

Abstract
The 300-kDa cation-independent mannose 6-phosphate receptor (CI-MPR), which contains multiple mannose 6-phosphate (Man-6-P) binding sites that map to domains 3, 5, and 9 within its 15-domain extracytoplasmic region, functions as an efficient carrier of Man-6-P-containing lysosomal enzymes. To determine the types of phosphorylated N-glycans recognized by each of the three carbohydrate binding sites of the CI-MPR, a phosphorylated glycan microarray was probed with truncated forms of the CI-MPR. Surface plasmon resonance analyses using lysosomal enzymes with defined N-glycans were performed to evaluate whether multiple domains are needed to form a stable, high affinity carbohydrate binding pocket. Like domain 3, adjacent domains increase the affinity of domain 5 for phosphomannosyl residues, with domain 5 exhibiting ∼60-fold higher affinity for lysosomal enzymes containing the phosphodiester Man-P-GlcNAc when in the context of a construct encoding domains 5–9. In contrast, domain 9 does not require additional domains for high affinity binding. The three sites differ in their glycan specificity, with only domain 5 being capable of recognizing Man-P-GlcNAc. In addition, domain 9, unlike domains 1–3, interacts with Man8GlcNAc2 and Man9GlcNAc2 oligosaccharides containing a single phosphomonoester. Together, these data indicate that the assembly of three unique carbohydrate binding sites allows the CI-MPR to interact with the structurally diverse phosphorylated N-glycans it encounters on newly synthesized lysosomal enzymes.
Introduction
The 300-kDa cation-independent mannose 6-phosphate receptor (CI-MPR),4 along with the smaller 46-kDa homodimeric cation-dependent mannose 6-phosphate receptor (CD-MPR), play an essential role in the degradative metabolism of cells by delivering ∼60 different mannose 6-phosphate (Man-6-P)-tagged lysosomal enzymes from the secretory pathway to the endosomal/lysosomal system. Newly synthesized lysosomal enzymes are modified with phosphomannosyl residues on their N-glycans as they traverse the Golgi (1–3). In early Golgi compartments, UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase (GlcNAc phosphotransferase; IUBMB accession number EC 2.7.8.17), adds N-acetylglucosamine (GlcNAc)-1-phosphate to the C-6 hydroxyl group of mannose residues to form a phosphodiester, Man-P-GlcNAc (Fig. 1) (4–7). The second enzyme, N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase (uncovering enzyme) (IUBMB accession number EC 3.1.4.45), removes the GlcNAc moiety in the trans Golgi network to generate a phosphomonoester (Fig. 1) (8–12). This recognition system is critical for normal development because a genetic defect in GlcNAc phosphotransferase causes the lysosomal storage disorders mucolipidosis II and III, with the more severe of these disorders, mucolipidosis II, resulting in death often within the first decade of life (13).
FIGURE 1.

Modification of N-glycans on lysosomal enzymes. Phosphorylation of mannose residues on lysosomal enzymes' N-linked oligosaccharides is carried out by the GlcNAc phosphotransferase, which transfers GlcNAc-1-phosphate from UDP-GlcNAc to the C-6 hydroxyl group of mannose to form a Man-P-GlcNAc phosphodiester. The N-glycans can be further modified by the uncovering enzyme, which removes the GlcNAc moiety to expose the Man-6-P phosphomonoester. The five potential sites of mannose phosphorylation are indicated in gray. The three arms of the N-glycan are labeled and their mannose residues designated A–I are shown.
The CI-MPR has been shown to be more efficient in targeting lysosomal enzymes to the lysosome than the CD-MPR, both in vitro (14, 15) and in vivo (16–19). The large extracellular region of CI-MPR contains 15 homologous, contiguous domains that are referred to as “MRH” domains (mannose 6-phosphate receptor homology domains) (20) due to their similar size (∼150 residues), conservation of residues, and spacing of conserved cysteine residues (6 or 8 cysteines) involved in disulfide bonding. In addition, the crystal structure of seven (i.e. domains 1–3, domains 11–14) of the 15 domains of the CI-MPR has been determined to date, which shows that each of these MRH domains shares a similar fold (21–23). Its ability to function as an efficient cargo transporter is likely due to the fact that the CI-MPR contains three Man-6-P binding sites: essential residues for carbohydrate recognition are located within domains 3, 5, and 9 (24–30) (see Fig. 2A). Biochemical and biophysical analyses of truncated forms of the CI-MPR have begun to reveal the distinct properties of each of these three Man-6-P binding sites. For example, domain 9 when expressed alone binds lysosomal enzymes with high affinity, whereas domain 3 requires the presence of domains 1 and 2 to form a high affinity carbohydrate binding site (31). The crystal structure of domains 1–3 reveals that residues in domain 1 and 2 do not directly contact Man-6-P, but rather provide stabilizing interactions to the loops of the binding pocket housed within domain 3 (32). However, it is not known whether adjacent domains also influence the binding affinity of domain 5 and/or domain 9.
FIGURE 2.

Man-6-P binding sites of the CI-MPR. A, schematic diagram of the full-length CI-MPR and CD-MPR. The MPRs are type I integral membrane glycoproteins, each with an N-terminal signal sequence, an extracytoplasmic region, a single transmembrane region, and a C-terminal cytoplasmic domain. The CI-MPR has a large extracytoplasmic region comprised of 15 contiguous domains, each ∼150 residues in length. The location of the Man-6-P and IGF-II binding sites are indicated. B, the truncated CI-MPR constructs used in this study are shown. Constructs that contain a C-terminal tag of six histidine residues are indicated. The sCI-MPR was purified from fetal bovine serum. The jagged arrow indicates that the C terminus of the sCI-MPR is not defined as this soluble form of the receptor is generated by proteolysis (62, 63). Inset, purified proteins were resolved on 12 (left panel) or 7.5% (right panel) nonreducing SDS-polyacrylamide gels and visualized by silver staining. The two bands observed for the Dom5His construct (lane 1) represent glycosylated (21 kDa) and non-glycosylated (18 kDa) species as determined by endoglycosidase H digestion (data not shown). The slower mobility of the P. pastoris-derived Dom1–3His construct (lane 3, 45–50 kDa) compared with the Sf9-derived Dom1–3 construct (lane 4, 43 kDa) is due to the presence of the six histidine residues and the larger (Man8-Man12) N-glycans produced in P. pastoris (36) compared with the smaller Man3GlcNAc2 N-glycans typically produced in Sf9 insect cells (36).
The phosphorylated N-glycans of lysosomal enzymes that serve as endogenous ligands for the MPRs are heterogeneous in structure with respect to the type of N-glycan (i.e. high mannose or hybrid), their size, presence of Man-6-P phosphomonoester or Man-P-GlcNAc phosphodiester residues, number of phosphorylated mannose residues (GlcNAc phosphotransferase modifies one or two mannose residues per N-glycan), and the location of phosphomannosyl residues on different branches of the N-glycan (i.e. five different mannose residues can be modified by GlcNAc phosphotransferase) (Fig. 1) (4, 5, 33, 34). An unanswered question is whether each of the three Man-6-P binding sites of the CI-MPR is capable of recognizing such a wide repertoire of phosphorylated N-glycans.
To understand how the CI-MPR functions as an efficient transporter for its diverse cargo, we have further characterized the carbohydrate specificity of its three Man-6-P binding sites and have evaluated whether adjacent domains influence the function of these sites by: 1) probing oligosaccharide microarrays containing purified high mannose-type glycans that have been modified in vitro using recombinant GlcNAc phosphotransferase followed by mild acid hydrolysis to generate glycans containing phosphodiesters or phosphomonoesters, respectively; 2) quantitative surface plasmon resonance (SPR) analyses using a lysosomal enzyme, acid α-glucosidase (GAA), modified in vitro with recombinant GlcNAc phosphotransferase, uncovering enzyme, and/or sweet potato purple acid phosphatase to generate lysosomal enzymes highly enriched in either phosphomonoesters or phosphodiesters. The results show that the three Man-6-P binding sites differ in their carbohydrate specificity, and when assembled together in full-length CI-MPR, account for the broad range of phosphorylated N-glycans structures recognized by the CI-MPR.
