

Abstract
Opioids display ligand-specific differences in the time course of ERK1/2 signaling. Whereas full agonists, like etorphine, induce only transient activation of ERK1/2, the partial agonist morphine mediates persistent stimulation of mitogenic signaling. Here we report that in stably δ-opioid receptor (DOR)-expressing HEK293 (HEK/DOR) cells, the transient nature of etorphine-induced ERK1/2 signaling is due to desensitization of epidermal growth factor (EGF) receptor-mediated activation of the Ras/Raf-1/ERK1/2 cascade. Desensitization of ERK1/2 activity by etorphine is associated with down-regulation of EGF receptors, an effect mediated by the ubiquitin ligase c-Cbl. In contrast, chronic morphine treatment failed to desensitize EGF receptors, resulting in unimpeded ERK1/2 signaling. The failure of morphine to desensitize ERK1/2 signaling is mediated by persistent activation of c-Src, which induces degradation of c-Cbl. The role of c-Src in opioid-specific ERK1/2 signaling is further demonstrated by pretreatment of the cells with PP2 and SKI-I as well as overexpression of a dominant negative c-Src mutant (c-Src(dn)) or a c-Src-resistant c-Cbl mutant (CblY3F), both of which facilitate desensitization of ERK1/2 signaling by morphine. Conversely, overexpression of c-Src as well as down-regulation of c-Cbl by small interfering RNA results in persistent etorphine-induced stimulation of ERK1/2 activity. Subcellular fractionation experiments finally attributed the ability of morphine to persistently activate c-Src to its redistribution from Triton X-100-insensitive membrane rafts to DOR and EGF receptor containing high density membrane compartments implicated in ERK1/2 signaling. These results demonstrate that agonist-specific differences in the temporal and spatial pattern of c-Src activation determine the kinetics of DOR-mediated regulation of ERK1/2 signaling.
Introduction
Activation of G protein-coupled receptors (GPCRs)2 results in stimulation of ERK 1/2, two members of the family of MAPKs implicated in cell growth, differentiation, and proliferation (1). The intracellular pathways mediating ERK1/2 stimulation include transactivation of receptor tyrosine kinases (RTK), e.g. the epidermal growth factor (EGF) receptor, which integrates a number of different GPCR-derived signals to activation of the well conserved Ras/Raf-1/ERK1/2 signal transduction module (2). Generally, the duration of ERK1/2 signaling by GPCRs is transient and desensitizes rapidly within minutes after receptor stimulation (3). This time course is best demonstrated for δ-opioid receptor (DOR) carrying cell lines and tissues, in which a number of full opioid agonists, like the alkaloid etorphine and the opioid peptide [d-Pen2,5]enkephalin, produce short term stimulation of ERK1/2 signaling that peaks after 5 min and completely reverts to control levels within 60 min of receptor stimulation (4, 5). Desensitization of GPCR-induced mitogenic signaling is mediated by degradation rather than phosphorylation and internalization of transactivated RTKs, because endocytosed EGF receptors are still able to mediate ERK1/2 activation (6, 7). Besides the induction of transient mitogenic signaling, the DOR system is also characterized by the ability of morphine to induce long lasting stimulation of the ERK1/2 pathway (8). Because stimulation of ERK1/2 activity by morphine is also mediated by transactivation of EGF receptors (4), it might be speculated that the ligand-specific property of morphine to produce persistent ERK1/2 stimulation is mediated by its failure to desensitize RTK signaling.
Morphine, a partial alkaloid agonist, differs from other opioids because it fails to induce opioid receptor desensitization and internalization (9). As a consequence, chronic morphine treatment is associated with a number of compensatory adaptations on a post-receptor level, including quantitative and qualitative changes in G proteins (10), effectors (11), and regulators of GPCR sensitivity (12, 13). Among the latter, protein kinase A- and protein kinase C-mediated phosphorylation of Gβ subunits, adenylyl cyclase type II, and G protein-coupled receptor kinase 2 (GRK2) has been shown to enhance opioid receptor signaling (12, 14). In addition, chronic morphine treatment also results in inhibition of β-arrestin1 function, an adapter protein that plays a central role in receptor sequestration and endocytosis. Because the β-arrestin1-dependent pathway is not specific for a certain receptor, chronic morphine treatment not only results in attenuation of homologous desensitization but also of heterologous desensitization of other inhibitory GPCRs, like the CB1 cannabinoid and M4 muscarinic receptor (8). Besides their role in GPCR signaling, β-arrestin1, GRK2, and Gβγ subunits have also been shown to regulate the function of diverse RTKs, like the insulin-like growth factor I receptor, platelet-derived growth factor receptor, and the EGF receptor (15–17). Thus, it is tempting to speculate whether chronic opioid treatment might also interfere with the regulation of RTK function. Indeed, chronic morphine treatment has been recently shown to promote insulin receptor signaling by increasing receptor abundance (18).
The function of EGF receptors is regulated by c-Cbl, an adaptor protein of the family of casitas B-lineage lymphoma proteins (19). In response to receptor activation, the primarily cytosolic protein is targeted to the autophosphorylated EGF receptor (Tyr1045) where it transfers ubiquitin to the C terminus and directs the receptor to proteasomal degradation (20). The activity of c-Cbl in turn is subject to regulation by the Src kinase family (19). Besides their role in regulation of EGF receptor activity, Src kinases are also involved in a number of different GPCR mechanisms (21, 22), providing a potential target for cross-regulation of c-Cbl function by GPCRs (23). In this respect, c-Src has been implicated in many aspects of DOR function, including the duration of opioid-stimulated ERK1/2 signaling (5). Thus, the aim of the present study was to investigate whether c-Src-regulated c-Cbl is involved in the termination of DOR-stimulated ERK1/2 signaling by full opioid agonists and whether morphine differs in this aspect. Our results demonstrate that in stably DOR-expressing HEK293 cells, chronic morphine treatment is associated with subcellular redistribution and persistent activation of c-Src, a ligand-specific property that prevents EGF receptor down-regulation and enables persistent ERK1/2 signaling by degradation of c-Cbl.
EXPERIMENTAL PROCEDURES
Cell Culture
HEK293 cells stably expressing the wild type or HA-tagged form of the mouse DOR (HEK/DOR and HEK/HA-DOR; 1.4 ± 0.2 and 1.7 ± 0.1 pmol receptors/mg membrane of protein, respectively) were generated as described (8). The cells were cultured under standard conditions in Dulbecco's modified Eagle's medium containing 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum in a humidified atmosphere of 5% CO2 in air at 37 °C. Where indicated, the cells were transfected to transiently overexpress c-Cbl, a c-Src-resistant mutant of c-Cbl (CblY3F), c-Src, and a dominant negative mutant of c-Src (c-Src(dn)), using the Metafectene ProTM transfection reagent (Biontex Laboratories, Martinsried, Germany). All of the cDNAs were subcloned into plasmid pcDNA3.1 (Invitrogen), and the cells were used for experimentation 24 h after transfection. The cells transfected with empty vector served as controls throughout. Knockdown of c-Cbl expression was achieved by transfection of HEK/DOR cells with three target-specific siRNAs (Santa Cruz Biotechnology, Santa Cruz, CA) according to the manufacturer's protocol. The cells transfected with control siRNA-A from the same company were used as the control.
