-derived growth factor receptor [137]. Because activated and proliferating synovial fibroblasts produce many of their growth factors, the inflammatory response in the synovial membrane induces a self-perpetuating cycle of synovial fibroblast activation and proliferation.
Activated synoviocytes, in particular in the pannus, produce matrix-degrading enzymes, such as aggrecanases and matrix metalloproteinases. Of particular relevance is the membrane type I matrix metalloproteinase, which has been shown to be a crucial promoter of synovial invasion [138]. Silencing of this enzyme reduced the invasiveness of synovial fibroblasts [139]. Matrix resorption and cartilage and bone invasion by synovial fibroblasts requires demineralization by osteoclasts [140]. Formation of osteoclasts is, therefore, an essential component of erosive RA. Osteoclast differentiation is in part driven by RANK ligand, which is expressed on tissue-residing CD4+ T cells and on synovial fibroblasts and is upregulated by a number of proinflammatory cytokines. By engaging RANK, RANK ligand induces the differentiation of monocytic cells into osteoclasts. Osteoclast differentiation can be inhibited by osteoprotegerin, which does not ameliorate the inflammatory signs of disease but can prevent structural damage to the joint.
Conclusion
The success of anti-cytokine therapy in RA has revolutionized the management of this disease and has provided a paradigm for novel therapeutic avenues in a variety of other inflammatory syndromes. The fact that blocking the action of TNF-α inhibits synovial inflammation and its destructive consequences is conclusive evidence that, at least at the effector stage, excess cytokines are of critical importance in RA. The past decade has seen the identification and molecular characterization of a multitude of cytokines, all of which may make their own contribution to the inflammatory battlefield. The latest in this collection is IL-17, which may or may not prove to be a valuable therapeutic target. Clinical studies in the next decade will decide which of these cytokines is acting at pivotal junctures in synovial inflammation and tissue damage. A selective approach will be beneficial only if cytokines do not act in parallel, because combination therapy blocking several cytokines appears to be unlikely due to the risk for unacceptable side effects as well as cost reasons.
Preventive and curative interventions in RA will depend on identifying mechanisms upstream of the synovial inflammation. The most promising finding paving the way for potential preventive therapy relates to the more recent concept of a systemic prodromal stage preceding synovitis. Several immune pathologies appear to be characteristic for this preclinical phase of RA, including acceleration of immune aging, loss of tolerance to neoantigens, and differentiation and accumulation of effector cells with high inflammatory capacity. The results from genetic association and linkage studies, as well as the recently described mouse models of spontaneous arthritis, suggest a role of signal calibration downstream of antigen recognition and triggering of cytokine receptors; understanding these abnormalities may inform novel strategies of stopping RA before it ever reaches its tissue targets.
Abbreviations
HLA: human leukocyte antigen; hTERT: human telomerase reverse transcriptase; IFN: interferon; IL: interleukin; MHC: major histocompatibility complex; NKG2D: natural-killer group 2, member D; RA: rheumatoid arthritis; RANK: receptor activator of nuclear factor-κB; SNP: single nucleotide polymorphism; TCR: T-cell receptor; TNF: tumor necrosis factor; TREC: T-cell receptor excision circle.
Competing interests
The authors declare that they have no competing interests.
Note
The Scientific Basis of Rheumatology: A Decade of Progress
This article is part of a special collection of reviews, The Scientific Basis of Rheumatology: A Decade of Progress, published to mark Arthritis Research & Therapy's 10th anniversary.
Other articles in this series can be found at: http://arthritis-research.com/sbr
Contributor Information
Jörg J Goronzy, Email: jgoronz@emory.edu.
Cornelia M Weyand, Email: cweyand@emory.edu.
Acknowledgements
This work was funded in part by grants from the National Institutes of Health (RO1 AR42527, RO1 AR41974, R01 AI44142, U19 AI57266, RO1 EY11916, and R01 AG15043). The authors thank Linda Arneson and Tamela Yeargin for help with preparing the manuscript.