EXPERIMENTAL PROCEDURES
Expression and Purification of CI-MPR Constructs
The constructs used in this study are shown in Fig. 2B. Generation of the Dom1–3 and Dom1–3His constructs (residues 1–432 of the bovine CI-MPR with or without a C-terminal His6 tag) (35), Dom7–11 construct (residues 1–50 and 932–1657 of the bovine CI-MPR) (35), Dom5His construct (residues 584–725 of the bovine CI-MPR followed by a His6 tag with Asn at position 711 was replaced with Gln to eliminate one of the two N-glycosylation sites and was shown to bind lysosomal enzymes with an affinity similar to the wild-type domain 5 construct (30)), and the Dom9His construct (residues 1184–1327 of the bovine CI-MPR followed by a His6 tag (27) in the pGAPZαA expression vector (Invitrogen)) were described previously.
The Dom1–3His cDNA construct (35) was cloned into the pGAPZαA expression vector. The Dom1–3His, Dom5His, and Dom9His constructs, which use the promoter from the glyceraldehyde-3-phosphate dehydrogenase gene for constitutive protein expression, were engineered in-frame with the 89-residue Saccharomyces cerevisiae α-factor signal sequence, resulting in proteins secreted from Pichia pastoris. Briefly, P. pastoris wild-type strain X-33 (Invitrogen) was transformed by electroporation, and Zeocin-resistant transformants were selected as described previously (36). Positive clones were inoculated in liquid medium (1% yeast extract, 2% peptone, and 2% dextrose), and cultures were harvested after 3 days of growth at 30°C. Following removal of the cells by centrifugation, the medium was dialyzed against nickel binding buffer (20 mm Tris, 10 mm imidazole, and 500 mm NaCl, pH 8.0). The dialyzed medium was passed over a nickel-nitrilotriacetic acid-agarose (Qiagen) column, washed, and then eluted with nickel binding buffer containing 50–100 mm imidazole.
The Dom5–9 construct (residues 1–50 and 584–1327 of the bovine CI-MPR) (25) was cloned into the pVL1392/3 baculovirus transfer vector (Pharmingen). Dom1–3, Dom5–9, and Dom7–11 were expressed in the fall army worm ovarian cell line Spodoptera frugiperda (Sf9, Expression Systems) as described previously (35), except that the cells were grown in shaker flasks using ESF921 serum-free medium (Expression Systems). The receptors were purified from the medium of transfected insect cells by pentamannosyl phosphate-agarose affinity chromatography (35).
Following affinity chromatography purification, all proteins were extensively dialyzed against MES buffer (50 mm MES, pH 6.5, 150 mm NaCl, 5 mm β-glycerophosphate, and 10 mm MnCl2) and concentrated by filtration using Vivaspin spin columns containing a polyethersulfone membrane with a 5-kDa nominal molecular mass limit. The Bradford protein assay (Bio-Rad) with bovine serum albumin as the standard was used to estimate protein yields.
Purification of Soluble CI-MPR from Fetal Bovine Serum
A soluble form of the CI-MPR (sCI-MPR) was purified from fetal bovine serum by phosphomannan affinity chromatography as described previously (37).
Generation and Purification of Phosphorylated Glycans Containing One or Two Phosphomonoesters or Phosphodiesters
High mannose-type N-linked oligosaccharides were obtained by peptide N-glycosidase F digestion of bovine RNase B and soybean agglutinin. The resulting mixture of reducing glycans, containing Man5GlcNAc2 through Man9GlcNAc2 were labeled at their reducing ends with the bifunctional fluorescent linker, 2-amino-N-(2-aminoethyl)-benzamide (AEAB) via direct conjugation through its arylamine group by reductive amination to form glycan-AEABs as described previously (38). The glycan-AEABs were separated according to their size and the 5 fractions (Man5–Man9) were separately incubated with recombinant GlcNAc phosphotransferase. The resulting phosphorylated glycans were purified by HPLC on a porous graphatized carbon column eluted with an acetonitrile gradient (15–35%), analyzed by MALDI-TOF, and identified by the masses of their molecular ions. Aliquots of the covered glycans (Man-P-GlcNAc phosphodiesters) were hydrolyzed with HCl (0.01 m HCl, 1 h at 100 °C) to remove the GlcNAc, generating the corresponding glycans that contain Man-6-P phosphomonoesters as described (see accompanying article, Song et al. (64)).
Glycan Microarray Analyses
The purified glycan-AEABs were covalently immobilized onto N-hydroxysuccinimide-activated glass slides via their free alkylamine as described previously (38). The slides were incubated sequentially for 1 h each with: 1) the soluble, bovine CI-MPR and the purified, soluble forms of bovine CI-MPR; 2) primary antibody (MPR-specific B14.5 or B3.5 rabbit polyclonal antibody, or mouse penta-His or mouse tetra-His monoclonal antibody); and 3) secondary fluorescent antibody (Alexa 488 goat anti-rabbit IgG or Alexa 488 goat anti-mouse IgG). All incubations were carried out at 25 °C in buffer containing 50 mm imidazole, pH 6.5, 150 mm NaCl, 10 mm MnCl2, as described for CD-MPR and CI-MPR in the accompanying article (64).
Generation of Recombinant GAA with GlcNAc1-P-6-Man (GAA Diester) and Man-6-P (GAA Monoester)
The GAA diester and the GAA monoester were prepared from recombinant human GAA with high-mannose type glycans using recombinant human GlcNAc phosphotransferase (39), uncovering enzyme (9), and/or sweet potato purple acid phosphatase (40) in vitro as described by Chavez et al. (30).
Purification of Human β-Glucuronidase
Human β-glucuronidase was collected from serum-free conditioned medium from cells that overexpress this lysosomal enzyme (MTX 3.2 cells were generously provided by Dr. William Sly, St. Louis University School of Medicine, St. Louis, MO). β-Glucuronidase was purified by affinity chromatography on a CI-MPR Affi-Gel-10 column as described previously (41).
Synthesis and Purification of N-[3H]Acetylglucosaminyl 6-Phosphomethylmannoside Diester
[3H]GlcNAc-6-phosphomethylmannoside diester was synthesized and purified following the method described by Mullis and Ketcham (42) except that recombinant soluble human GlcNAc phosphotransferase (39) was used as the enzyme source instead of a lysate derived from the soil amoeba Acathamoeba castellanii. Briefly, the molecule was synthesized from UDP-[3H]GlcNAc and methyl α-d-mannoside (Sigma) using GlcNAc phosphotransferase and purified by size exclusion chromatography using Sephadex G-25 equilibrated in 7% (v/v) propanol. The concentration of [3H]GlcNAc-6-phosphomethylmannoside diester was determined by carbohydrate composition analysis.
Biosensor Studies
All SPR measurements were performed at 25 °C using a BIAcore 3000 instrument (GE Healthcare). CM5 research grade sensor chips, surfactant P20, and amine coupling kits were also obtained from GE Healthcare. Purified proteins (β-glucuronidase, GAA phosphomonoester, GAA phosphodiester, Dom1–3, Dom1–3His, and sCI-MPR) were immobilized on CM5 sensor chips following activation of the surface using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and N-hydroxysuccinimide as recommended by the manufacturer. Briefly, the proteins were injected onto the activated dextran surface at a concentration of 10–20 μg/ml in 10 mm sodium acetate buffer, pH 5.0 (except β-glucuronidase and GAA phosphodiester in which 10 mm sodium acetate buffer, pH 4.5, was used), using immobilization buffer (10 mm MES, pH 6.5, 150 mm NaCl, and 0.005% (v/v) P20) as the running buffer. After coupling, unreacted N-hydroxysuccinimide ester groups were blocked with ethanolamine. The reference surface was treated in the same way except that protein was omitted. Samples of purified proteins (Dom5His, Dom9His, Dom1–3His, Dom1–3, Dom5–9, Dom7–11, sCI-MPR, β-glucuronidase, GAA phosphomonoester, GAA phosphodiester, insulin-like growth factor II (IGF-II), Glu-plasminogen (plasminogen, which contains an intact N-terminal peptide)) were prepared in running buffer (50 mm MES, pH 6.5, 150 mm NaCl, 10 mm MnCl2, 5 mm β-glycerophosphate, and 0.005% (v/v) P20) and were injected in a volume of 80 μl over the coupled and reference flow cells at a flow rate of 40 μl/min. After 2 or 3 min, the solutions containing the purified proteins were replaced with buffer and the complexes allowed to dissociate for 3 min. The sensor chip surface was regenerated with a 10-μl injection of 10 mm HCl or 10 mm Man-6-P at a flow rate of 10 μl/min. The surface was allowed to re-equilibrate in running buffer for 1 min prior to subsequent injections. Concentration curves were fit to a 1:1 Langmuir binding model using the BIAevaluation software package (version 4.0.1; GE Healthcare). KD values were calculated as the ratio of off (kd) and on (ka) rate constants. In some experiments, Dom5His (500 nm) or Dom5–9 (50 nm) were flowed over sensor surfaces immobilized with either GAA monoester or GAA diester in the presence of increasing concentrations of glucose 6-phosphate (Glc-6-P) (Sigma), Man-6-P (Sigma), or N-acetylglucosaminyl 6-phosphomethylmannoside diester. In other experiments, GAA monoester (20 nm) or GAA diester (20 nm) were flowed over the sensor surface immobilized with Dom1–3 in the presence of increasing concentrations of Glc-6-P (Sigma), Man-6-P (Sigma), or N-acetylglucosaminyl 6-phosphomethylmannoside diester. The response at equilibrium (Req) for each concentration of protein was determined by averaging the response over a 10-s period within the steady state region of the sensorgram using the BIAevaluation software package. The Req was plotted versus the concentration of inhibitor and was fit by nonlinear regression to a one-site inhibition model (SigmaPlot version 10.0, Systat Software, Inc.). All response data were double or triple referenced (43), where controls for the contribution of the change in refractive index were performed in parallel with flow cells derivatized in the absence of protein and subtracted from all binding sensorgrams.