Cell Treatment
Naive or transiently transfected HEK/DOR cells were seeded onto 12-well tissue culture plates and allowed to grow overnight. Subconfluent monolayers were then washed and equilibrated to serum-reduced conditions (Dulbecco's modified Eagle's medium containing 0.1% fetal calf serum) for 2 h at 37 °C. The cells were treated with etorphine, morphine HCl, EGF, and phorbol 12-myristate 13-acetate for the times and concentrations indicated in the text. In some experiments, the function of EGF receptors and c-Src was inhibited by preincubation of the cells for 15 min with 4-(3-chloroanillino)-6,7-dimethoxyquinazoline (AG1478) (34), 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4 d]pyrimidine (PP2) (24), and 4-phenoxyanilino-6,7-dimethoxy-quinazoline (SKI-I) (25) as described before (24, 25). All of the reactions were stopped by aspiration of the incubation medium and subsequent solubilization of the cells using 150 μl of ice-cold Laemmli sample buffer (62.5 mm Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 50 mm dithiothreitol, and 0.01% phenol red). The cell lysates were centrifuged (10 min; 15,000 × g) before the supernatants were heated for 5 min to 95 °C.
c-Cbl Redistribution
HEK/DOR cells were plated on 22-mm coverslips and transfected with c-Cbl subcloned into pEGFP-C3 (BD Clontech, Heidelberg, Germany). The next day, the cells were washed and then cultured for 2 h in the absence of serum before redistribution of c-Cbl was initiated by the addition of 100 nm etorphine and 1 μm morphine for 5 min at 37 °C. Subsequently the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline, mounted on glass slides, and analyzed by confocal microscopy (Carl Zeiss, Jena, Germany). The images shown were acquired using a 63×/1.4 oil immersion objective.
Subcellular Fractionation
HEK/HA-DOR cells were grown to confluence in 10-cm2 dishes, washed twice, and incubated for 1 h under serum-free conditions. The cells were kept for an additional hour in the absence (control) or presence of 100 nm etorphine and 1 μm morphine, before they were scraped into ice-cold phosphate-buffered saline, washed twice by centrifugation at 300 × g for 10 min, and subjected to subcellular fractionation after solubilization with 1% Triton X-100 as previously reported (26). All of the steps were performed at 4 °C. The cells were homogenized in 0.6 ml of TME lysis buffer (50 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1% (v/v) Triton X-100, and Complete® protease inhibitors), containing 40% (m/v) sucrose, by 10 passages through a 22-gauge needle. The homogenate was placed on the bottom of a 2.2-ml ultracentrifuge tube and overlaid with of 0.8 ml of 30% sucrose and 0.8 ml of 5% sucrose in TME lysis buffer to form a discontinuous gradient. The samples were centrifuged at 200,000 × g for 18 h using a S-55S swinging bucket rotor in a Sorvall RC M120GX ultracentrifuge (Sorvall Deutschland GmbH, Bad Homburg, Germany). Twelve fractions (∼183 μl each) were collected from the top and analyzed for the presence of the membrane raft marker caveolin (fractions 5–7) and the non-raft marker transferrin receptor (fractions 11 and 12) by Western blot. These fractions were pooled, mixed with 5× Laemmli sample buffer, and designated as low density membrane rafts or high density membranes.
SDS-PAGE and Western Blot
Whole cell proteins or sucrose density gradient samples were resolved by electrophoresis over 8 (EGF receptor), 10 (c-Cbl, c-Src, ERK1/2, HA-DOR, transferrin receptor), or 12% (caveolin, H-Ras) SDS-polyacrylamide gels, before proteins were blotted onto polyvinylidene difluoride membranes (Millipore, Billerica, MA). The blots were blocked for 1 h with 5% casein in Tris-buffered saline, containing 0.1% Tween 20 (TBS/T) and incubated overnight at 4 °C with antibodies recognizing the following proteins: caveolin, EGF receptor, H-Ras, ERK1/2, phospho(Thr202/Tyr204)ERK1/2, phospho(Tyr527)c-Src (Cell Signaling Technology, Danvers, MA), c-Cbl, c-Src (Santa Cruz Biotechnology, Santa Cruz, CA), transferrin receptor (BD Transduction Laboratories, San Jose, CA), and HA tag (generated in our own laboratory). All of the antibodies were diluted in TBS/T containing 0.1% casein and used at the concentrations recommended by the supplier. The membranes were washed three times, and bound antibodies were labeled by incubation for 1 h with anti-rabbit or anti-mouse-IgG coupled to horseradish peroxidase (Promega, Mannheim, Germany). After extensive washing with TBS/T, the blots were developed using the enhanced chemiluminescence system (Amersham Biosciences ECL, GE Healthcare Lifescience, Munich, Germany), and the density of the immunoreactive bands was quantified by video densitometry.
Statistical Analysis
The data are presented as the means ± S.D. and were tested for statistical significance by an unpaired Student's t test.
RESULTS
Morphine Induces Sustained EGF Receptor Signaling
Treatment of HEK/DOR cells with a maximal effective concentration of etorphine (100 nm) results in transient stimulation of ERK1/2 activity, which is maximal after 5 min and rapidly desensitizes within 60 min of receptor activation (Fig. 1A). Morphine (1 μm) displays a fundamental different kinetics, because it produces persistent ERK1/2 stimulation, which is still maximal even after 60 min of cell treatment. The failure of morphine to desensitize DOR-stimulated ERK1/2 signaling is further demonstrated by the ability of etorphine (5 min at 100 nm) to fully stimulate ERK1/2 activation in cells pretreated for 60 min with morphine but not with etorphine (Fig. 1A). Because opioid-induced stimulation of ERK1/2 signaling is mediated by transactivation of EGF receptors (Fig. 1B), we investigated next whether the differences in ERK1/2 signaling observed might be possibly because of opioid-specific regulation of RTK activity. For this, HEK/DOR cells were pretreated for 60 min with etorphine or morphine before the effect of EGF on MAPK stimulation was determined. As shown in Fig. 1C (lower panel), the addition of EGF (10 ng/ml for 5 min) to naïve cells results in strong stimulation of ERK1/2 phosphorylation. Most interestingly, pretreatment of the cells with a maximum effective concentration of etorphine (100 nm) obtained in preliminary dose-response experiments (not shown) completely abolished this effect without affecting the overall abundance of these MAPKs (Fig. 1C, upper panel). On the other hand, exposure of HEK/DOR cells for 60 min to morphine (1 μm) fails to interfere with EGF-induced ERK1/2 activation. These findings indicate that in HEK/DOR cells, chronic etorphine treatment results in rapid desensitization of EGF-stimulated ERK1/2 signaling, whereas morphine does not.