References
- Smolen JS, Aletaha D. Developments in the clinical understanding of rheumatoid arthritis. Arthritis Res Ther. 2009;11:204. doi: 10.1186/ar2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–3545. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–196. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nat Clin Pract Rheumatol. 2006;2:619–626. doi: 10.1038/ncprheum0338. [DOI] [PubMed] [Google Scholar]
- Waldburger JM, Firestein GS. Garden of therapeutic delights: new targets in rheumatic diseases. Arthritis Res Ther. 2009;11:206. doi: 10.1186/ar2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielen MM, van Schaardenburg D, Reesink HW, Stadt RJ van de, Horst-Bruinsma IE van der, de Koning MH, Habibuw MR, Vandenbroucke JP, Dijkmans BA. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–386. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, Sundin U, van Venrooij WJ. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- Turesson C, Matteson EL. Vasculitis in rheumatoid arthritis. Curr Opin Rheumatol. 2009;21:35–40. doi: 10.1097/BOR.0b013e32831c5303. [DOI] [PubMed] [Google Scholar]
- Silman AJ, MacGregor AJ, Thomson W, Holligan S, Carthy D, Farhan A, Ollier WE. Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol. 1993;32:903–907. doi: 10.1093/rheumatology/32.10.903. [DOI] [PubMed] [Google Scholar]
- Seldin MF, Amos CI, Ward R, Gregersen PK. The genetics revolution and the assault on rheumatoid arthritis. Arthritis Rheum. 1999;42:1071–1079. doi: 10.1002/1529-0131(199906)42:6<1071::AID-ANR1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Plenge RM, Criswell LA. Genetic variants that predict response to anti-tumor necrosis factor therapy in rheumatoid arthritis: current challenges and future directions. Curr Opin Rheumatol. 2008;20:145–152. doi: 10.1097/BOR.0b013e3282f5135b. [DOI] [PubMed] [Google Scholar]
- Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada R, Yamamoto K. Mechanisms of disease: genetics of rheumatoid arthritis: ethnic differences in disease-associated genes. Nat Clin Pract Rheumatol. 2007;3:644–650. doi: 10.1038/ncprheum0592. [DOI] [PubMed] [Google Scholar]
- Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- Huizinga TW, Amos CI, Helm-van Mil AH van der, Chen W, van Gaalen FA, Jawaheer D, Schreuder GM, Wener M, Breedveld FC, Ahmad N, Lum RF, de Vries RR, Gregersen PK, Toes RE, Criswell LA. Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum. 2005;52:3433–3438. doi: 10.1002/art.21385. [DOI] [PubMed] [Google Scholar]
- Helm-van Mil AH van der, Verpoort KN, Breedveld FC, Huizinga TW, Toes RE, de Vries RR. The HLA-DRB1 shared epitope alleles are primarily a risk factor for anti-cyclic citrullinated peptide antibodies and are not an independent risk factor for development of rheumatoid arthritis. Arthritis Rheum. 2006;54:1117–1121. doi: 10.1002/art.21739. [DOI] [PubMed] [Google Scholar]
- MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, Silman AJ. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000;43:30–37. doi: 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oene M, Wintle RF, Liu X, Yazdanpanah M, Gu X, Newman B, Kwan A, Johnson B, Owen J, Greer W, Mosher D, Maksymowych W, Keystone E, Rubin LA, Amos CI, Siminovitch KA. Association of the lymphoid tyrosine phosphatase R620W variant with rheumatoid arthritis, but not Crohn's disease, in Canadian populations. Arthritis Rheum. 2005;52:1993–1998. doi: 10.1002/art.21123. [DOI] [PubMed] [Google Scholar]
- Wesoly J, Helm-van Mil AH van der, Toes RE, Chokkalingam AP, Carlton VE, Begovich AB, Huizinga TW. Association of the PTPN22 C1858T single-nucleotide polymorphism with rheumatoid arthritis phenotypes in an inception cohort. Arthritis Rheum. 2005;52:2948–2950. doi: 10.1002/art.21294. [DOI] [PubMed] [Google Scholar]
- Gregersen PK. Pathways to gene identification in rheumatoid arthritis: PTPN22 and beyond. Immunol Rev. 2005;204:74–86. doi: 10.1111/j.0105-2896.2005.00243.x. [DOI] [PubMed] [Google Scholar]
- Ikari K, Momohara S, Inoue E, Tomatsu T, Hara M, Yamanaka H, Kamatani N. Haplotype analysis revealed no association between the PTPN22 gene and RA in a Japanese population. Rheumatology (Oxford) 2006;45:1345–1348. doi: 10.1093/rheumatology/kel169. [DOI] [PubMed] [Google Scholar]
- Mori M, Yamada R, Kobayashi K, Kawaida R, Yamamoto K. Ethnic differences in allele frequency of autoimmune-disease-associated SNPs. J Hum Genet. 2005;50:264–266. doi: 10.1007/s10038-005-0246-8. [DOI] [PubMed] [Google Scholar]
- Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, Tang J, Jeffery D, Mortara K, Sampang J, Williams SR, Buggy J, Clark JM. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem. 2006;281:11002–11010. doi: 10.1074/jbc.M600498200. [DOI] [PubMed] [Google Scholar]
- Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med. 1999;189:111–121. doi: 10.1084/jem.189.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vang T, Congia M, Macis MD, Musumeci L, Orrú V, Zavattari P, Nika K, Tautz L, Taskén K, Cucca F, Mustelin T, Bottini N. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. 2005;37:1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- Bottini N, Vang T, Cucca F, Mustelin T. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol. 2006;18:207–213. doi: 10.1016/j.smim.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, Ohtsuki M, Furukawa H, Yoshino S, Yukioka M, Tohma S, Matsubara T, Wakitani S, Teshima R, Nishioka Y, Sekine A, Iida A, Takahashi A, Tsunoda T, Nakamura Y, Yamamoto K. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34:395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Ikari K, Nakamura T, Kuwahara M, Toyama Y, Tomatsu T, Momohara S, Kamatani N. Association between PADI4 and rheumatoid arthritis: a meta-analysis. Rheumatology (Oxford) 2006;45:804–807. doi: 10.1093/rheumatology/kel023. [DOI] [PubMed] [Google Scholar]
- Kang CP, Lee HS, Ju H, Cho H, Kang C, Bae SC. A functional haplotype of the PADI4 gene associated with increased rheumatoid arthritis susceptibility in Koreans. Arthritis Rheum. 2006;54:90–96. doi: 10.1002/art.21536. [DOI] [PubMed] [Google Scholar]
- Barton A, Bowes J, Eyre S, Spreckley K, Hinks A, John S, Worthington J. A functional haplotype of the PADI4 gene associated with rheumatoid arthritis in a Japanese population is not associated in a United Kingdom population. Arthritis Rheum. 2004;50:1117–1121. doi: 10.1002/art.20169. [DOI] [PubMed] [Google Scholar]
- Caponi L, Petit-Teixeira E, Sebbag M, Bongiorni F, Moscato S, Pratesi F, Pierlot C, Osorio J, Chapuy-Regaud S, Guerrin M, Cornelis F, Serre G, Migliorini P. ECRAF. A family based study shows no association between rheumatoid arthritis and the PADI4 gene in a white French population. Ann Rheum Dis. 2005;64:587–593. doi: 10.1136/ard.2004.026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Valdivia A, Pascual-Salcedo D, Lamas JR, Fernandez-Arquero M, Balsa A, Fernandez-Gutierrez B, de la Concha EG, Urcelay E. PADI4 polymorphisms are not associated with rheumatoid arthritis in the Spanish population. Rheumatology (Oxford) 2005;44:1263–1266. doi: 10.1093/rheumatology/kei008. [DOI] [PubMed] [Google Scholar]
- Plenge RM, Padyukov L, Remmers EF, Purcell S, Lee AT, Karlson EW, Wolfe F, Kastner DL, Alfredsson L, Altshuler D, Gregersen PK, Klareskog L, Rioux JD. Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. Am J Hum Genet. 2005;77:1044–1060. doi: 10.1086/498651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, Li W, Masters SL, Booty MG, Carulli JP, Padyukov L, Alfredsson L, Klareskog L, Chen WV, Amos CI, Criswell LA, Seldin MF, Kastner DL, Gregersen PK. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007;357:977–986. doi: 10.1056/NEJMoa073003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Remmers EF, Le JM, Kastner DL, Bae SC, Gregersen PK. Association of STAT4 with rheumatoid arthritis in the Korean population. Mol Med. 2007;13:455–460. doi: 10.2119/2007-00072.Lee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson W, Barton A, Ke X, Eyre S, Hinks A, Bowes J, Donn R, Symmons D, Hider S, Bruce IN. Wellcome Trust Case Control Consortium; Wilson AG, Marinou I, Morgan A, Emery P. YEAR Consortium; Carter A, Steer S, Hocking L, Reid DM, Wordsworth P, Harrison P, Strachan D, Worthington J. Rheumatoid arthritis association at 6q23. Nat Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, Pe'er I, Burtt NP, Blumenstiel B, DeFelice M, Parkin M, Barry R, Winslow W, Healy C, Graham RR, Neale BM, Izmailova E, Roubenoff R, Parker AN, Glass R, Karlson EW, Maher N, Hafler DA, Lee DM, Seldin MF, Remmers EF, Lee AT, Padyukov L, Alfredsson L, Coblyn J. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, McNally E, Pickart C, Ma A. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurreeman FA, Padyukov L, Marques RB, Schrodi SJ, Seddighzadeh M, Stoeken-Rijsbergen G, Helm-van Mil AH van der, Allaart CF, Verduyn W, Houwing-Duistermaat J, Alfredsson L, Begovich AB, Klareskog L, Huizinga TW, Toes RE. A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med. 2007;4:e278. doi: 10.1371/journal.pmed.0040278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, Liew A, Khalili H, Chandrasekaran A, Davies LR, Li W, Tan AK, Bonnard C, Ong RT, Thalamuthu A, Pettersson S, Liu C, Tian C, Chen WV, Carulli JP, Beckman EM, Altshuler D, Alfredsson L, Criswell LA, Amos CI, Seldin MF, Kastner DL, Klareskog L, Gregersen PK. TRAF1-C5 as a risk locus for rheumatoid arthritis: a genomewide study. N Engl J Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Rowland CM, Garcia VE, Schrodi SJ, Catanese JJ, Helm-van Mil AH van der, Ardlie KG, Amos CI, Criswell LA, Kastner DL, Gregersen PK, Kurreeman FA, Toes RE, Huizinga TW, Seldin MF, Begovich AB. A large-scale rheumatoid arthritis genetic study identifies association at chromosome 9q33.2. PLoS Genet. 2008;4:e1000107. doi: 10.1371/journal.pgen.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Remmers EF, Lee AT, Hackett R, Guiducci C, Burtt NP, Gianniny L, Korman BD, Padyukov L, Kurreeman FA, Chang M, Catanese JJ, Ding B, Wong S, Helm-van Mil AH van der, Neale BM, Coblyn J, Cui J, Tak PP, Wolbink GJ, Crusius JB, Horst-Bruinsma IE van der, Criswell LA, Amos CI, Seldin MF, Kastner DL, Ardlie KG, Alfredsson L, Costenbader KH, Altshuler D. Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat Genet. 2008;40:1216–1223. doi: 10.1038/ng.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmdahl R, Andersson M, Goldschmidt TJ, Gustafsson K, Jansson L, Mo JA. Type II collagen autoimmunity in animals and provocations leading to arthritis. Immunol Rev. 1990;118:193–232. doi: 10.1111/j.1600-065X.1990.tb00817.x. [DOI] [PubMed] [Google Scholar]
- Glant TT, Finnegan A, Mikecz K. Proteoglycan-induced arthritis: immune regulation, cellular mechanisms, and genetics. Crit Rev Immunol. 2003;23:199–250. doi: 10.1615/CritRevImmunol.v23.i3.20. [DOI] [PubMed] [Google Scholar]
- Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/S0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, Mathis D, Benoist C. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002;3:360–365. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- Caton AJ, Stark SE, Kavaler J, Staudt LM, Schwartz D, Gerhard W. Many variable region genes are utilized in the antibody response of BALB/c mice to the influenza virus A/PR/8/34 hemagglutinin. J Immunol. 1991;147:1675–1686. [PubMed] [Google Scholar]
- Rankin AL, Reed AJ, Oh S, Cozzo Picca C, Guay HM, Larkin J 3rd, Panarey L, Aitken MK, Koeberlein B, Lipsky PE, Tomaszewski JE, Naji A, Caton AJ. CD4+ T cells recognizing a single self-peptide expressed by APCs induce spontaneous autoimmune arthritis. J Immunol. 2008;180:833–841. doi: 10.4049/jimmunol.180.2.833. [DOI] [PubMed] [Google Scholar]
- Atsumi T, Ishihara K, Kamimura D, Ikushima H, Ohtani T, Hirota S, Kobayashi H, Park SJ, Saeki Y, Kitamura Y, Hirano T. A point mutation of Tyr-759 in interleukin 6 family cytokine receptor subunit gp130 causes autoimmune arthritis. J Exp Med. 2002;196:979–990. doi: 10.1084/jem.20020619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S, Sakaguchi S. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003;426:454–460. doi: 10.1038/nature02119. [DOI] [PubMed] [Google Scholar]
- Hirota K, Hashimoto M, Yoshitomi H, Tanaka S, Nomura T, Yamaguchi T, Iwakura Y, Sakaguchi N, Sakaguchi S. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med. 2007;204:41–47. doi: 10.1084/jem.20062259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C, Ilic A, Koelsch K, Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–277. doi: 10.1016/S0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- Calzascia T, Pellegrini M, Lin A, Garza KM, Elford AR, Shahinian A, Ohashi PS, Mak TW. CD4 T cells, lymphopenia, and IL-7 in a multistep pathway to autoimmunity. Proc Natl Acad Sci USA. 2008;105:2999–3004. doi: 10.1073/pnas.0712135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope AP. Altered signalling thresholds in T lymphocytes cause autoimmune arthritis. Arthritis Res Ther. 2004;6:112–116. doi: 10.1186/ar1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R, Turner M, Cope AP. High avidity autoreactive T cells with a low signalling capacity through the T-cell receptor: central to rheumatoid arthritis pathogenesis? Arthritis Res Ther. 2008;10:210. doi: 10.1186/ar2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goronzy JJ, Weyand CM. Rheumatoid arthritis. Immunol Rev. 2005;204:55–73. doi: 10.1111/j.0105-2896.2005.00245.x. [DOI] [PubMed] [Google Scholar]