RESULTS
Expression and Purification of Truncated Forms of the CI-MPR
To probe the lectin properties and structural requirements of the three carbohydrate binding sites of the CI-MPR, constructs encompassing the three sites were generated. We previously reported that both P. pastoris yeast and insect cells are highly effective heterologous host systems for the generation of functional, soluble forms of the bovine MPRs (28–31, 36). Truncated, soluble forms of the bovine CI-MPR composed of extracellular domain 5 alone (Dom5His), domain 9 alone (Dom9His), and domains 1–3 (Dom1–3His), each containing a C-terminal tag of six histidine residues to facilitate purification (Fig. 2B), were expressed in P. pastoris yeast as secreted proteins, and purified from the culture medium using nickel-nitrilotriacetic acid chromatography. Untagged, truncated versions of the CI-MPR encompassing domains 1–3 (Dom1–3), domains 5–9 (Dom5–9), and domains 7–11 (Dom7–11) (Fig. 2B) were secreted by Sf9 insect cells following infection with recombinant baculovirus, and purified from the medium using phosphomannan affinity chromatography. The purified MPRs migrated on SDS-polyacrylamide gels with the expected size (Fig. 2, inset).
Fetal bovine serum has been shown to contain a soluble form of the CI-MPR (sCI-MPR) that, based on apparent mobility (∼260 kDa), contains the majority of the extracellular region of the receptor (37). The sCI-MPR was purified from fetal bovine serum using phosphomannan affinity chromatography (Fig. 2, inset). To assess the activity of the purified protein, the sCI-MPR was immobilized to the surface of a sensor chip and subjected to SPR analyses, which showed that the sCI-MPR binds the lysosomal enzyme β-glucuronidase, plasminogen, and IGF-II with high affinity (KD = 40, 30, and 1 nm, respectively). Because the plasminogen binding site has been mapped to domain 1 (44), the data indicate that the N terminus of the sCI-MPR is intact. The IGF-II binding site has been mapped to domain 11, with sequences in domain 13 enhancing the affinity 10-fold (45–48). The observation that the sCI-MPR binds IGF-II with an affinity similar to that observed for a recombinant CI-MPR construct encoding domains 1–15 (Kd = 14 nm (48)), along with its apparent size of 260 kDa (Fig. 2, inset), indicates that this purified preparation of the sCI-MPR contains, at a minimum, domains 1–13. The various recombinant forms of the CI-MPR (Fig. 2B) were then assayed for their ability to bind purified glycans and lysosomal enzymes, and their properties compared with that of the sCI-MPR (see below).
Generation of Phosphomannosyl-containing Glycan Microarray
Analysis of phosphorylated glycans found in total cellular extracts or derived from lysosomal enzymes demonstrate a wide range of phosphorylated species (4, 5, 33, 34). To generate a panel of non-phosphorylated and phosphorylated oligosaccharides, N-glycans were released from bovine RNase B and soybean agglutinin by enzymatic digestion with peptide N-glycosidase F. High mannose-type oligosaccharides containing five through eight mannose residues (Man5GlcNAc2 through Man8GlcNAc2; M5 through M8) were isolated from RNase B and high mannose-type oligosaccharide containing nine mannose residues (Man9GlcNAc2; M9) was isolated from soybean agglutinin. The free glycans were labeled at the reducing end with the bifunctional fluorescent linker, AEAB, and an aliquot of each glycan (M5 through M9) was treated with GlcNAc phosphotransferase and the resulting phosphodiester-containing glycans with one or two Man-P-GlcNAc were separated from each other by HPLC. A portion of the isolated diester-containing glycans was subjected to mild acid hydrolysis to remove the GlcNAc residue, generating the corresponding set of glycans that have one or two phosphomonoesters. The purified glycans (non-phosphorylated and phosphorylated species) were assigned structures based on their chromatographic behavior relative to standards and their mass as determined by MALDI-TOF analysis and covalently immobilized onto N-hydroxysuccinimide-activated glass slides via their free alkylamine (38). The slides were incubated with recombinant MPR proteins or sCI-MPR and the interaction detected by MPR-specific antibodies. Incubation with different concentrations of MPR permitted an estimation of binding affinities based on half-maximal binding (see Table 1 and supplemental Fig. S1).
TABLE 1.
Estimated, relative binding affinities of CI-MPR and truncated forms of sCI-MPR based on half-maximal binding to the phosphorylated glycan microarray
| Glycan No. | Designation | CI-MPRa | Dom1–3-His | Dom5-His | Dom5–9 | Dom9His | Dom7–11 |
|---|---|---|---|---|---|---|---|
| 9 | GPM6 | 6.2 × 10−10 | DNBb | 8.7 × 10−6 | 3.7 × 10−9 | DNB | DNB |
| 10 | 2(GP)M6 | 8.0 × 10−11 | DNB | 6.9 × 10−6 | 3.3 × 10−9 | 1.1 × 10−6 | 5.48 × 10−7 |
| 11 | GPM7(3) | 5.9 × 10−9 | DNB | 1.8 × 10−5 | 8.9 × 10−9 | DNB | DNB |
| 12 | GPM7(2) | >5.8 × 10−8 | DNB | DNB | >2.8 × 10−7 | DNB | DNB |
| 13 | 2(GP)M7(1)+(3) | 6.0 × 10−11 | DNB | 4.3 × 10−6 | 3.0 × 10−9 | DNB | DNB |
| 14 | GPM8(1) | 1.1 × 10−10 | DNB | 1.6 × 10−5 | 4.0 × 10−9 | DNB | DNB |
| 15 | GPM9 | 1.9 × 10−10 | DNB | 1.8 × 10−5 | 5.7 × 10−9 | DNB | DNB |
| 16 | 2(GP)M9 | 7.0 × 10−11 | DNB | 4.8 × 10−6 | 4.1 × 10−9 | DNB | DNB |
| 17 | PM6 | 7.0 × 10−11 | 1.1 × 10−7 | DNB | 1.4 × 10−8 | 6.6 × 10−8 | 5.5 × 10−8 |
| 18 | 2(P)M6 | 8.0 × 10−11 | 1.0 × 10−7 | DNB | 1.9 × 10−8 | 5.7 × 10−8 | 6.7 × 10−8 |
| 19 | PM7(3) | 9.0 × 10−11 | 1.3 × 10−7 | DNB | 2.5 × 10−8 | 6.8 × 10−8 | 8.1 × 10−8 |
| 20 | PM7(2) | 5.2 × 10−9 | >1.0 × 10−6 | DNB | >2.8 × 10−7 | >1.2 × 10−6 | >6.0 × 10−7 |
| 21 | 2(P)M7(1)+(3) | 8.0 × 10−11 | 8.7 × 10−8 | DNB | 1.7 × 10−8 | 5.5 × 10−8 | 4.8 × 10−8 |
| 22 | PM8(1) | 1.7 × 10−10 | >1.0 × 10−6 | DNB | 4.2 × 10−8 | 1.9 × 10−7 | 2.6 × 10−7 |
| 23 | PM9 | 1.2 × 10−10 | >1.0 × 10−6 | DNB | 5.7 × 10−8 | 1.5 × 10−7 | 2.7 × 10−7 |
| 24 | 2(P)M9 | 8.0 × 10−11 | 1.4 × 10−7 | DNB | 2.6 × 10−8 | 5.2 × 10−8 | 8.6 × 10−8 |
a The half-maximal binding concentration (μg/ml) for each MPR construct was estimated from graphs of the data shown in supplemental Fig. S1. The estimated Kd value are shown in molar concentrations using the following molecular mass assignments: sCI-MPR (dimer), 520,000 Da; Dom1–3His, 50,000 Da; Dom5His, 21,000 Da; Dom5–9, 90,000 Da; Dom9His (dimer), 42,000 Da; and Dom7–11, 84,000 Da.
b DNB, did not bind.