FIGURE 1.

Etorphine, but not morphine, desensitizes EGF receptor activation of ERK1/2. A, serum-starved HEK/DOR cells were exposed to 1 μm morphine or 100 nm etorphine for 5 and 60 min to stimulate ERK1/2 signaling. In some experiments, the cells were first pretreated with morphine (1 μm; mor60) or etorphine (100 nm; eto60) for 60 min, washed, and subsequently stimulated with the opioids indicated for 5 min. Controls (cn) were left untreated. The cells were lysed by the addition of sample buffer and examined for ERK1/2 activation by Western blot using of a phospho-specific antibody (p-ERK). B, HEK/DOR cells were pretreated with the EGF receptor inhibitor AG1478 (5 μm; 15 min) before 100 nm etorphine (eto) and 1 μm morphine (mor) were added for 5 min to determine ERK1/2 phosphorylation. Controls (cn) were left untreated. The cell lysates were prepared and analyzed for total (ERK) and phosphorylated ERK1/2 (p-ERK). C, serum-starved HEK/DOR cells were pretreated with etorphine (100 nm; eto) and morphine (1 μm; mor) for 60 min, washed, and subsequently stimulated with EGF (10 ng/ml) for 5 min. Controls (cn) received EGF alone. D, serum-starved HEK/DOR cells were incubated with 100 nm etorphine and 1 μm morphine for 60 min before ERK1/2 activation by cell exposure to 100 nm phorbol 12-myristate 13-acetate (PMA) for 5 min was evaluated. The cells of the same passage treated with the vehicle alone served as controls (cn). Stimulation of ERK1/2 activity was measured by Western blot using a phospho-specific antibody. Equal protein load was verified by analyzing the samples with an overall reactive ERK1/2 antibody. In each experiment, the intensity of phospho-ERK1/2 immunoreactivity was scanned and quantitated by video densitometry. ERK1/2 activation is expressed as the percentage of change from maximum stimulation, which was set to 100%. The data shown are the mean values ± S.D. from at least three independent experiments. ***, p < 0.001 versus nontreated controls (cn).
To elucidate whether attenuation of EGF-stimulated ERK1/2 signaling is because of desensitization of EGF receptor activity or to downstream adaptations within the Ras/Raf-1/ERK1/2 signaling pathway, the effect of opioid pretreatment on protein kinase C-stimulated ERK1/2 activity was tested. Phorbol 12-myristate 13-acetate (100 nm; 5 min) is an activator of protein kinase C, which directly stimulates Raf-1 (27) and thus may be used as a tool to investigate EGF receptor-independent stimulation of the ERK1/2 pathway (28). As shown in Fig. 1D, both chronic etorphine and morphine failed to affect the ability of phorbol 12-myristate 13-acetate to activate ERK1/2. This finding indicates that chronic etorphine pretreatment attenuates EGF-induced ERK1/2 stimulation by desensitization of EGF receptor function.
Chronic Morphine Induces Down-regulation of c-Cbl
Desensitization of GPCR-stimulated ERK1/2 signaling is mediated by EGF receptor degradation (7). This mechanism is associated with agonist-induced redistribution of c-Cbl to the plasma membrane, a cytosolic adaptor protein directing activated RTKs from lipid rafts to proteasomal degradation (20, 29). In HEK/DOR cells transiently overexpressing an EGFP-tagged form of c-Cbl, only etorphine (100 nm), but not morphine (1 μm) treatment is able to induce a punctuate accumulation of the construct at the plasma membrane (Fig. 2A). This observation raises the possibility as to whether in HEK/DOR cells etorphine treatment might possibly cross-regulate EGF receptor function. Because functional desensitization of EGF receptor activity occurs before their degradation in the endosomal compartment (30), the overall abundance of EGF receptors in whole cell preparations was determined as a measure for chronic opioid-induced RTK down-regulation. As shown in Fig. 2B, exposure of HEK/DOR cells to etorphine (100 nm) results in a time-dependent loss of EGF receptor immunoreactivity in whole cell preparations, which is complete after 90 min of cell exposure. In contrast, chronic morphine treatment for up to 90 min had no effect on EGF receptor abundance. These results demonstrate that sustained DOR activation by etorphine, but not morphine, results in substantial EGF receptor down-regulation within 60 min of drug exposure.
FIGURE 2.

Regulation of c-Cbl and EGF receptors by etorphine and morphine. A, HEK/DOR cells transfected with EGFP-tagged c-Cbl (green) were left untreated (cn) or were stimulated with 100 nm etorphine and 1 μm morphine for 5 min at 37 °C. The cells were fixed and cellular distribution of c-Cbl was analyzed by confocal microscopy. In unstimulated controls (cn), fluorescence associated with c-Cbl is distributed homogenously throughout the cytoplasm (* marks the nucleus). The addition of etorphine, but not morphine, results in a punctate accumulation of c-Cbl at the plasma membrane (arrows) and the cytosol. The images show representative cells taken from one of three independent experiments (space bar, 10 μm). B, serum-starved HEK/DOR cells were treated with etorphine (100 nm) and morphine (1 μm) for 5–60 or 90 min before the abundance of the EGF receptor (left panel) and of c-Cbl (right panel) was determined in whole cell lysates by Western blot technique. The abundance of EGF receptors and c-Cbl was quantitated by video densitometry and is expressed as a percentage of change from nontreated controls, which was set to 100%. The data shown are the mean values ± S.D. from at least three independent experiments. ***, p < 0.001; ** p < 0.01 versus non-opioid incubated control (0 min).
We next investigated whether the failure of morphine to down-regulate the EGF receptor is because of regulatory changes at the level of c-Cbl. For this, HEK/DOR cells were exposed for up to 60 min to maximum effective concentrations of either etorphine or morphine and were subsequently analyzed for c-Cbl abundance. Immunoblot analysis of whole cell lysates obtained from morphine-treated cells revealed a time-dependent decrease in c-Cbl immunoreactivity. In contrast, the relative amount of c-Cbl remained unchanged after etorphine treatment (Fig. 2B). Thus, long term exposure of HEK/DOR cells to morphine, but not to etorphine, is associated with down-regulation of c-Cbl.