- Goronzy JJ, Weyand CM. Aging, autoimmunity and arthritis: T-cell senescence and contraction of T-cell repertoire diversity-catalysts of autoimmunity and chronic inflammation. Arthritis Res Ther. 2003;5:225–234. doi: 10.1186/ar974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodes RJ, Hathcock KS, Weng NP. Telomeres in T and B cells. Nat Rev Immunol. 2002;2:699–706. doi: 10.1038/nri890. [DOI] [PubMed] [Google Scholar]
- Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 2000;97:9203–9208. doi: 10.1073/pnas.97.16.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmegna I, Diaz-Borjon A, Fujii H, Schaefer L, Goronzy JJ, Weyand CM. Defective proliferative capacity and accelerated telomeric loss of hematopoietic progenitor cells in rheumatoid arthritis. Arthritis Rheum. 2008;58:990–1000. doi: 10.1002/art.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonland SO, Lopez C, Widmann T, Zimmer J, Bryl E, Goronzy JJ, Weyand CM. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc Natl Acad Sci USA. 2003;100:13471–13476. doi: 10.1073/pnas.2233561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci USA. 2009;106:4360–4365. doi: 10.1073/pnas.0811332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med. 2009;206:1435–1449. doi: 10.1084/jem.20082251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douek DC, McFarland RD, Keiser PH, Gage EA, Massey JM, Haynes BF, Polis MA, Haase AT, Feinberg MB, Sullivan JL, Jamieson BD, Zack JA, Picker LJ, Koup RA. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–695. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- Dool C van den, de Boer RJ. The effects of age, thymectomy, and HIV Infection on alpha and beta TCR excision circles in naive T cells. J Immunol. 2006;177:4391–4401. doi: 10.4049/jimmunol.177.7.4391. [DOI] [PubMed] [Google Scholar]
- Goronzy JJ, Weyand CM. Thymic function and peripheral T-cell homeostasis in rheumatoid arthritis. Trends Immunol. 2001;22:251–255. doi: 10.1016/S1471-4906(00)01841-X. [DOI] [PubMed] [Google Scholar]
- Wagner UG, Koetz K, Weyand CM, Goronzy JJ. Perturbation of the T cell repertoire in rheumatoid arthritis. Proc Natl Acad Sci USA. 1998;95:14447–14452. doi: 10.1073/pnas.95.24.14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boekel MA, Vossenaar ER, Hoogen FH van den, van Venrooij WJ. Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res. 2002;4:87–93. doi: 10.1186/ar395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavi A, Axford JS. Sweet and sour: the impact of sugars on disease. Rheumatology (Oxford) 2008;47:760–770. doi: 10.1093/rheumatology/ken081. [DOI] [PubMed] [Google Scholar]
- Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol. 2003;4:248–254. doi: 10.1038/ni895. [DOI] [PubMed] [Google Scholar]
- Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 2005;3:e356. doi: 10.1371/journal.pbio.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Deshpande P, Pryshchep S, Weyand CM, Goronzy JJ. Increased signal amplification in the Raf-MEK-ERK module of RA T cells. Arthritis Rheum. 2008;58:S293. [Google Scholar]
- Churchman SM, Ponchel F. Interleukin-7 in rheumatoid arthritis. Rheumatology (Oxford) 2008;47:753–759. doi: 10.1093/rheumatology/ken053. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Leung BP, Sturrock RD, Field M, Liew FY. Inter-leukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nat Med. 1997;3:189–195. doi: 10.1038/nm0297-189. [DOI] [PubMed] [Google Scholar]
- Ettinger R, Kuchen S, Lipsky PE. Interleukin 21 as a target of intervention in autoimmune disease. Ann Rheum Dis. 2008;67(suppl 3):iii83–iii86. doi: 10.1136/ard.2008.098400. [DOI] [PubMed] [Google Scholar]
- Fitzgerald JE, Ricalton NS, Meyer AC, West SG, Kaplan H, Behrendt C, Kotzin BL. Analysis of clonal CD8+ T cell expansions in normal individuals and patients with rheumatoid arthritis. J Immunol. 1995;154:3538–3547. [PubMed] [Google Scholar]
- Waase I, Kayser C, Carlson PJ, Goronzy JJ, Weyand CM. Oligoclonal T cell proliferation in patients with rheumatoid arthritis and their unaffected siblings. Arthritis Rheum. 1996;39:904–913. doi: 10.1002/art.1780390606. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Martens PB, Weyand CM, Goronzy JJ. The repertoire of CD4+ CD28- T cells in rheumatoid arthritis. Mol Med. 1996;2:608–618. [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Goronzy JJ, Weyand CM. CD4+ CD7- CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest. 1996;97:2027–2037. doi: 10.1172/JCI118638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Colmegna I, He X, Weyand CM, Goronzy JJ. Synoviocyte stimulation by the LFA-1-intercellular adhesion molecule-2-Ezrin-Akt pathway in rheumatoid arthritis. J Immunol. 2008;180:1971–1978. doi: 10.4049/jimmunol.180.3.1971. [DOI] [PubMed] [Google Scholar]