Domains 3, 5, and 9 of the CI-MPR Each Recognize a Distinct Repertoire of Phosphorylated Glycans
MPR binding to the phosphorylated glycan microarray was analyzed at decreasing concentrations of proteins to evaluate their relative affinities for the different glycans. Comparison of data obtained at 50 μg/ml is shown in Fig. 3, A–F, whereas the relative affinities are better observed upon inspection of the data showing the binding as a function of protein concentration in supplemental Fig. S1. From previous reports in which linear high mannose oligosaccharides are poor inhibitors for the CI-MPR (49, 50), it is not surprising that no appreciable binding is detected to the non-phosphorylated high mannose glycans (glycans 1–8) by soluble CI-MPR or its different recombinant, truncated forms (Fig. 3, A–F). In contrast, the receptor constructs interact specifically with the phosphorylated glycans. Dom9His is highly specific for phosphomonoesters and recognizes to a similar extent M6 to M9 species bearing one or two phosphomonoesters, with the exception of the M7 isomer, PM7(2) (glycan 20), which binds Dom9His poorly (Fig. 3F). Dom9His binds PM8 (glycan 22) and PM9 (glycan 23) with ∼3–4-fold lower affinity when compared with the other phosphomonoesters (Table 1 and supplemental Fig. S1). The influence of additional domains was evaluated using the Dom7–11 construct. The presence of additional flanking domains (i.e. domains 7, 8, 10, and 11) had no appreciable effect on carbohydrate binding: Dom7–11 is highly specific for phosphomonoesters and exhibits a similar pattern of glycan binding specificity as observed for Dom9His at all concentrations analyzed (Fig. 3E, Table 1, and supplemental Fig. S1). Low, but detectable levels of binding were observed for the diphosphorylated diester 2(GP)M6 (glycan 10) by both Dom9His and Dom7–11 at the highest concentration tested.
FIGURE 3.

Interaction of CI-MPR constructs with the phosphorylated glycan microarray. The phosphorylated glycan microarray was printed as described in the accompanying article (64) and the individual glycans are identified by their glycan number (glycan number) as indicated in Table 1 of the accompanying article (64). Glycans 1–8 are non-phosphorylated high mannose-type oligosaccharides; 9–16, phosphodiesters of high mannose-type oligosaccharides; and 17–24, phosphomonoesters of high mannose-type oligosaccharides. The structural assignments are made according to Table 1 in the accompanying article (64) and are shown in Fig. 3. CI-MPR constructs were applied to the array at 50 μg/ml and detected with polyclonal rabbit antibody 14.5 as described under “Experimental Procedures.” A, sCI-MPR is the same data shown in Fig. 4D of the accompanying article (64); B, Dom1–3; C, Dom5; D, Dom5–9; E, Dom7–11; and F, Dom9. Error bars represent the mean ± S.D. of 6 replicates after removing the high and low values.
Recently, we have determined that domain 5, unlike the CD-MPR or domain 9 of the CI-MPR, preferentially binds lysosomal enzymes containing phosphodiesters (28, 30). However, the structures of the phosphodiester-bearing glycans on the lysosomal enzyme recognized by domain 5 were not known. In addition, this study did not evaluate whether adjacent domains influenced the binding affinity of domain 5. Analysis of the Dom5His construct on the glycan microarray shows relatively low affinity binding (compare RFU of Dom5 and other truncated forms (Fig. 3)), but clearly demonstrates that domain 5 of the CI-MPR recognizes Man-P-GlcNAc-containing glycans, with little or no binding detected to glycans containing phosphomonoesters. Dom5His exhibits the highest affinity to diphosphodiester species 2(GP)M7(1)+(3) (glycan 13) and 2(GP)M9 (glycan 16), and the lowest affinity for GPM7(2) (glycan 12). Analysis of the Dom5–9 construct demonstrates that it binds both phosphomonoesters and phosphodiesters with higher affinity (Fig. 3, supplemental Fig. S1, and Table 1), consistent with it containing two Man-6-P binding sites (i.e. domains 5 and 9). The types of monoester glycans recognized by Dom5–9 is similar to that of Dom9His and Dom7–11, indicating that additional domains (i.e. domains 5–8, 10, and 11) do not significantly influence the binding pocket or glycan specificity of domain 9 (see Fig. 3, Table 1, and supplemental Fig. S1). In contrast, the extent of binding displayed toward the phosphodiester species is dramatically increased compared with Dom5His, indicating that Dom5–9 has a significantly higher affinity for Man-P-GlcNAc than Dom5His (see Fig. 3, Table 1, and supplemental Fig. S1). These results suggest that additional domains act to stabilize the binding pocket in domain 5, similar to that observed for the effect of domains 1 and 2 on the binding affinity of domain 3 (31). As with Dom5His, the lowest affinity observed for the phosphodiester species is with GPM7(2) (glycan 12). However, in comparison to Dom5His, a higher affinity was observed by the Dom5–9 construct for GPM7(3) (glycan 11), GPM8 (glycan 14), and GPM9 (glycan 15). It is interesting to note that the Dom5–9 construct displays a higher affinity overall for the phosphodiester glycans than for the phosphomonoester glycans (see Fig. 3, Table 1, and supplemental Fig. S1).
The N-terminal binding site of the CI-MPR has not been characterized previously for its ability to recognize phosphodiesters. The results show for the first time that Dom1–3 is highly specific for phosphomonoesters, with little appreciable binding to any of the phosphodiester glycans. In contrast to Dom9His or Dom5–9, little interaction is observed with PM8 (glycan 22) and PM9 (glycan 23) (see Fig. 3, Table 1, and supplemental Fig. S1). Thus, the Man-6-P binding site located within domain 9 has broader glycan specificity than the N-terminal Man-6-P binding site of the CI-MPR. Similar results were obtained for the His-tagged version of domains 1–3 expressed and purified from P. pastoris yeast (data not shown). In addition, detection of Dom5His (supplemental Fig. S2), Dom9His (data not shown), and Dom1–3His (data not shown) constructs using either His-specific antibodies or CI-MPR-specific antibodies did not change the pattern of glycans recognized by each construct, further demonstrating the specificity of the interaction.
The sCI-MPR purified from fetal bovine serum binds both phosphomonoesters and phosphodiesters (Fig. 3A). However, unlike Dom5–9, sCI-MPR binds various phosphomonoester species with a similar affinity as the phosphodiester species (see accompanying article, Ref. 64). Taken together, the results indicate that CI-MPR utilizes its three different carbohydrate binding sites to recognize a wide range of phosphorylated high mannose-type glycans.
Domain 9 Interacts Specifically with Lysosomal Enzymes Containing Phosphomonoesters
To determine whether a similar carbohydrate preference would be observed when phosphorylated glycans are presented to the CI-MPR in the context of a lysosomal enzyme, quantitative measurements of binding affinities were performed by SPR using a lysosomal enzyme, GAA, containing either phosphomonoesters or phosphodiesters. Human GAA is synthesized as a 110-kDa precursor that undergoes a series of proteolytic processing events yielding major species of 76- and 70-kDa, each of which exists as a complex of four peptides (51). Human GAA contains seven N-glycosylation sites, six of which are efficiently utilized. Previously, we described the generation of GAA containing either phosphomonoester (GAA monoester) or phosphodiester (GAA diester) moieties by in vitro enzymatic reactions using GlcNAc phosphotransferase, uncovering enzymes, and/or sweet potato acid phosphatase. The glycans on the recombinant human GAA were exclusively of the high-mannose type, with 8.35 mannose residues, on average, per oligosaccharide. In addition, 1 mol of GAA diester contains 5.5 mol of phosphodiester, whereas 1 mol of GAA monoester contains 7.1 mol of phosphomonoester (30).