Overexpression of c-Cbl Restores the Ability of Morphine to Down-regulate the EGF Receptor and to Desensitize ERK1/2 Signaling
The duration of EGF receptor-stimulated ERK1/2 signaling is prolonged in c-Cbl-deficient HeLa cells (31). To determine whether down-regulation of c-Cbl might be responsible for the ability of morphine to induce persistent ERK1/2 stimulation, morphine-stimulated MAPK signaling was analyzed in transiently c-Cbl-overexpressing HEK/DOR cells. In control cells carrying endogenous levels of this adaptor protein, exposure to morphine for both 5 and 60 min results in a pronounced stimulation of ERK1/2 phosphorylation. After moderate overexpression of c-Cbl, morphine gains the ability to desensitize ERK1/2 signaling after 60 min of treatment, an effect that is accompanied by down-regulation of EGF receptor abundance (Fig. 3). Heterologous expression of higher c-Cbl concentrations results in the reduction of basal levels of EGF receptor abundance and further decreases ERK1/2 phosphorylation after 5 min of morphine treatment. Under these conditions, MAPK signaling is completely desensitized after 60 min of morphine exposure (Fig. 3). These results illustrate that overexpression of c-Cbl converts morphine into an opioid agonist that is now able to down-regulate EGF receptors and to desensitize ERK1/2 signaling.
FIGURE 3.

Overexpression of c-Cbl induces desensitization of morphine-stimulated ERK1/2 signaling. A, HEK/DOR cells were transfected with plasmid encoding c-Cbl (0.5/1 μg) and treated with 1 μm morphine for 5 and 60 min. The reactions were stopped, and cell lysates were subjected to Western blot analysis using antibodies against the EGF receptor (EGFR), c-Cbl, ERK1/2, and phospho-ERK1/2. The intensity of ERK1/2 bands was quantitated by video densitometry and is expressed as percentage change from maximum stimulation, which was set to 100%. B, HEK/DOR cells overexpressing c-Cbl were kept in the absence (0) or presence of morphine for 5 and 60 min before cells were lysed, and EGF receptors were determined by Western blotting. The density of immunoreactive bands was quantitated by video densitometry. The data were normalized to untreated controls (0 min), which were set to 100%. The data shown are the mean values ± S.D. from at least three independent experiments. ***, p < 0.001 versus non-morphine treated control (0 min).
Down-regulation of c-Cbl by Morphine Is Mediated by c-Src
Because proteasomal destruction of c-Cbl is under the control of c-Src (32), regulation of this tyrosine kinase by the opioids was investigated next. Under basal conditions, c-Src is inactivated by constitutive phosphorylation at Tyr527 (33). Stimulation of the cells for 5–60 min with morphine (1 μm) results in rapid and sustained dephosphorylation of Tyr527, without affecting total c-Src abundance. In contrast, exposure of the cells to etorphine (100 nm) failed to affect the phosphorylation state of c-Src. These findings demonstrate that morphine is able to mediate prolonged activation of c-Src, whereas etorphine does not (Fig. 4A).
FIGURE 4.

Chronic morphine-induced down-regulation of c-Cbl and EGF receptors is mediated by prolonged c-Src activation. A, serum-starved HEK/DOR cells were incubated with 100 nm etorphine and 1 μm morphine for up to 60 min. The controls were left untreated (0 min). Subsequently, the cells were lysed by the addition of sample buffer and examined for total (Src) and Tyr527-phosphorylated c-Src (p-Src) by Western blotting. B, serum-starved HEK/DOR cells either transfected with the dominant negative c-Src mutant c-Src(dn) CblY3F or pretreated with PP2 (1 μm) and SKI-I (10 μm) were treated with morphine (1 μm) for up to 60 min before cells lysates were prepared and examined for EGF receptor, c-Cbl, and ERK1/2 regulation by Western blot. Mock transfected cells served as controls (mock). The blots shown were repeated at least three times yielding similar results. ERK1/2 activation was quantitated by video densitometry and is expressed as a percentage of change from maximum stimulation (100%). C, quantification of EGF receptor abundance. The density of EGF receptor immunoreactive bands was quantified by video densitometry. In each experiment, the data were normalized to untreated cells (0 min), which were set to 100%. The data represented are the mean values ± S.D. ***, p < 0.001; **, p < 0.01; *, p < 0.05 versus acute morphine-treated cells (5 min).
To further evaluate whether morphine-specific regulation of c-Src activity possibly contributes to sustained MAPK activation by this opiate, the effect of the Src inhibitors PP2 and SKI-I on chronic morphine-induced ERK1/2 signaling was examined. At concentrations specific for inhibition of c-Src, pretreatment of the cells with both inhibitors completely blocked chronic morphine-induced down-regulation of c-Cbl. As a consequence, both c-Src inhibitors facilitated down-regulation of EGF receptors and desensitization of ERK1/2 signaling by chronic morphine. A similar observation is made in morphine-treated cells after overexpression of c-Src(dn), a dominate negative mutant that competes with endogenous c-Src (Fig. 4B). These results demonstrate that persistent activation of c-Src by chronic morphine results in down-regulation of c-Cbl, which prevents desensitization of EGF receptors and ERK1/2 signaling.
Besides its regulatory role on c-Cbl stability, c-Src also interferes with a number of additional steps in GPCR-stimulated ERK1/2 signaling (34). To exclude such a possibility, HEK/DOR cells were transiently transfected with a c-Src-resistant mutant of c-Cbl (CblY3F) (31) and subsequently analyzed for chronic morphine-induced regulation of ERK1/2 signaling. Indeed, CblY3F is able to bind to and ubiquitinate the EGF receptor (35) but is resistant to c-Src mediated degradation (32). In cells overexpressing this c-Src resistant mutant, chronic morphine treatment results in time-dependent degradation of EGF receptors (Fig. 4C), indicating that c-Src-mediated activation and degradation of c-Cbl indeed prevents desensitization of ERK1/2 signaling.
Overexpression of c-Src Promotes Persistent ERK1/2 Activation by Etorphine
The finding that persistent activation of c-Src by chronic morphine treatment is responsible for c-Cbl degradation means that overexpression of c-Src should block the ability of etorphine to terminate ERK1/2 signaling. To test this hypothesis, the effect of chronic etorphine treatment on mitogenic signaling was evaluated in the presence of overexpressed c-Src kinase. As shown in Fig. 5A, overexpression of c-Src in HEK/DOR cells is associated with an increase in basal ERK1/2 activity, which is further stimulated after DOR activation by etorphine. However, in contrast to control cells, the ability of etorphine to desensitize ERK1/2 signaling is completely lost. Overexpression of c-Src by itself reduces basal levels of c-Cbl abundance. Under these conditions, exposure of the cells to etorphine for 60 min results in substantial down-regulation of c-Cbl, leaving EGF receptor density unaffected. To further examine whether degradation of c-Cbl underlies persistent ERK1/2 signaling by morphine, HEK/DOR cells were transfected with c-Cbl siRNA. Although expression of c-Cbl may not be completely abrogated by this technology, the proteins remaining should be insufficient to promote etorphine-induced EGF receptor degradation. Indeed, the amount of EGF receptors remained unaffected after treatment of the cells for 5–60 min with etorphine (100 nm). Moreover, knockdown of c-Cbl by siRNA now prevents etorphine-induced desensitization of ERK1/2 signaling. Together, these results demonstrate that etorphine is unable to produce persistent stimulation of c-Src activity sufficient for down-regulation of c-Cbl and termination of EGF receptor-mediated ERK1/2 signaling.
