

Abstract
This study quantifies the effects of naturally occurring X-linked variation on immune response in Drosophila melanogaster to assess associations between immunity genotypes and innate immune response. We constructed a set of 168 X-chromosomal extraction lines, incorporating X chromosomes from a natural population into co-isogenic autosomal backgrounds, and genotyped the lines at 88 SNPs in 20 X-linked immune genes. We find that genetic variation in many of the genes is associated with immune response phenotypes, including bacterial load and immune gene expression. Many of the associations act in a sex-specific or sexually antagonistic manner, supporting the theory that with the selective pressures facing genes on the X chromosome, sexually antagonistic variation may be more easily maintained.
THE deep evolutionary conservation of many specific genes in innate immunity underscores the potent forces of natural selection maintaining this vital function. While it is widely accepted as the ancestral form of immune response, its role in the activation of adaptive immune response further motivates investigation into variation in its function (Medzhitov and Janeway 1997). Drosophila has been used as a valuable model organism to identify and characterize functions of the components of innate immune pathways as well as the evolutionary patterns present among the genes comprising these pathways (reviewed in Brennan and Anderson 2004; Irving et al. 2004; Ferrandon et al. 2007). The humoral response, resulting in the production of antimicrobial peptides in response to bacterial or fungal infection, relies mainly on Toll and imd signal transduction pathways, both of which are highly homologous to pathways in mammalian immunity (reviewed in Kimbrell and Beutler 2001). The cellular component, on the other hand, incorporates phagocytic engulfment as well as melanization and encapsulation of infecting particles. While less well defined in the Drosophila model, portions of other systems also appear to affect the effectiveness of immune response, including JAK/STAT and JNK signaling pathways, hematopoesis, and iron metabolism.
Population genetic analysis can be used to determine whether sequence polymorphism and divergence patterns among Drosophila genes in innate immune pathways are consistent with signatures of selection acting within and between species of flies. If, for example, the innate immune pathways are involved in an evolutionary “arms race” with pathogenic organisms, genes in these pathways would be expected to show signs of positive selection driven by evolutionary pressure to counter virulence mechanisms of invading microbes. When signs of selection (as inferred from sequence comparisons within Drosophila simulans populations and between D. simulans and D. melanogaster) in immune genes and nonimmune genes were evaluated, immune genes as a group were found to have higher KA/KS ratios than nonimmune genes, providing evidence for elevated adaptive evolution (Schlenke and Begun 2003). Since receptor, effector, and signaling proteins function in different portions of the immune response pathways, these may be exposed to differing levels of contact with invading microbes and may display nonuniform levels of functional redundancy or pleiotropy. Thus, genes from different functional groups may be exposed to distinct selective pressures. Antimicrobial peptides, which might be expected to encounter unique selective pressures due to their direct interactions with invading microbes, have shown little sign of positive selection, bearing low levels of amino acid divergence (Clark and Wang 1997; Date et al. 1998; Ramos-Onsins and Aguadé 1998; Lazzaro and Clark 2003). Furthermore, sequence analyses of immune-related receptors have shown evidence for purifying selection in peptidoglycan recognition proteins (PGRPs), while others, including some scavenger receptors (SRs), appear to be rapidly evolving under pressures consistent with positive selection (Jiggins and Hurst 2003; Lazzaro 2005). On a deeper evolutionary timescale, sequence comparisons between immune genes in multiple Drosophila species (based on full-genome sequence data) have shown striking differences among functional groups of immune genes, with recognition molecules showing much more positive selection than either signaling or effector genes (Sackton et al. 2007).
Beyond using sequence data and the analysis of polymorphism and divergence to infer levels and modes of selection that have previously acted on immune genes (either individually or in functional groups), other studies have investigated correlations between autosomal variation in genotype and immune response phenotype in natural populations of Drosophila (Lazzaro et al. 2004, 2006). These experiments tested associations between naturally occurring genetic variation in immune-related genes and postinfection bacterial load. In these studies, genetic variation in many of the immune genes was found to associate significantly with one or more of the bacterial load phenotypes. Specifically, polymorphisms in autosomal genes encoding recognition and signaling proteins (but not antimicrobial peptides) associate consistently with bacterial load phenotypes, suggesting that not all functional classes of immune-related genes harbor equally influential genetic variation.
The focus of this study is X-linked immune genes, which may be under unique regulatory and selective pressures simply because they are hemizygous in males, are dosage compensated, and face elevated influence of random genetic drift due to their smaller effective population size. As a consequence, the X chromosome should favor the more rapid fixation of beneficial recessive alleles and more rapid loss of harmful recessive alleles compared to the autosomes (Charlesworth et al. 1987; Singh et al. 2008). Thus, with different selective pressures compared to autosomal genes, X-linked immunity genes are expected to bear different standing levels of variation, and segregating polymorphisms in these genes may have different impacts on phenotype.