SPR analyses were performed in which similar amounts of GAA monoester and GAA diester were immobilized to separate flow cells and increasing concentrations of the different CI-MPR constructs, and sCI-MPR was flowed over the sensor surface. Due to nonspecific interaction with the reference surface, Dom1–3 was immobilized on the surface of the flow cell and increasing concentrations of GAA monoester or GAA diester were flowed over the sensor surface. Representative sensorgrams are shown in Fig. 4. The data were analyzed by nonlinear regression and the affinity constants summarized in Table 2. SPR analyses were initially performed on all constructs using the lysosomal enzyme, β-glucuronidase, that was purified from a cell line that overexpresses and secretes this acid hydrolase into the medium. The results confirmed that Dom5His binds β-glucuronidase with low affinity (KD = ∼40 μm) compared with the sCI-MPR or constructs that contain domains 1–3 or domain 9 (KD = ∼ 1 to 200 nm) (Table 2).
FIGURE 4.

SPR analysis of MPR constructs binding to GAA phosphomonoester or GAA phosphodiester. Similar amounts (2500 and 2300 response units, respectively) of GAA monoester and GAA diester were immobilized on the surface of a CM5 sensor chip (panels A–D and F). MPR constructs were injected in a volume of 80 μl over the coupled and reference flow cells at a rate of 40 μl/min. After 2 min, the solutions containing the MPRs were replaced with buffer and the complexes allowed to dissociate for 3 min. Shown are representative sensorgrams for: A, Dom9His at 10 nm, 100 nm, and 1 μm; B, Dom7–11 at 10 nm, 100 nm, and 1 μm; C, Dom5His at 10 nm, 100 nm, and 1 μm; D, Dom5–9 at 10 nm, 100 nm, and 1 μm; and F, sCI-MPR at 10 nm, 100 nm, and 1 μm comparing the response on GAA phosphomonoester (blue lines) and GAA phosphodiester (red lines) surfaces. E, Dom1–3 was immobilized on the surface of a CM5 sensor chip and GAA monoester or GAA diester were injected in a volume of 80 μl over the Dom1–3 and reference flow cells at a rate of 40 μl/min. After 3 min, the solutions containing the GAA lysosomal enzymes were replaced with buffer and the complexes were allowed to dissociate for 3 min. Shown are representative sensorgrams of the GAA phosphomonoester (blue lines) and GAA phosphodiester (red lines) at 10 nm, 100 nm, and 1 μm. Kinetic parameters were determined by global fitting of the sensorgrams to a 1:1 binding model using BIAevaluation version 4.0.1 software and summarized in Table 1.
TABLE 2.
Summary of dissociation constants for CI-MPR constructs
“Analyte” refers to the protein that is flowed over the sensor surface. The binding affinities (KD) represent the mean ± S.E. of three independent experiments involving at least two different coupled surfaces except where noted.
| Analyte |
KD |
||
|---|---|---|---|
| GAAa monoester | GAA diester | β-Glucuronidase | |
| nm | |||
| Dom9His | 71 ± 20 | NBb | 70 ± 8 |
| Dom7–11 | 102 ± 30 | NB | 180 ± 70 |
| Dom5His | 47,000 ± 50,000 | 10,000 ± 8,000 | 37,000 ± 13,000 |
| Dom5–9 | 72 ± 17 | 170 ± 100 | 56 ± 30 |
| Dom1–3c (n = 1) | 0.4 | 3.5 | NDd |
| Dom1–3His | 2 nme | 98 nme | 1.0 ± 0.5f |
| sCI-MPR | 7 ± 5 | 45 ± 18 | 40 ± 20 |
a GAA, acid α-glucosidase.
b NB, no detectable binding above background at concentrations up to 10 μm.
c Dom1–3 and Dom1–3His constructs were coupled to the surface of a CM5 sensor chip.
d ND, not determined.
e Average of two trials.
f Average of four trials.
The results show that Dom9His is highly specific for the GAA monoester, as no detectable interaction was observed between Dom9His and the GAA diester surface. Additional domains did not affect either the affinity for the GAA monoester (Dom9His KD = ∼70 nm; Dom7–11 KD = ∼100 nm) or specificity, as the Dom7–11 construct did not exhibit any appreciable binding to the GAA diester. The Dom5–9 construct also displayed a similar affinity for the GAA monoester (KD = ∼70 nm), further demonstrating that adjacent domains have little influence on the binding pocket located within domain 9 of the CI-MPR.
Additional Domains Enhance the Binding Affinity of Domain 5 for Phosphodiesters
We had previously reported that domain 5 (Dom5His construct) exhibits a 14–18-fold higher affinity for the disaccharide Man-P-GlcNAc than the monosaccharide Man-6-P (30). Consistent with this observation, we show that Dom5His displays ∼5-fold higher affinity for GAA diester than GAA monoester (KD = 10 μm versus KD = 47 μm) (see Fig. 4 and Table 2). It should be noted that these affinity determinations were calculated from rate constants (ka and kd) that approach the limits of accuracy of the BIAcore 3000 instrument. To assess whether additional domains influence the binding affinity of domain 5, the Dom5–9 construct was generated. The results demonstrate that Dom5–9 binds with ∼60-fold higher affinity to the GAA diester (KD = 170 nm) than Dom5His (KD = 10 μm).
To directly measure the binding affinity of Dom5His and Dom5–9 to Man-P-GlcNAc, the MPR constructs were passed over the sensor surface containing immobilized GAA diester in the presence of increasing concentrations of Man-6-P, GlcNAc- P-methyl mannoside, or Glc-6-P. Glc-6-P was chosen as a control because it had been shown previously to bind with low affinity (Kd = 1–8 × 10−2 m) to the full-length CD-MPR and CI-MPR (52). The results demonstrate that Dom5–9 binds with a 10-fold higher affinity to GlcNAc-P-methyl mannoside than Dom5His (Ki = 0.1 mm versus Ki = 1 mm (Fig. 5). Minimal inhibition was observed with Man-6-P, consistent with the binding preference of domain 5 for the phosphodiester, whereas no appreciable inhibition was observed at the highest concentration of Glc-6-P (10 mm) (Fig. 5). Taken together, the results suggest that, like the N-terminal carbohydrate site in domain 3, domain 5 utilizes other domains of the receptor to stabilize its binding pocket.
FIGURE 5.

Inhibition of Dom5His and Dom5–9 binding to GAA by Man-6-P, Glc-6-P, and N-acetylglucosaminyl 6-phosphomethylmannoside diester. Aliquots of 500 nm Dom5His (A) and 50 nm Dom5–9 (B) were incubated with increasing concentrations of Glc-6-P (▿), Man-6-P (●), or N-acetylglucosaminyl 6-phosphomethylmannoside diester (○) and injected over a flow cell coupled with GAA diester. The Req from the resulting sensorgrams was plotted as a percent of the maximal response against the log of the inhibitor concentration. The Ki was determined using nonlinear regression (SigmaPlot, version 10.0).
The N-terminal Binding Site Is Specific for Lysosomal Enzymes Containing Phosphomonoesters
The phosphomonoester preference of the N-terminal binding site of the CI-MPR was further evaluated by SPR analyses. Consistent with the above glycan microarray data, Dom1–3 is highly specific for the GAA monoester and binds with high affinity (KD = 0.4 nm) (see Fig. 4 and Table 2). Although binding was observed to the GAA diester, the interaction was inhibited with Man-6-P (Ki = 12 μm), but not significantly by GlcNAc-P-methyl mannoside (Ki = 1 mm) (Fig. 6), and is indicative of low levels of phosphomonoester on the GAA diester. Similar results were obtained with the His-tagged version of domains 1–3 expressed and purified from P. pastoris yeast (KD = 2 nm GAA monoester, KD = 98 nm GAA diester). A striking difference compared with the other constructs is the slow off rate displayed by Dom1–3 (Fig. 4). Taken together, the results demonstrate that only domain 5 of the CI-MPR is capable of interacting with Man-P-GlcNAc. Furthermore, as observed with the glycan microarray analyses, the sCI-MPR exhibits a similar affinity for the GAA monoester (KD = ∼7 nm) and the GAA diester (KD = ∼45 nm) (Table 2).
FIGURE 6.