FIGURE 5.

Overexpression of c-Src and siRNA-induced knockdown of c-Cbl both prevent etorphine-mediated desensitization of ERK1/2 signaling. HEK/DOR cells were transfected either with c-Src or with siRNA for c-Cbl and were incubated with 100 nm etorphine for the times indicated. Mock transfected cells (mock) or cells transfected with control siRNA-A were incubated with etorphine in parallel and served as controls. The cell lysates were subjected to SDS-PAGE and examined for expression of c-Src, EGF receptor, and c-Cbl as well as regulation of the ERK1/2 activity by Western blot. ERK1/2 activation (A) and EGF receptor abundance (B) was quantitated by video densitometry and is expressed as the percentage of change from the maximum effect, which was set to 100%. The data shown are the mean values ± S.D. from at least three independent experiments. ***, p < 0.001; **, p < 0.01 versus acute etorphine-treated cells (5 min).
Chronic Morphine Alters the Subcellular Localization of c-Src
We finally investigated the mechanism by which morphine is able to bring about persistent activation of c-Src, whereas etorphine does not. In this respect, a recent report demonstrated that in polarized Madin-Darby canine kidney cells there are two distinct subcellular pools of c-Src responsible for the induction of different intracellular signal transduction pathways (26). One compartment is associated with heavy endosomal membranes and mediates rapid activation of the ERK1/2 cascade. The other pool is associated with a subset of Triton X-100-insoluble “membrane rafts” and controls sustained activation of the phosphatidylinositol 3-kinase/Akt pathway. To explore whether etorphine and morphine might possibly activate different pools of c-Src, subcellular distribution of key components involved in DOR-mediated ERK1/2 activation was analyzed by sucrose density fractionation of naïve and opioid-treated HEK/HA-DOR (Fig. 6). After homogenization of the cells in the presence of 1% (v/v) Triton X-100, the entire amount of DOR and EGF receptor immunoreactivity was found in a Triton X-100-soluble “non-raft” fraction comprised of high density endosomal and plasma membranes, which are characterized by the presence of the transferrin receptor (36). Under these experimental conditions, H-Ras and c-Src were found both in the Triton X-100-sensitive non-raft as well as to a lower extent also in a Triton X-100-insensitive low density membrane fraction, which is characterized by the presence of the membrane raft marker caveolin. Chronic etorphine treatment largely reduced the amount of EGF receptors in the non-raft fraction because of protein degradation, whereas it had no effect on HA-DOR immunoreactivity. Although chronic morphine treatment failed to recruit HA-DOR and EGF receptors to membrane rafts, it completely shifted the proportion of c-Src located in the membrane rafts to the high density membrane compartment. These data verify that the DOR is extracted out of membrane rafts by the presence of 1% (v/v) Triton X-100 and that chronic morphine recruits the entire amount of c-Src to a Triton X-100-soluble membrane compartment also containing DOR and EGF receptors and implicated in mediating activation of the ERK1/2 pathway.
FIGURE 6.
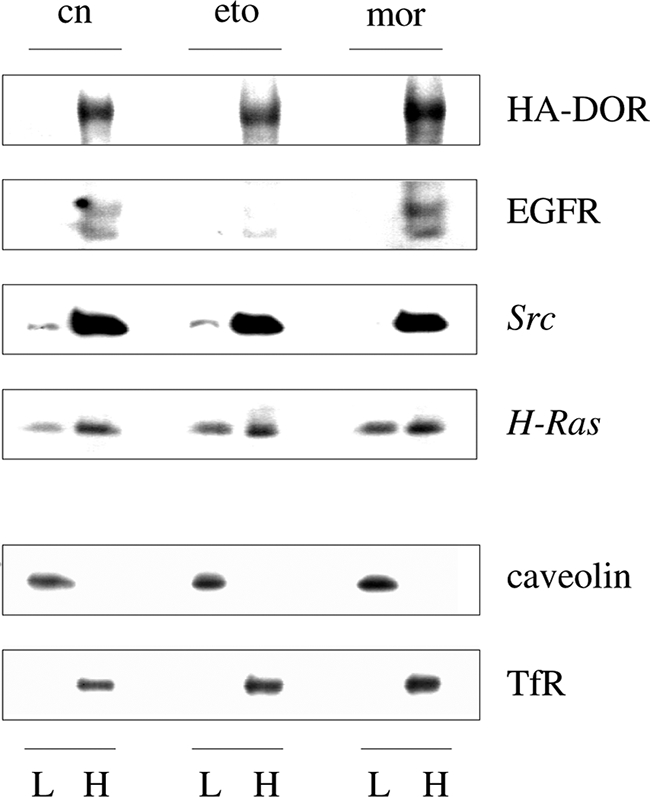
Chronic morphine redistributes c-Src from membrane rafts to detergent soluble non-raft membranes. HEK/HA-DOR cells kept for 1 h either in the absence (cn) or presence of 100 nm etorphine (eto) or 1 μm morphine (mor) were homogenized in the presence of 1% (v/v) Triton X-100, before low density membrane rafts (L) and high density non-raft membranes (H) were separated by sucrose density gradient ultracentrifugation. Equal fraction volumes were subjected to Western blot analysis using antibodies recognizing the HA-DOR, EGF receptor (EGFR), c-Src, and H-Ras. The quality of membrane fractions was verified by staining the blots with the membrane raft and non-raft markers caveolin and transferrin receptor (TfR), respectively. Note the disappearance of c-Src from the low density, detergent-insoluble fraction after chronic morphine treatment. Representative blots of three different experiments are shown.
DISCUSSION
Prolonged DOR activation by morphine and etorphine results in differential kinetics of ERK1/2 signaling by an as yet unknown cellular mechanism. Because opioid-induced ERK1/2 stimulation is mediated by transactivation of EGF receptors (4), the present study focused on ligand-specific differences in the regulation of EGF receptor activity. Here we demonstrate that in DOR-expressing HEK293 cells chronic morphine treatment, but not etorphine treatment, results in c-Src-mediated down-regulation of c-Cbl, an E3 ubiquitin ligase required for the initiation of EGF receptor degradation. Thus, down-regulation of c-Cbl by morphine ultimately prevents EGF receptor degradation, a regulatory mechanism contributing to persistent stimulation of the Ras/Raf-1/ERK1/2 signaling cascade. These results provide the first example for ligand-specific differences in DOR-mediated cross-regulation of RTK activity, which might explain some of the individual properties of morphine in the regulation of cell proliferation (37), survival (38), and the development of tolerance and addiction (39).