Different exposures of X-linked genes to selection in males and females can also contribute to sexual dimorphism. Rice (1984) suggested that X-linked sexually antagonistic alleles may more freely influence sexually dimorphic traits than can those on autosomes. In fact, the X chromosome appears to favor the maintenance of sexually antagonistic variation (Gibson et al. 2002); if a given allele is slightly deleterious in one sex, it may be maintained in the population by being beneficial to the other sex. Immune-related genes may be particularly prone to bearing sexual dimorphism in Drosophila, since males and females have been shown to have different evolutionary optima for energetic expenditure on immune response, and thus their respective immune responses may differ on the basis of conditions such as food or reproductive resource availability (McKean and Nunney 2001, 2005). If sexually antagonistic traits are responsible for some of the observed sexual dimorphism, variation in X-linked genes could contribute to phenotypic differences, and so X-linked variation in immune genes could face unique selective pressures.
In this report we investigate the standing levels of variation in X-linked immune genes in natural populations of D. melanogaster and quantify the impacts of that variation on immune response phenotypes. We genotyped 168 lines at single-nucleotide polymorphisms (SNPs) across 20 X-linked immunity loci and quantified postinfection bacterial load and immune gene expression phenotypes. We found significant variation across the lines for bacterial load after infection, and we were able to identify polymorphisms in immune-related genes that associate with immune response phenotypes individually and in interacting pairs of SNPs. Additionally, some of the genetic variation was found to associate with a sex difference in immune competence, with alleles acting in either a sex-specific or a sexually antagonistic manner. This provides evidence for X-linked genetic variation in immune-related loci associating with both phenotypic variation among lines and sex differences in these phenotypes.
MATERIALS AND METHODS
Construction of lines:
D. melanogaster females were collected from apple orchards near Ithaca, New York by Todd Schlenke and Brian Lazzaro in 2004. Isofemale lines were established and kept under laboratory conditions for fewer than five generations prior to isogenization. X chromosomes were isogenized in these lines, by individually mating males from each line to females of the highly inbred balancer stock FM7a, B1 sc8 vOf wa y31d. From each of these crosses, three female offspring were individually mated to FM7a males. Since the balancer chromosome bears the codominant marker Bar, heterozygous female offspring could be selected for the crosses each generation. The crossing scheme was repeated for each line in triplicate for a total of seven generations to replace the background autosomes from the natural population. This resulted in 168 lines, each homozygous (or hemizygous) for a unique X chromosome from nature and all co-isogenic for the replaced autosomes. The degree of background replacement was quantified by subsequent SNP genotyping, finding concordance between the marker background and the isogenized lines in 99.6% of all assays (1191 tests of 1196 examined; see supporting information, Table S1 for full autosomal genotyping results).
Genotyping of SNPs across lines:
Candidate immune-related genes were selected for genotyping on the basis of previously indicated connections to immune responses in genetic studies and/or large-scale expression assays (Table 1). These genes include well-characterized members of the Toll and imd pathways, as well as genes involved in other aspects of the response to infection, including hematopoesis and iron metabolism. There is a significant overrepresentation of the genes in the JAK/STAT pathway among the X-linked immunity genes (χ2, P = 7.7 × 10−5), and several of these were included in our study. Notably, none of the 20 genes encoding antimicrobial peptides genomewide exist on the X chromosome in D. melanogaster, so our investigation lacks any genotyped members of this class of immune genes.
TABLE 1.
Genes selected for genotyping
| Functional group | Gene name | Cytological position | Sequence length | n | S | π | θW | D | SNPs |
|---|---|---|---|---|---|---|---|---|---|
| Recognition | PGRP-LE | 13F1 | 1027 | 8 | 4 | 0.0014 | 0.0015 | −0.2218 | 1 |
| PGRP-SA | 10C6 | 1414 | 8 | 0 | 0 | 0 | NC | 0 | |
| Signal transduction | domeless | 18D13–E1 | 1484 | 8 | 7 | 0.0028 | 0.0025 | 0.3364 | 6 |
| Dredd | 1B12–13 | 2452 | 8 | 13 | 0.0018 | 0.0021 | −1.3748 | 4 | |
| hemipterous | 11D10 | 5444 | 8 | 65 | 0.0067 | 0.0073 | −1.1266 | 14 | |
| hopscotch | 10B5–6 | 5388 | 8 | 41 | 0.0040 | 0.0040 | −0.5257 | 7 | |
| pole hole | 3A1 | 3834 | 8 | 47 | 0.0059 | 0.0058 | −0.8160 | 9 | |