- Zhang X, Nakajima T, Goronzy JJ, Weyand CM. Tissue trafficking patterns of effector memory CD4+ T cells in rheumatoid arthritis. Arthritis Rheum. 2005;52:3839–3849. doi: 10.1002/art.21482. [DOI] [PubMed] [Google Scholar]
- Sawai H, Park YW, Roberson J, Imai T, Goronzy JJ, Weyand CM. T cell costimulation by fractalkine-expressing synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2005;52:1392–1401. doi: 10.1002/art.21140. [DOI] [PubMed] [Google Scholar]
- Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci USA. 2003;100:9452–9457. doi: 10.1073/pnas.1632807100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa T, Snyder MR, Yen JH, Goehring BE, Leibson PJ, Weyand CM, Goronzy JJ. Killer cell activating receptors function as costimulatory molecules on CD4+ CD28null T cells clonally expanded in rheumatoid arthritis. J Immunol. 2000;165:1138–1145. doi: 10.4049/jimmunol.165.2.1138. [DOI] [PubMed] [Google Scholar]
- Yen JH, Moore BE, Nakajima T, Scholl D, Schaid DJ, Weyand CM, Goronzy JJ. Major histocompatibility complex class I-recognizing receptors are disease risk genes in rheumatoid arthritis. J Exp Med. 2001;193:1159–1167. doi: 10.1084/jem.193.10.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park W, Weyand CM, Schmidt D, Goronzy JJ. Co-stimulatory pathways controlling activation and peripheral tolerance of human CD4+ CD28- T cells. Eur J Immunol. 1997;27:1082–1090. doi: 10.1002/eji.1830270507. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Goek O, Zhang X, Kopecky SL, Frye RL, Goronzy JJ, Weyand CM. De novo expression of killer immunoglobulin-like receptors and signaling proteins regulates the cytotoxic function of CD4 T cells in acute coronary syndromes. Circ Res. 2003;93:106–113. doi: 10.1161/01.RES.0000082333.58263.58. [DOI] [PubMed] [Google Scholar]
- Goronzy JJ, Matteson EL, Fulbright JW, Warrington KJ, Chang-Miller A, Hunder GG, Mason TG, Nelson AM, Valente RM, Crowson CS, Erlich HA, Reynolds RL, Swee RG, O'Fallon WM, Weyand CM. Prognostic markers of radiographic progression in early rheumatoid arthritis. Arthritis Rheum. 2004;50:43–54. doi: 10.1002/art.11445. [DOI] [PubMed] [Google Scholar]
- Martens PB, Goronzy JJ, Schaid D, Weyand CM. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997;40:1106–1114. doi: 10.1002/art.1780400615. [DOI] [PubMed] [Google Scholar]
- Warrington KJ, Kent PD, Frye RL, Lymp JF, Kopecky SL, Goronzy JJ, Weyand CM. Rheumatoid arthritis is an independent risk factor for multi-vessel coronary artery disease: a case control study. Arthritis Res Ther. 2005;7:R984–R991. doi: 10.1186/ar1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson AK, Li C, Brennan FM. Recent developments in the immunobiology of rheumatoid arthritis. Arthritis Res Ther. 2008;10:204. doi: 10.1186/ar2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gorman CL, Vermi AC, Monaco C, Foey A, Owen S, Amjadi P, Vallance A, McClinton C, Marelli-Berg F. TCRzetadim lymphocytes define populations of circulating effector cells that migrate to inflamed tissues. Blood. 2007;109:4328–4335. doi: 10.1182/blood-2006-12-064170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Cope AP. Studies of T-cell activation in chronic inflammation. Arthritis Res. 2002;4(suppl 3):S197–S211. doi: 10.1186/ar557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen ME, Young SP, Michell RH, Bacon PA. Altered T lymphocyte signaling in rheumatoid arthritis. Eur J Immunol. 1995;25:1547–1554. doi: 10.1002/eji.1830250612. [DOI] [PubMed] [Google Scholar]
- Brennan FM, Hayes AL, Ciesielski CJ, Green P, Foxwell BM, Feldmann M. Evidence that rheumatoid arthritis synovial T cells are similar to cytokine-activated T cells: involvement of phosphatidylinositol 3-kinase and nuclear factor kappaB pathways in tumor necrosis factor alpha production in rheumatoid arthritis. Arthritis Rheum. 2002;46:31–41. doi: 10.1002/1529-0131(200201)46:1<31::AID-ART10029>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Bryl E, Vallejo AN, Matteson EL, Witkowski JM, Weyand CM, Goronzy JJ. Modulation of CD28 expression with anti-tumor necrosis factor alpha therapy in rheumatoid arthritis. Arthritis Rheum. 2005;52:2996–3003. doi: 10.1002/art.21353. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Schellekens GA, de Jong BA, Hoogen FH van den, Putte LB van de, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–281. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossenaar ER, Despres N, Lapointe E, Heijden A van der, Lora M, Senshu T, van Venrooij WJ, Menard HA. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res Ther. 2004;6:R142–R150. doi: 10.1186/ar1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axford JS, Sumar N, Alavi A, Isenberg DA, Young A, Bodman KB, Roitt IM. Changes in normal glycosylation mechanisms in autoimmune rheumatic disease. J Clin Invest. 1992;89:1021–1031. doi: 10.1172/JCI115643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klareskog L, Stolt P, Lundberg K, Källberg H, Bengtsson C, Grunewald J, Rönnelid J, Harris HE, Ulfgren AK, Rantapää-Dahlqvist S, Eklund A, Padyukov L, Alfredsson L. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54:38–46. doi: 10.1002/art.21575. [DOI] [PubMed] [Google Scholar]
- Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet. 2009;373:659–672. doi: 10.1016/S0140-6736(09)60008-8. [DOI] [PubMed] [Google Scholar]
- Lee HS, Irigoyen P, Kern M, Lee A, Batliwalla F, Khalili H, Wolfe F, Lum RF, Massarotti E, Weisman M, Bombardier C, Karlson EW, Criswell LA, Vlietinck R, Gregersen PK. Interaction between smoking, the shared epitope, and anti-cyclic citrullinated peptide: a mixed picture in three large North American rheumatoid arthritis cohorts. Arthritis Rheum. 2007;56:1745–1753. doi: 10.1002/art.22703. [DOI] [PubMed] [Google Scholar]
- Linn-Rasker SP, Helm-van Mil AH van der, van Gaalen FA, Kloppenburg M, de Vries RR, le Cessie S, Breedveld FC, Toes RE, Huizinga TW. Smoking is a risk factor for anti-CCP antibodies only in rheumatoid arthritis patients who carry HLA-DRB1 shared epitope alleles. Ann Rheum Dis. 2006;65:366–371. doi: 10.1136/ard.2005.041079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger I, Balandraud N, Rak J, Lambert N, Martin M, Roudier J. New autoantigens in rheumatoid arthritis (RA): screening 8268 protein arrays with sera from patients with RA. Ann Rheum Dis. 2009;68:591–594. doi: 10.1136/ard.2008.096917. [DOI] [PubMed] [Google Scholar]
- Goeb V, Thomas-L'Otellier M, Daveau R, Charlionet R, Fardellone P, Le Loet X, Tron F, Gilbert D, Vittecoq O. Candidate autoantigens identified by mass spectrometry in early rheumatoid arthritis are chaperones and citrullinated glycolytic enzymes. Arthritis Res Ther. 2009;11:R38. doi: 10.1186/ar2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Puente A, Knowler WC, Pettitt DJ, Bennett PH. The incidence of rheumatoid arthritis is predicted by rheumatoid factor titer in a longitudinal population study. Arthritis Rheum. 1988;31:1239–1244. doi: 10.1002/art.1780311004. [DOI] [PubMed] [Google Scholar]
- Majka DS, Deane KD, Parrish LA, Lazar AA, Baron AE, Walker CW, Rubertone MV, Gilliland WR, Norris JM, Holers VM. Duration of preclinical rheumatoid arthritis-related autoantibody positivity increases in subjects with older age at time of disease diagnosis. Ann Rheum Dis. 2008;67:801–807. doi: 10.1136/ard.2007.076679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlson EW, Chibnik LB, Tworoger SS, Lee IM, Buring JE, Shadick NA, Manson JE, Costenbader KH. Biomarkers of inflammation and development of rheumatoid arthritis in women from two prospective cohort studies. Arthritis Rheum. 2009;60:641–652. doi: 10.1002/art.24350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JA, Bell DA, Brintnell W, Yue D, Wehrli B, Jevnikar AM, Lee DM, Hueber W, Robinson WH, Cairns E. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. J Exp Med. 2008;205:967–979. doi: 10.1084/jem.20072051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, Holers VM. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. 2006;116:961–973. doi: 10.1172/JCI25422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uysal H, Bockermann R, Nandakumar KS, Sehnert B, Bajtner E, Engstrom A, Serre G, Burkhardt H, Thunnissen MM, Holmdahl R. Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J Exp Med. 2009;206:449–462. doi: 10.1084/jem.20081862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzky V, Hannawi S, Thomas R. Cells of the synovium in rheumatoid arthritis. Dendritic cells. Arthritis Res Ther. 2007;9:219. doi: 10.1186/ar2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R, Lipsky PE. Human peripheral blood dendritic cell subsets. Isolation and characterization of precursor and mature antigen-presenting cells. J Immunol. 1994;153:4016–4028. [PubMed] [Google Scholar]
- Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O'Fallon WM, Goronzy JJ, Weyand CM. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- Weyand CM, Kurtin PJ, Goronzy JJ. Ectopic lymphoid organogenesis: a fast track for autoimmunity. Am J Pathol. 2001;159:787–793. doi: 10.1016/S0002-9440(10)61751-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyler TM, Park YW, Takemura S, Bram RJ, Kurtin PJ, Goronzy JJ, Weyand CM. BLyS and APRIL in rheumatoid arthritis. J Clin Invest. 2005;115:3083–3092. doi: 10.1172/JCI25265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder AE, Greiner A, Seyfert C, Berek C. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 1996;93:221–225. doi: 10.1073/pnas.93.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–970. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Miossec P. Interleukin-17 in fashion, at last: ten years after its description, its cellular source has been identified. Arthritis Rheum. 2007;56:2111–2115. doi: 10.1002/art.22733. [DOI] [PubMed] [Google Scholar]
- Klimiuk PA, Yang H, Goronzy JJ, Weyand CM. Production of cytokines and metalloproteinases in rheumatoid synovitis is T cell dependent. Clin Immunol. 1999;90:65–78. doi: 10.1006/clim.1998.4618. [DOI] [PubMed] [Google Scholar]
- Rezzonico R, Burger D, Dayer JM. Direct contact between T lymphocytes and human dermal fibroblasts or synoviocytes down-regulates types I and III collagen production via cell-associated cytokines. J Biol Chem. 1998;273:18720–18728. doi: 10.1074/jbc.273.30.18720. [DOI] [PubMed] [Google Scholar]
- Beech JT, Andreakos E, Ciesielski CJ, Green P, Foxwell BM, Brennan FM. T-cell contact-dependent regulation of CC and CXC chemokine production in monocytes through differential involvement of NFkappaB: implications for rheumatoid arthritis. Arthritis Res Ther. 2006;8:R168. doi: 10.1186/ar2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai H, Park YW, He X, Goronzy JJ, Weyand CM. Fractalkine mediates T cell-dependent proliferation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. 2007;56:3215–3225. doi: 10.1002/art.22919. [DOI] [PubMed] [Google Scholar]
- Burger D, Dayer JM. The role of human T-lymphocyte-monocyte contact in inflammation and tissue destruction. Arthritis Res. 2002;4(suppl 3):S169–S176. doi: 10.1186/ar558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravallese EM, Goldring SR. Cellular mechanisms and the role of cytokines in bone erosions in rheumatoid arthritis. Arthritis Rheum. 2000;43:2143–2151. doi: 10.1002/1529-0131(200010)43:10<2143::AID-ANR1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev. 2008;223:252–270. doi: 10.1111/j.1600-065X.2008.00648.x. [DOI] [PubMed] [Google Scholar]
- Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, Takeichi M, Brenner MB. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315:1006–1010. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- Kiener HP, Lee DM, Agarwal SK, Brenner MB. Cadherin-11 induces rheumatoid arthritis fibroblast-like synoviocytes to form lining layers in vitro. Am J Pathol. 2006;168:1486–1499. doi: 10.2353/ajpath.2006.050999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Makarov SS. An essential role of NF-kappaB in the 'tumor-like' phenotype of arthritic synoviocytes. Proc Natl Acad Sci USA. 2006;103:17432–17437. doi: 10.1073/pnas.0607939103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paniagua RT, Sharpe O, Ho PP, Chan SM, Chang A, Higgins JP, Tomooka BH, Thomas FM, Song JJ, Goodman SB, Lee DM, Genovese MC, Utz PJ, Steinman L, Robinson WH. Selective tyrosine kinase inhibition by imatinib mesylate for the treatment of autoimmune arthritis. J Clin Invest. 2006;116:2633–2642. doi: 10.1172/JCI28546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MC, Manning HB, Jain A, Troeberg L, Dudhia J, Essex D, Sandison A, Seiki M, Nanchahal J, Nagase H, Itoh Y. Membrane type 1 matrix metalloproteinase is a crucial promoter of synovial invasion in human rheumatoid arthritis. Arthritis Rheum. 2009;60:686–697. doi: 10.1002/art.24331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkauskaite E, Volkmer D, Shigeyama Y, Schedel J, Pap G, Müller-Ladner U, Meinecke I, Alexander D, Gay RE, Drynda S, Neumann W, Michel BA, Aicher WK, Gay S, Pap T. Retroviral gene transfer of an antisense construct against membrane type 1 matrix metalloproteinase reduces the invasiveness of rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2005;52:2010–2014. doi: 10.1002/art.21156. [DOI] [PubMed] [Google Scholar]
- Schett G. Erosive arthritis. Arthritis Res Ther. 2007;9(suppl 1):S2. doi: 10.1186/ar2166. [DOI] [PMC free article] [PubMed] [Google Scholar]