Morphine is a partial agonist with low affinity for the DOR that displays fundamental differences in its signaling properties as compared with full opioid agonists. Most notably, morphine fails to induce agonist-induced receptor desensitization and internalization, regulatory mechanisms currently thought to contribute to the phenomenon of persistent ERK1/2 signaling observed after chronic morphine treatment (5). However, our previous work has indicated that blockade of DOR internalization by sucrose and concanavalin A is not associated with the prevention of rapid desensitization of the ERK1/2 signaling pathway by chronic etorphine treatment (4). Thus, the transient nature of ERK1/2 signaling must originate from downstream adaptations rather than from GPCR desensitization. Using the DOR system, the present study provides evidence for chronic etorphine-mediated EGF receptor down-regulation, a RTK connecting DOR-derived signals to activation of the well conserved Ras/Raf-1/ERK1/2 signaling module (40). A similar down-regulation of EGF receptors has been previously reported after 5-HT2A receptor activation in HEK293 cells (7) and after μ-opioid receptor activation in astrocytes (41). In both systems, GPCR-induced desensitization of EGF receptor activity is also associated with termination of ERK1/2 signaling. Our finding that only etorphine is able to specifically down-regulate EGF receptors strongly suggests that chronic morphine treatment produces long lasting mitogenic signaling by circumventing desensitization of RTK activity.
Desensitization of EGF receptor function is a multistep process in which ubiquitination and subsequent proteasomal degradation of the receptor by the adaptor protein c-Cbl plays a critical role (42). In fact, knock-out BT20 cells lacking c-Cbl are unable to undergo EGF receptor degradation (43). The current finding that chronic morphine treatment leads to a substantial decrease in the amount of c-Cbl correlates well with the failure of morphine to down-regulate EGF receptors. Thus, persistent mitogenic signaling induced by morphine is likely to be because of down-regulation of c-Cbl, a regulatory event preventing the degradation of EGF receptors and desensitization of ERK1/2 signaling. Such a mechanism appears plausible, because c-Cbl deficiency is associated with prolonged EGF receptor-mediated ERK1/2 signaling in HeLa cells (31).
Overexpression of c-Cbl converts morphine into an agonist that produces only transient stimulation ERK1/2 signaling, like etorphine. Because c-Cbl specifically regulates the EGF receptor rather than the DOR (44), the present finding further supports the notion that rapid desensitization of GPCR-induced mitogenic signaling is mediated by attenuation of EGF receptor function. A similar effect has been previously observed for Chinese hamster ovary cells, in which overexpression of c-Cbl also accelerated degradation of growth factor-activated EGF receptors (45). In addition, because chronic morphine treatment is known to down-regulate the ubiquitin ligase UCH-L1 in the nucleus accumbens (46, 47), the present findings are in line with the notion that down-regulation of c-Cbl is associated with the ligand-specific property of morphine to induce persistent EGF receptor signaling. Besides the c-Cbl pathway, desensitization of EGF receptor signaling might also be accomplished by GRK2-induced serine phosphorylation (17) and subsequent degradation of the RTK (48). Because DOR activation by a full agonist enhances GRK2 activity (49, 50), the contribution of such a mechanism to etorphine-mediated EGF receptor desensitization and subsequent degradation remains to be explored.
Degradation of c-Cbl is mediated by activation of c-Src (32), a tyrosine kinase previously suggested to contribute to the regulation of DOR-induced ERK1/2 signaling (5). Together with the observation that in HEK/DOR cells chronic morphine treatment, but not etorphine treatment, results in activation of c-Src, ligand-specific differences in the regulation of this tyrosine kinase may be decisive for the time course of MAPK signaling. Besides the control of c-Cbl, c-Src is also implicated in many other aspects of GPCR-mediated EGF receptor transactivation (51). As shown previously (5), inactivation of c-Src failed to prevent acute ERK1/2 activation. This finding indicates that in HEK/DOR cells c-Src is not involved in acute stimulation of the Ras/Raf-1/ERK1/2 signaling module. Indeed, DOR activation has been recently reported to transactivate EGF receptor-mediated ERK1/2 signaling by a protein kinase C-dependent (40), but c-Src-independent mechanism (52).
Regardless of the mechanism involved in EGF receptor transactivation, the present study indicates that c-Src plays a critical role in ligand-specific regulation of ERK1/2 signaling after sustained DOR activation. This notion is strengthened by the observation that both chemical inhibition of c-Src activity as well as overexpression of a dominant negative c-Src mutant (c-Src(dn)) and of a c-Src resistant c-Cbl mutant (CblY3F) result in chronic morphine-induced desensitization of ERK1/2 signaling and loss of EGF receptors. In addition, overexpression of c-Src mimicked the effect of morphine and enabled etorphine to down-regulate c-Cbl, leaving the amount of EGF receptors unchanged. Overexpression of the wild type as well as the constitutive active form of c-Src is known to diminish EGF receptor ubiquitination and degradation. This effect is mediated by Tyr371 phosphorylation and subsequent down-regulation of c-Cbl (32, 53). Thus, it is conceivable that the phenomenon of chronic morphine-induced degradation of c-Cbl is mediated by persistent activation of c-Src.
The question remains why chronic morphine and etorphine differentially regulate c-Src activity. The answer is probably linked to the failure of the partial agonist morphine to desensitize and internalize the receptor (9). This ligand inherent deficiency to terminate receptor activity in the continued presence of the agonist is prone to induce a series of compensatory adaptations on the post-receptor level, leading to an altered opioid receptor signaling (12, 14) or to the induction of alternative intracellular signaling pathways (21). With respect to mitogenic signaling, a recent report demonstrated that in μ-opioid receptor-transfected HEK293 cells, chronic morphine treatment stimulates ERK1/2 activity in a G protein-sensitive manner, whereas etorphine switches to a β-arrestin-dependent pathway (54). The authors explained the shift in the intracellular pathway mediating ERK1/2 activation by ligand-specific recruitment of the etorphine-activated μ-opioid receptor to “non-lipid” rafts, whereas the morphine-activated receptor remained in membrane rafts (55). Membrane rafts are specialized subcellular compartments enriched in cholesterol and certain sphingolipids that function as organizing centers for cellular signal transduction (26, 36). Although a fraction of the DOR has been previously identified in membrane rafts (56), which is even increased after agonist treatment (57), our experiments failed to identify such a subcellular distribution of the DOR in HEK/HA-DOR cells. This discrepancy is most likely because of the use of deviating experimental protocols. Whereas the DOR is only associated with membrane rafts when these are prepared in the absence of detergent, our studies were conducted with Triton X-100-solubilized cell homogenates, which results in the isolation of highly enriched and detergent-insoluble membrane rafts. Under these conditions, the entire proportion of DOR immunoreactivity is found in detergent-soluble non-raft membranes, and there is no indication of an agonist-induced shift of the receptor to membrane rafts, confirming previous data (58). Despite the failure of regulating DOR localization, our studies revealed that chronic morphine treatment results in the recruitment of the entire pool of membrane raft located c-Src to non-raft membranes. Here it co-localizes with the HA-DOR, EGF receptor, and H-Ras (Fig. 6 and Refs. 58 and 59) to form a potential subcellular compartment mediating ERK1/2 activation (26). These results indicate that chronic morphine treatment of HEK/HA-DOR cells results in the recruitment of c-Src to HA-DOR containing non-raft membranes, a regulatory mechanism possibly underlying the ability of morphine to persistently activate the ERK1/2 pathway.