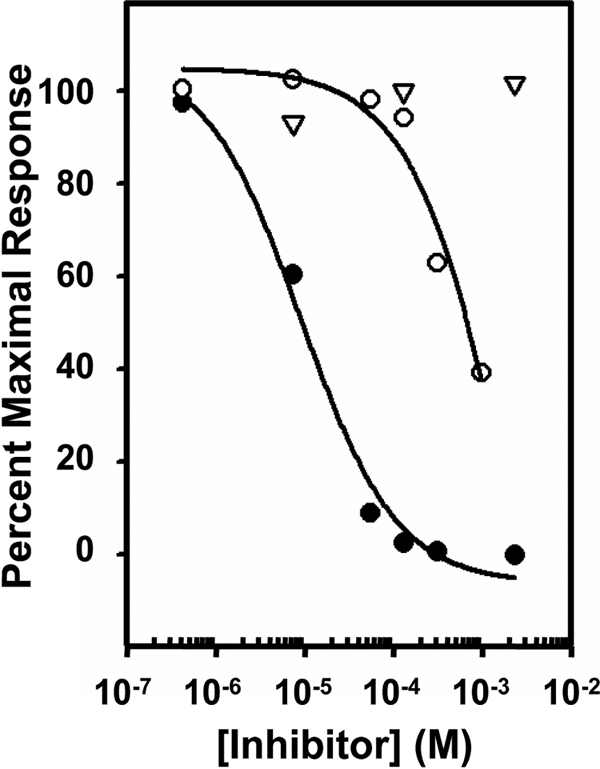
Inhibition of Dom1–3 binding to GAA by Man-6-P, Glc-6-P, and N-acetylglucosaminyl 6-phosphomethylmannoside diester. Aliquots of 20 nm GAA diester were incubated with increasing concentrations of Glc-6-P (▿), Man-6-P (●), or N-acetylglucosaminyl 6-phosphomethylmannoside diester (○) and injected over a flow cell coupled with Dom1–3. The Req was plotted against the log of the inhibitor concentration. The Ki was determined using nonlinear regression (SigmaPlot, version 10.0).
To rule out the possibility that the presence of aggregated protein in the purified MPR preparations used to flow over the sensor chip influences the affinity determination of receptor binding to ligand, the recombinant proteins and sCI-MPR were subjected to gel filtration. The results demonstrate that all recombinant proteins elute predominantly as a monomeric species, with the exception of Dom9His, which elutes at a size consistent with a dimer (supplemental Fig. S3). To further evaluate the oligomeric state of Dom5His, which eluted as a broad peak (26–33 kDa, supplemental Fig. S3C) on a Superdex 75 gel filtration column, NMR spectroscopy was performed on 15N-labeled Dom5 using pulse field gradient self-diffusion measurements (53), which confirmed that Dom5 exists in a monomeric form (data not shown). Consistent with previous reports that indicate that the CI-MPR exists as a dimer (54–56), the ∼260 kDa sCI-MPR isolated from serum elutes as a 550-kDa species on a Superdex 200 column (supplemental Fig. S3F). The inability of Dom1–3, Dom5–9, and Dom7–11 to migrate as a dimer on a size exclusion column suggests that these regions of the receptor do not play a significant role in establishing the oligomeric state of the CI-MPR and supports previous biochemical (56) and crystallographic (22) studies that implicate domain 12 as playing a role in dimerization of the CI-MPR. Additional studies are needed to evaluate whether the His tag of the Dom9His construct plays any role in facilitating dimerization of this construct. Although no detectable aggregated species were observed for the sCI-MPR, Dom5His, or Dom7–11 constructs, the amount of higher order oligomeric species detected by gel filtration or dynamic light scattering measurements for the Dom9His and Dom5–9 constructs was observed to vary from preparation to preparation. To eliminate the possible influence of these aggregated species in binding affinity measurements, the peak fractions from the gel filtration columns corresponding to the monomeric (Dom1–3, Dom5His, Dom5–9, and Dom7–11) or dimeric (Dom9His and sCI-MPR) species of the receptors were isolated and used immediately, without additional concentration, in SPR studies in which these proteins were flowed over the sensor surface. The results demonstrate that the affinity does not change appreciably when the receptors were subjected to further purification by gel filtration, with differences ranging from 10% to 7-fold of the values obtained for the proteins that have not been subjected to gel filtration.
DISCUSSION
A major function of the CI-MPR is the delivery of ∼60 different soluble lysosomal enzymes from their site of synthesis to the endosomal/lysosomal system. Newly synthesized lysosomal enzymes are modified post-translationally on their N-glycans as they travel through the Golgi to acquire first phosphodiesters, which can then be converted to phosphomonoesters by the action of the uncovering enzyme in the trans Golgi network. The structures of these phosphorylated high mannose-type glycans are diverse and the accompanying paper (64) demonstrates that the CI-MPR recognizes a wide repertoire of phosphorylated glycans. The large extracytoplasmic region of the CI-MPR is composed of 15 contiguous MRH domains that display a similar size (∼150 residues) and cysteine distribution with each other. The three carbohydrate binding sites map to domains 3, 5, and 9 and recent studies showed that domain 5 differed from domain 9 in that it preferentially bound phosphodiester-containing lysosomal enzymes (30). In the current report, we used a unique phosphorylated glycan microarray (see Ref. 64) to further probe the glycan binding specificities of the three carbohydrate recognition sites of the CI-MPR. In addition, SPR analyses of the interaction of truncated forms of the CI-MPR with lysosomal enzymes enriched in phosphomonoesters or phosphodiesters were performed to assess whether adjacent MRH domains influence the binding affinity and specificity of the individual carbohydrate binding sites.
The generation of the phosphorylated glycan microarray has allowed, for the first time, the ability to probe the detailed binding specificity of the three phosphomannosyl recognition sites, located in domains 3, 5, and 9 of the CI-MPR. None of the three sites is capable of recognizing the phosphodiester GPM7(2) (glycan 12) or the corresponding phosphomonoester PM7(2) (glycan 20), indicating that a phosphate group on either the B arm (mannose “E”) or C arm (mannose “F”) of the glycan is not accommodated by the three binding pockets (see Figs. 1, 3, and supplemental S1). In previous studies, we captured the crystal structure of domains 1–3 in which the binding pocket of domain 3 was occupied with a non-phosphorylated branched oligosaccharide (Manα(1,3)[Manα(1,6)]Manα(1,4)GlcNAcβ(1,4)GlcNAc (21), with all three mannose residues interacting with the receptor. Because the α1,3-linked mannose occupied the known Man-6-P binding pocket of domain 3 (32), we hypothesize that at least for domain 3, it is the phosphate group on the α1,6-linked mannose (C arm, mannose F), that does not support high affinity binding.
The results from the phosphorylated glycan microarray clearly demonstrate that each carbohydrate recognition site of the CI-MPR exhibits a different profile of preferred glycans. The N-terminal binding site (domains 1–3) and domain 9 are quite specific for glycans containing phosphomonoesters, with only weak binding detected for the diphosphodiester-containing species 2(GP)M6 (glycan 10) (see Fig. 3, Table 1, and supplemental S1). However, a striking difference between these two binding sites is that domains 1–3 are unable to interact with PM8(1) (glycan 22) and PM9 (glycan 23), both of which have a single phosphate on the terminal α1,2-linked mannose of the C arm (Fig. 3). We have shown previously that an α1,2-linked disaccharide (Manα1,2Man) is a 2–3-fold better inhibitor of domain 1–3 binding to a lysosomal enzyme than either Manα1,3Man or Manα1,6Man disaccharides (21). Therefore, additional studies will be needed to determine the structural basis for the lack of binding of PM8(1) (glycan 22) and PM9 (glycan 23) to domain 3. One possibility is that the presence of the terminal mannose (mannose “G”) on the A arm inhibits access of the phosphorylated C arm into the binding pocket of domain 3 (see Fig. 1).
Previously, we showed that domain 5 of CI-MPR exhibits 14–18-fold higher affinity for Man-P-GlcNAc than Man-6-P, whereas no detectable binding to the phosphodiester was detected by domain 9 (30). We have extended these studies to show that the N-terminal binding site of the CI-MPR (domain 3), like domain 9, does not bind phosphodiesters. Furthermore, we have shown that a construct encoding domains 5–9 interacts with phosphodiesters with a significantly higher affinity than a construct encoding domain 5 alone (see Tables 1 and 2). To complement the phosphorylated glycan microarray studies, SPR analyses were performed to evaluate the ability of the CI-MPR constructs to recognize glycans when presented to the receptor in the context of a lysosomal enzyme. We used a recombinant GAA, which is a heavily glycosylated protein that was modified in vitro using GlcNAc phosphotransferase, uncovering enzyme, and/or sweet potato acid phosphatase to contain on average 5.5 mol of phosphodiester (GAA diester) or 7.1 mol of phosphomonoester (GAA monoester) (30). The results demonstrate that the three-dimensional presentation of phosphodiester- or phosphomonoester-containing glycans in the context of a lysosomal enzyme does not change the binding preference of domains 3, 5, or 9 (see Figs. 4 and 6 and Table 2). In addition, the SPR analyses further support the hypothesis that domain 5 requires the interaction of one or more MRH domains to stabilize the binding pocket because the construct encoding domains 5–9 binds phosphodiesters (GAA diester) with ∼60-fold higher affinity than domain 5 alone (see Fig. 4 and Table 2). This effect on binding affinity is reminiscent of domain 3: a construct encoding domain 3 alone exhibits ∼1,000-fold lower affinity for a lysosomal enzyme than domains 1–3 (31). The crystal structure of domains 1–3 revealed that these three MRH domains assemble into a compact structure, with residues in domains 1 and 2 stabilizing the loops of domain 3 involved in carbohydrate recognition (21, 32). Recent crystallographic studies by Brown et al. (22) have revealed that domains 11–14 are assembled differently than the three N-terminal MRH domains, and exist in a more extended “beads on a string” arrangement. Additional studies will be required to determine how the surrounding (one or more) MRH domains influence the binding affinity of domain 5.