Because c-Cbl represents an ubiquitously expressed regulator of diverse RTKs (19), c-Src-mediated down-regulation of c-Cbl could explain a number of well documented chronic morphine effects mediated by diverse RTK systems in both neuronal and non-neuronal tissues. For example, down-regulation of c-Cbl could explain persistent stimulation of ERK1/2 activity in neuronal tissues expressing the fibroblast growth factor receptor (60) or in brains of chronically morphine treated mice (61, 62). Because of its critical role in neurite outgrowth, down-regulation of c-Cbl could also contribute to chronic morphine-induced apoptosis of neuronal cells (19, 63). In non-neuronal tissues, degradation of c-Cbl could finally contribute to chronic morphine-induced glucose tolerance (18), impaired B-cell and monocyte responses (18, 64), and tumor cell survival (38, 65).
Acknowledgments
We are grateful to Dr. W. Y. Langdon (Australia), Dr. H. Y. Wong (Hong Kong), and Dr. A. Tsygankov (United States) for providing plasmids containing cDNAs for c-Cbl, c-Src, and c-Src(dn) as well as the Src-resistant c-Cbl mutant CblY3F. We also thank A. Blaschke for excellent technical assistance.
Footnotes
- GPCR
- G protein-coupled receptor
- DOR
- δ-opioid receptor
- EGF
- epidermal growth factor
- ERK
- extracellular signal-regulated protein kinase
- HEK
- human embryonic kidney
- RTK
- receptor tyrosine kinase
- GRK
- G protein-coupled receptor kinase
- c-Src kinase
- cellular sarcoma kinase
- dn
- dominant negative
- siRNA
- small interfering RNA
- MAPK
- mitogen-activated protein kinase
- HA
- hemagglutinin.
REFERENCES
- 1.Yoon S., Seger R. (2006) Growth Factors 24, 21–44 [DOI] [PubMed] [Google Scholar]
- 2.Pierce K. L., Luttrell L. M., Lefkowitz R. J. (2001) Oncogene 20, 1532–1539 [DOI] [PubMed] [Google Scholar]
- 3.Gurevich V. V., Gurevich E. V. (2008) Mol. Pharmacol. 74, 312–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisinger D. A., Schulz R. (2004) J. Pharmacol. Exp. Ther. 309, 776–785 [DOI] [PubMed] [Google Scholar]
- 5.Audet N., Paquin-Gobeil M., Landry-Paquet O., Schiller P. W., Piñeyro G. (2005) J. Biol. Chem. 280, 7808–7816 [DOI] [PubMed] [Google Scholar]
- 6.Xue L., Lucocq J. (1998) Cell Signal. 10, 339–348 [DOI] [PubMed] [Google Scholar]
- 7.Grewal J. S., Luttrell L. M., Raymond J. R. (2001) J. Biol. Chem. 276, 27335–27344 [DOI] [PubMed] [Google Scholar]
- 8.Eisinger D. A., Ammer H., Schulz R. (2002) J. Neurosci. 22, 10192–10200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keith D. E., Murray S. R., Zaki P. A., Chu P. C., Lissin D. V., Kang L., Evans C. J., von Zastrow M. (1996) J. Biol. Chem. 271, 19021–19024 [DOI] [PubMed] [Google Scholar]
- 10.Ammer H., Schulz R. (1993) Biochem. J. 295, 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chakrabarti S., Liu N. J., Gintzler A. R. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 13686–13691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakrabarti S., Oppermann M., Gintzler A. R. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 4209–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrer-Alcón M., La Harpe R., García-Sevilla J. A. (2004) Brain Res. Mol. Brain Res. 121, 114–122 [DOI] [PubMed] [Google Scholar]
- 14.Chakrabarti S., Gintzler A. R. (2003) Brain Res. Mol. Brain Res. 119, 144–151 [DOI] [PubMed] [Google Scholar]
- 15.Lin F. T., Daaka Y., Lefkowitz R. J. (1998) J. Biol. Chem. 273, 31640–31643 [DOI] [PubMed] [Google Scholar]
- 16.Kim J., Ahn S., Guo R., Daaka Y. (2003) Biochemistry 42, 2887–2894 [DOI] [PubMed] [Google Scholar]
- 17.Freedman N. J., Kim L. K., Murray J. P., Exum S. T., Brian L., Wu J. H., Peppel K. (2002) J. Biol. Chem. 277, 48261–48269 [DOI] [PubMed] [Google Scholar]
- 18.Sood A., Thakur V. S., Karmarkar M. G., Ahuja M. M. (2001) Endocr. Res. 27, 215–221 [DOI] [PubMed] [Google Scholar]
- 19.Swaminathan G., Tsygankov A. Y. (2006) J. Cell. Physiol. 209, 21–43 [DOI] [PubMed] [Google Scholar]
- 20.Duan L., Miura Y., Dimri M., Majumder B., Dodge I. L., Reddi A. L., Ghosh A., Fernandes N., Zhou P., Mullane-Robinson K., Rao N., Donoghue S., Rogers R. A., Bowtell D., Naramura M., Gu H., Band V., Band H. (2003) J. Biol. Chem. 278, 28950–28960 [DOI] [PubMed] [Google Scholar]
- 21.Zhang L., Zhao H., Qiu Y., Loh H. H., Law P. Y. (2009) J. Biol. Chem. 284, 1990–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmerman B., Simaan M., Lee M. H., Luttrell L. M., Laporte S. A. (2009) Cell Signal. 21, 103–110 [DOI] [PubMed] [Google Scholar]
- 23.Jacob C., Cottrell G. S., Gehringer D., Schmidlin F., Grady E. F., Bunnett N. W. (2005) J. Biol. Chem. 280, 16076–16087 [DOI] [PubMed] [Google Scholar]
- 24.Hanke J. H., Gardner J. P., Dow R. L., Changelian P. S., Brissette W. H., Weringer E. J., Pollok B. A., Connelly P. A. (1996) J. Biol. Chem. 271, 695–701 [DOI] [PubMed] [Google Scholar]
- 25.Tian G., Cory M., Smith A. A., Knight W. B. (2001) Biochemistry 40, 7084–7091 [DOI] [PubMed] [Google Scholar]
- 26.de Diesbach P., Medts T., Carpentier S., D‘Auria L., Van Der Smissen P., Platek A., Mettlen M., Caplanusi A., van den Hove M. F., Tyteca D., Courtoy P. J. (2008) Exp. Cell Res. 314, 1465–1479 [DOI] [PubMed] [Google Scholar]