The carbohydrate binding sites of the CI-MPR (domains 3, 5, and 9) and CD-MPR share a common feature: four residues (Gln, Arg, Glu, and Tyr) are conserved in the linear sequence and mutation of any one of these four residues demonstrates that they are critical for Man-6-P binding (27, 29, 30). Furthermore, the crystal structure of the CD-MPR (57, 58) and domains 1–3 of the CI-MPR (32) demonstrates that these four residues are located in identical positions in the binding pocket and contact the 2-, 3-, and 4-hydroxyl groups of the mannose ring. From these mutational studies and the finding that 7 of the 15 MRH domains of the CI-MPR exhibit a similar overall fold (21, 22), it is predicted that domains 5 and 9 will adopt a similar fold, with these four residues occupying similar positions in the binding pocket. Although the residues used to contact mannose are conserved, the diversity observed among the MPR binding pockets in ligand recognition has been shown to be generated, at least in part, by alterations in the receptor binding site architecture surrounding the phosphate moiety: the CD-MPR (57, 58) uses Asp-103, Asn-104, and His-105, whereas domain 3 of the CI-MPR (32) uses Ser-386 and an ordered water molecule to coordinate the phosphate group of Man-6-P. The ability of domain 5 to bind GlcNAc phosphodiesters indicates that the architecture of its binding pocket surrounding the phosphate moiety must be significantly different from the other Man-6-P binding sites to accommodate and form specific contacts with the GlcNAc sugar. Additional structural studies will be required to understand the mechanism of carbohydrate recognition by domains 5 and 9 of the CI-MPR.
In summary, the CI-MPR has evolved to contain three different Man-6-P binding sites that complement the two-step post-translational modification of lysosomal enzymes (Fig. 1) and provide the receptor the capacity to recognize a wide diversity of phosphomonoester- and phosphodiester-containing glycans. The presence of a phosphodiester Man-P-GlcNAc binding site may facilitate the targeting of newly synthesized lysosomal enzymes that are not efficiently modified by the uncovering enzyme in the trans Golgi network. Support for this role is the observation that the majority of the N-glycans of human β-glucuronidase isolated from spleen had a single phosphate that was in a diester linkage (34). These studies have health-related implications. For example, an understanding of the molecular basis by which the CI-MPR recognizes phosphorylated glycans may lead to the development of new and improved therapies for the treatment of lysosomal storage disorders, because several of the Food and Drug Administration-approved enzyme replacement therapies target the CI-MPR for the uptake of the infused Man-6-P-containing enzyme, which is deficient in a particular disorder (e.g. Fabry disease, mucopolysaccharidosis I, and Pompe disease) (59). Furthermore, the diversity of identified extracellular ligands, including growth factors (e.g. transforming growth factor-β) and pathogenic organisms (e.g. varicella-zoster virus) (60, 61), which interact with the CI-MPR in a Man-6-P-dependent manner suggests numerous potential therapeutic applications.
Acknowledgments
We thank Drs. Marielle Boonen and Stuart Kornfeld for preparing phosphorylated high mannose-type N-glycans. We also thank Amanda Otieno for technical assistance in the purification and initial SPR analyses of Dom5–9. The BIAcore 3000 instrument (Protein and Nucleic Acid Core Facility, Medical College of Wisconsin) was purchased through a grant from the Advancing a Healthier Wisconsin program.
This work was supported, in whole or in part, by National Institutes of Health Grants R01DK42667 (to N. M. D.) and GM085448 (to D. F. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- CI-MPR
- cation-independent mannose 6-phosphate receptor
- Man-6-P
- mannose 6-phosphate
- sCI-MPR
- soluble form of the CI-MPR isolated from fetal bovine serum
- CD-MPR
- cation-dependent mannose 6-phosphate receptor
- GlcNAc phosphotransferase
- UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase
- Man-P-GlcNAc
- mannose 6-phosphate N-acetylglucosamine ester
- uncovering enzyme
- N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase
- MRH
- mannose 6-phosphate receptor homology
- SPR
- surface plasmon resonance
- GAA
- acid α-glucosidase
- AEAB
- 2-amino-N-(2-aminoethyl)-benzamide
- Glc-6-P
- glucose 6-phosphate
- IGF-II
- insulin-like growth factor II
- MES
- 4-morpholineethanesulfonic acid
- HPLC
- high pressure liquid chromatography
- MALDI-TOF
- matrix-assisted laser desorption ionization time-of-flight
- Req
- response at equilibrium
- PM
- phosphomonoester-containing high mannose glycan
- GPM
- GlcNAc phosphodiester-containing high mannose glycan.
REFERENCES
- 1.Ghosh P., Dahms N. M., Kornfeld S. (2003) Nat. Rev. Mol. Cell Biol. 4, 202–212 [DOI] [PubMed] [Google Scholar]
- 2.Mullins C., Bonifacino J. S. (2001) Bioessays 23, 333–343 [DOI] [PubMed] [Google Scholar]
- 3.Dell'Angelica E. C., Payne G. S. (2001) Cell 106, 395–398 [DOI] [PubMed] [Google Scholar]
- 4.Hasilik A., Klein U., Waheed A., Strecker G., von Figura K. (1980) Proc. Natl. Acad. Sci. U.S.A. 77, 7074–7078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varki A., Kornfeld S. (1980) J. Biol. Chem. 255, 10847–10858 [PubMed] [Google Scholar]
- 6.Bao M., Booth J. L., Elmendorf B. J., Canfield W. M. (1996) J. Biol. Chem. 271, 31437–31445 [DOI] [PubMed] [Google Scholar]
- 7.Kudo M., Bao M., D'Souza A., Ying F., Pan H., Roe B. A., Canfield W. M. (2005) J. Biol. Chem. 280, 36141–36149 [DOI] [PubMed] [Google Scholar]
- 8.Kornfeld R., Bao M., Brewer K., Noll C., Canfield W. (1999) J. Biol. Chem. 274, 32778–32785 [DOI] [PubMed] [Google Scholar]
- 9.Do H., Lee W. S., Ghosh P., Hollowell T., Canfield W., Kornfeld S. (2002) J. Biol. Chem. 277, 29737–29744 [DOI] [PubMed] [Google Scholar]
- 10.Varki A., Sherman W., Kornfeld S. (1983) Arch. Biochem. Biophys. 222, 145–149 [DOI] [PubMed] [Google Scholar]
- 11.Waheed A., Hasilik A., von Figura K. (1981) J. Biol. Chem. 256, 5717–5721 [PubMed] [Google Scholar]
- 12.Rohrer J., Kornfeld R. (2001) Mol. Biol. Cell 12, 1623–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kornfeld S., Sly W. S. (2001) in Metabolic and Molecular Bases of Inherited Diseases (Scriver C. R., Beaudet A. L., Sly W. S., Valle D. eds) 8th Ed., pp. 3469–3482, McGraw Hill, New York [Google Scholar]
- 14.Hoflack B., Fujimoto K., Kornfeld S. (1987) J. Biol. Chem. 262, 123–129 [PubMed] [Google Scholar]