- 27.Kolch W., Heidecker G., Kochs G., Hummel R., Vahidi H., Mischak H., Finkenzeller G., Marmé D., Rapp U. R. (1993) Nature 364, 249–252 [DOI] [PubMed] [Google Scholar]
- 28.Rubio I., Rennert K., Wittig U., Beer K., Dürst M., Stang S. L., Stone J., Wetzker R. (2006) Biochem. J. 398, 243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Melker A. A., van der Horst G., Calafat J., Jansen H., Borst J. (2001) J. Cell Sci. 114, 2167–2178 [DOI] [PubMed] [Google Scholar]
- 30.Burke P., Schooler K., Wiley H. S. (2001) Mol. Biol. Cell 12, 1897–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Timpson P., Lynch D. K., Schramek D., Walker F., Daly R. J. (2005) Cancer Res. 65, 3273–3280 [DOI] [PubMed] [Google Scholar]
- 32.Bao J., Gur G., Yarden Y. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 2438–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper J. A., King C. S. (1986) Mol. Cell. Biol. 6, 4467–4477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rozengurt E. (2007) J. Cell. Physiol. 213, 589–602 [DOI] [PubMed] [Google Scholar]
- 35.Levkowitz G., Waterman H., Ettenberg S. A., Katz M., Tsygankov A. Y., Alroy I., Lavi S., Iwai K., Reiss Y., Ciechanover A., Lipkowitz S., Yarden Y. (1999) Mol. Cell. 4, 1029–1240 [DOI] [PubMed] [Google Scholar]
- 36.Patel H. H., Murray F., Insel P. A. (2008) Annu. Rev. Pharmacol. Toxicol. 48, 359–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta K., Kshirsagar S., Chang L., Schwartz R., Law P. Y., Yee D., Hebbel R. P. (2002) Cancer Res. 62, 4491–4498 [PubMed] [Google Scholar]
- 38.Tegeder I., Geisslinger G. (2004) Pharmacol Rev. 56, 351–369 [DOI] [PubMed] [Google Scholar]
- 39.Martini L., Whistler J. L. (2007) Curr. Opin Neurobiol. 17, 556–564 [DOI] [PubMed] [Google Scholar]
- 40.Eisinger D. A., Ammer H. (2008) Cell Signal. 20, 2324–2331 [DOI] [PubMed] [Google Scholar]
- 41.Belcheva M. M., Tan Y., Heaton V. M., Clark A. L., Coscia C. J. (2003) Mol. Pharmacol. 64, 1391–1401 [DOI] [PubMed] [Google Scholar]
- 42.Roepstorff K., Grøvdal L., Grandal M., Lerdrup M., van Deurs B. (2008) Histochem. Cell Biol. 129, 563–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pennock S., Wang Z. (2008) Mol. Cell. Biol. 28, 3020–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanowitz M., Von Zastrow M. (2002) J. Biol. Chem. 277, 50219–50222 [DOI] [PubMed] [Google Scholar]
- 45.Levkowitz G., Waterman H., Zamir E., Kam Z., Oved S., Langdon W. Y., Beguinot L., Geiger B., Yarden Y. (1998) Genes Dev. 12, 3663–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li K. W., Jimenez C. R., van der Schors R. C., Hornshaw M. P., Schoffelmeer A. N., Smit A. B. (2006) Proteomics 6, 2003–2008 [DOI] [PubMed] [Google Scholar]
- 47.Liu Y., Fallon L., Lashuel H. A., Liu Z., Lansbury P. T., Jr. (2002) Cell 111, 209–218 [DOI] [PubMed] [Google Scholar]
- 48.Oksvold M. P., Thien C. B., Widerberg J., Chantry A., Huitfeldt H. S., Langdon W. Y. (2003) Oncogene 22, 8509–8518 [DOI] [PubMed] [Google Scholar]
- 49.Zhang J., Ferguson S. S., Law P. Y., Barak L. S., Caron M. G. (1999) J. Recept. Signal. Transduct Res. 19, 301–313 [DOI] [PubMed] [Google Scholar]
- 50.Marie N., Aguila B., Hasbi A., Davis A., Jauzac P., Allouche S. (2008) Cell Signal. 20, 1209–1220 [DOI] [PubMed] [Google Scholar]
- 51.Daub H., Wallasch C., Lankenau A., Herrlich A., Ullrich A. (1997) EMBO J. 16, 7032–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kodama H., Fukuda K., Takahashi T., Sano M., Kato T., Tahara S., Hakuno D., Sato T., Manabe T., Konishi F., Ogawa S. (2002) J. Mol. Cell Cardiol. 34, 139–150 [DOI] [PubMed] [Google Scholar]
- 53.Yokouchi M., Kondo T., Sanjay A., Houghton A., Yoshimura A., Komiya S., Zhang H., Baron R. (2001) J. Biol. Chem. 276, 35185–35193 [DOI] [PubMed] [Google Scholar]
- 54.Zheng H., Loh H. H., Law P. Y. (2008) Mol. Pharmacol. 73, 178–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng H., Chu J., Qiu Y., Loh H. H., Law P. Y. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 9421–9426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang P., Xu W., Yoon S. I., Chen C., Chong P. L., Liu-Chen L. Y. (2007) Biochem. Pharmacol. 73, 534–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.André A., Gaibelet G., Le Guyader L., Welby M., Lopez A., Lebrun C. (2008) Biochim. Biophys. Acta 1778, 1483–1492 [DOI] [PubMed] [Google Scholar]
- 58.Levitt E. S., Clark M. J., Jenkins P. M., Martens J. R., Traynor J. R. (2009) J. Biol. Chem. 284, 22108–22122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y. T., Song L., Templeton D. M. (2007) J. Cell. Physiol. 211, 205–212 [DOI] [PubMed] [Google Scholar]
- 60.Belcheva M. M., Haas P. D., Tan Y., Heaton V. M., Coscia C. J. (2002) J. Pharmacol. Exp. Ther. 303, 909–918 [DOI] [PubMed] [Google Scholar]
- 61.Berhow M. T., Hiroi N., Nestler E. J. (1996) J. Neurosci. 16, 4707–4715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Narita M., Ioka M., Suzuki M., Narita M., Suzuki T. (2002) Neurosci. Lett. 324, 97–100 [DOI] [PubMed] [Google Scholar]
- 63.Mao J., Sung B., Ji R. R., Lim G. (2002) J. Neurosci. 22, 7650–7661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carr D. J., Serou M. (1995) Immunopharmacology 31, 59–71 [DOI] [PubMed] [Google Scholar]
- 65.Adachi T., Kar S., Wang M., Carr B. I. (2002) J. Cell. Physiol. 192, 151–159 [DOI] [PubMed] [Google Scholar]