- 15.Sleat D. E., Lobel P. (1997) J. Biol. Chem. 272, 731–738 [DOI] [PubMed] [Google Scholar]
- 16.Ludwig T., Munier-Lehmann H., Bauer U., Hollinshead M., Ovitt C., Lobel P., Hoflack B. (1994) EMBO J. 13, 3430–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pohlmann R., Boeker M. W., von Figura K. (1995) J. Biol. Chem. 270, 27311–27318 [DOI] [PubMed] [Google Scholar]
- 18.Kasper D., Dittmer F., von Figura K., Pohlmann R. (1996) J. Cell Biol. 134, 615–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sohar I., Sleat D., Gong Liu C., Ludwig T., Lobel P. (1998) Biochem. J. 330, 903–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munro S. (2001) Curr. Biol. 11, R499–501 [DOI] [PubMed] [Google Scholar]
- 21.Olson L. J., Yammani R. D., Dahms N. M., Kim J. J. (2004) EMBO J. 23, 2019–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown J., Delaine C., Zaccheo O. J., Siebold C., Gilbert R. J., van Boxel G., Denley A., Wallace J. C., Hassan A. B., Forbes B. E., Jones E. Y. (2008) EMBO J. 27, 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uson I., Schmidt B., von Bulow R., Grimme S., von Figura K., Dauter M., Rajashankar K. R., Dauter Z., Sheldrick G. M. (2003) Acta Crystallogr. Sect. D Biol. Crystallogr. 59, 57–66 [DOI] [PubMed] [Google Scholar]
- 24.Westlund B., Dahms N. M., Kornfeld S. (1991) J. Biol. Chem. 266, 23233–23239 [PubMed] [Google Scholar]
- 25.Dahms N. M., Rose P. A., Molkentin J. D., Zhang Y., Brzycki M. A. (1993) J. Biol. Chem. 268, 5457–5463 [PubMed] [Google Scholar]
- 26.Olson L. J., Hancock M. K., Dix D., Kim J. J., Dahms N. M. (1999) J. Biol. Chem. 274, 36905–36911 [DOI] [PubMed] [Google Scholar]
- 27.Hancock M. K., Haskins D. J., Sun G., Dahms N. M. (2002) J. Biol. Chem. 277, 11255–11264 [DOI] [PubMed] [Google Scholar]
- 28.Reddy S. T., Chai W., Childs R. A., Page J. D., Feizi T., Dahms N. M. (2004) J. Biol. Chem. 279, 38658–38667 [DOI] [PubMed] [Google Scholar]
- 29.Sun G., Zhao H., Kalyanaraman B., Dahms N. M. (2005) Glycobiology 15, 1136–1149 [DOI] [PubMed] [Google Scholar]
- 30.Chavez C. A., Bohnsack R. N., Kudo M., Gotschall R. R., Canfield W. M., Dahms N. M. (2007) Biochemistry 46, 12604–12617 [DOI] [PubMed] [Google Scholar]
- 31.Hancock M. K., Yammani R. D., Dahms N. M. (2002) J. Biol. Chem. 277, 47205–47212 [DOI] [PubMed] [Google Scholar]
- 32.Olson L. J., Dahms N. M., Kim J. J. (2004) J. Biol. Chem. 279, 34000–34009 [DOI] [PubMed] [Google Scholar]
- 33.Varki A., Kornfeld S. (1983) J. Biol. Chem. 258, 2808–2818 [PubMed] [Google Scholar]
- 34.Natowicz M., Baenziger J. U., Sly W. S. (1982) J. Biol. Chem. 257, 4412–4420 [PubMed] [Google Scholar]
- 35.Marron-Terada P. G., Hancock M. K., Haskins D. J., Dahms N. M. (2000) Biochemistry 39, 2243–2253 [DOI] [PubMed] [Google Scholar]
- 36.Reddy S. T., Dahms N. M. (2002) Protein Expr. Purif. 26, 290–300 [DOI] [PubMed] [Google Scholar]
- 37.Valenzano K. J., Remmler J., Lobel P. (1995) J. Biol. Chem. 270, 16441–16448 [DOI] [PubMed] [Google Scholar]
- 38.Song X., Xia B., Stowell S. R., Lasanajak Y., Smith D. F., Cummings R. D. (2009) Chem. Biol. 16, 36–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kudo M., Canfield W. M. (2006) J. Biol. Chem. 281, 11761–11768 [DOI] [PubMed] [Google Scholar]
- 40.Hefler S. K., Averill B. A. (1987) Biochem. Biophys. Res. Commun. 146, 1173–1177 [DOI] [PubMed] [Google Scholar]
- 41.Marron-Terada P. G., Brzycki-Wessell M. A., Dahms N. M. (1998) J. Biol. Chem. 273, 22358–22366 [DOI] [PubMed] [Google Scholar]
- 42.Mullis K. G., Ketcham C. M. (1992) Anal. Biochem. 205, 200–207 [DOI] [PubMed] [Google Scholar]
- 43.Myszka D. G. (2000) Methods Enzymol. 323, 325–340 [DOI] [PubMed] [Google Scholar]
- 44.Leksa V., Godár S., Cebecauer M., Hilgert I., Breuss J., Weidle U. H., Horejsí V., Binder B. R., Stockinger H. (2002) J. Biol. Chem. 277, 40575–40582 [DOI] [PubMed] [Google Scholar]
- 45.Dahms N. M., Wick D. A., Brzycki-Wessell M. A. (1994) J. Biol. Chem. 269, 3802–3809 [PubMed] [Google Scholar]
- 46.Schmidt B., Kiecke-Siemsen C., Waheed A., Braulke T., von Figura K. (1995) J. Biol. Chem. 270, 14975–14982 [DOI] [PubMed] [Google Scholar]
- 47.Devi G. R., Byrd J. C., Slentz D. H., MacDonald R. G. (1998) Mol. Endocrinol. 12, 1661–1672 [DOI] [PubMed] [Google Scholar]
- 48.Linnell J., Groeger G., Hassan A. B. (2001) J. Biol. Chem. 276, 23986–23991 [DOI] [PubMed] [Google Scholar]
- 49.Distler J. J., Guo J. F., Jourdian G. W., Srivastava O. P., Hindsgaul O. (1991) J. Biol. Chem. 266, 21687–21692 [PubMed] [Google Scholar]
- 50.Tomoda H., Ohsumi Y., Ichikawa Y., Srivastava O. P., Kishimoto Y., Lee Y. C. (1991) Carbohydr. Res. 213, 37–46 [DOI] [PubMed] [Google Scholar]
- 51.Moreland R. J., Jin X., Zhang X. K., Decker R. W., Albee K. L., Lee K. L., Cauthron R. D., Brewer K., Edmunds T., Canfield W. M. (2005) J. Biol. Chem. 280, 6780–6791 [DOI] [PubMed] [Google Scholar]
- 52.Tong P. Y., Kornfeld S. (1989) J. Biol. Chem. 264, 7970–7975 [PubMed] [Google Scholar]
- 53.Veldkamp C. T., Peterson F. C., Pelzek A. J., Volkman B. F. (2005) Protein Sci. 14, 1071–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.York S. J., Arneson L. S., Gregory W. T., Dahms N. M., Kornfeld S. (1999) J. Biol. Chem. 274, 1164–1171 [DOI] [PubMed] [Google Scholar]
- 55.Byrd J. C., Park J. H., Schaffer B. S., Garmroudi F., MacDonald R. G. (2000) J. Biol. Chem. 275, 18647–18656 [DOI] [PubMed] [Google Scholar]
- 56.Kreiling J. L., Byrd J. C., MacDonald R. G. (2005) J. Biol. Chem. 280, 21067–21077 [DOI] [PubMed] [Google Scholar]
- 57.Roberts D. L., Weix D. J., Dahms N. M., Kim J. J. P. (1998) Cell 93, 639–648 [DOI] [PubMed] [Google Scholar]
- 58.Olson L. J., Zhang J., Lee Y. C., Dahms N. M., Kim J. J. P. (1999) J. Biol. Chem. 274, 29889–29896 [DOI] [PubMed] [Google Scholar]
- 59.Brady R. O. (2006) Annu. Rev. Med. 57, 283–296 [DOI] [PubMed] [Google Scholar]
- 60.Dahms N. M., Hancock M. K. (2002) Biochim. Biophys. Acta 1572, 317–340 [DOI] [PubMed] [Google Scholar]
- 61.Gary-Bobo M., Nirdé P., Jeanjean A., Morère A., Garcia M. (2007) Curr. Med. Chem. 14, 2945–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bobek G., Scott C. D., Baxter R. C. (1991) Endocrinology 128, 2204–2206 [DOI] [PubMed] [Google Scholar]
- 63.Clairmont K. B., Czech M. P. (1991) J. Biol. Chem. 266, 12131–12134 [PubMed] [Google Scholar]
- 64.Song X., Lasanajak Y., Olson L. J., Boonen M., Dahms N. M., Kornfeld S., Cummings R. D., Smith D. F. (2009) J. Biol. Chem. 284, 35201–35214 [DOI] [PMC free article] [PubMed] [Google Scholar]