| Tak1 | 19D2 | 6318 | 8 | 115 | 0.0086 | 0.0083 | −0.0373 | 5 | |
| Traf2 | 7D16 | 3327 | 8 | 70 | 0.0092 | 0.0091 | 0.3354 | 3 | |
| Traf3 | 14C4 | 2704 | 8 | 31 | 0.0058 | 0.0063 | −1.3593 | 2 | |
| Other | Dsor1 | 8D2–3 | 1171 | 6 | 6 | 0.0043 | 0.0043 | NC | 2 |
| lozenge | 8D5–6 | 3452 | 8 | 44 | 0.0066 | 0.0069 | 0.9657 | 4 | |
| multi sex combs | 8D2 | 2738 | 8 | 21 | 0.0043 | 0.0040 | −1.3101 | 2 | |
| Ntf2 | 19E7 | 2969 | 8 | 52 | 0.0078 | 0.0082 | −0.7046 | 2 | |
| outstretched | 17A5 | 2953 | 8 | 13 | 0.0026 | 0.0028 | 0.2036 | 1 | |
| Pvf1 | 17E1–6 | 5372 | 8 | 110 | 0.0118 | 0.0117 | NC | 10 | |
| Rps6 | 7C2 | 2278 | 8 | 54 | 0.0103 | 0.0106 | 0.3699 | 3 | |
| Ser7 | 9A2 | 2339 | 8 | 15 | 0.0023 | 0.0027 | −0.0835 | 3 | |
| Transferrin 1 | 17A9 | 3135 | 8 | 28 | 0.0034 | 0.0037 | −1.4206 | 6 | |
| unpaired 2 | 17A3 | 2524 | 8 | 21 | 0.0043 | 0.0039 | −0.4479 | 3 | |
|
unpaired 3 |
17A4 |
5261 |
8 |
190 |
0.0164 |
0.0186 |
−0.8004 |
4 |
n, number of lines sequenced; S, segregating sites; D, Tajima's D (NC, not calculated); SNPs, number genotyped.
To identify SNPs for genotyping, the entire gene regions for each gene, including roughly 1 kb upstream and downstream, were resequenced in eight of the X-extraction lines (see Table S2 for list of primers used). Table 1 reports summary statistics for these sequence alignments, calculated using DnaSP (Rozas and Rozas 1995), except for Tajima's D, which was calculated with VariScan so as not to exclude all sites with missing data (Vilella et al. 2005). Once polymorphism data were collected for all genes, SNPs were chosen from among those present at relatively intermediate frequencies in the samples and spaced ∼500–1000 bp apart within the genes. SNPs in high linkage disequilibrium (LD) with one another were generally avoided. Wherever possible, nonsynonymous SNPs were included; however, the selection of SNPs genotyped included those from exonic, intronic, 5′- and 3′-untranslated, and intergenic regions. In total, 91 SNPs were chosen from among these 20 genes for genotyping across all 168 lines. PGRP-SA was not included due to a complete absence of detectable variation found in the resequenced sample.
To identify the genotype for each line at each selected SNP, the SNPlex system (Applied Biosystems, Foster City, CA) was used. Oligos were designed and synthesized to query the genotype of all 91 SNPs (see Figure S1 for oligo and SNP information). The associated GeneMapper software was used to make the initial SNPlex allele calls, and these were followed by manual inspection. Eighty-eight of the 91 SNP assays in the SNPlex system yielded useful genotypic information across the 168 lines (see Table S3 for genotype calls at each site for all lines).
Bacterial cultures and infections:
Bacterial stocks were chosen on the basis of previous use for immune challenges in D. melanogaster. The strain of gram-positive bacterium Enterococcus faecalis was derived from that used by Lazzaro et al. (2006) (identified via 16S rDNA sequence and results of API 20Strep substrate utilization testing). We also selected gram-negative Serratia marcescens, derived from ATCC strain 13880, which also had been used in previous studies (Lazzaro et al. 2004, 2006). Bacterial cultures for infections were grown from freezer stocks, and cultures were grown overnight at 37° to a final concentration of OD600 ≈ 1.0 for each day of infections.
Bacterial load quantification:
Bacterial clearing ability of the lines was measured through quantification of bacterial load after infection with bacteria, following Lazzaro et al. (2004). D. melanogaster were individually infected by pricking their thoraces with 0.1-mm tungsten needles (Fine Science Tools, Foster City, CA) dipped in bacterial culture. For each bacterium, a block design of infections was used: each round of infections was repeated three times over 6 days in a 2-week span, with half the lines infected on a given day. For each round of infections, 12 males and 12 females from each line, aged ∼3–10 days, were infected (for technical feasibility, several people served as infectors on each day, but lines were randomized among infectors from day to day). Approximately 26–30 hr after infection, three groups of three flies per line were homogenized in 500 μl of LB broth and were then plated onto LB agar plates using a spiral plater (Spiral Biotech, Bethesda, MD). Homogenates with E. faecalis bacteria were diluted 1:1000 before plating to achieve a countable level of colonies. Plates were kept at either room temperature or 37° to allow bacterial colonies to grow until they could be counted by a colony counter. These counts allowed inference of the concentration of bacteria in each homogenate sample. Plates were visually inspected to confirm that colonies counted were consistent with size and morphology expected. Thus, for each line, both sexes were infected with each of two bacteria, and each round of infections included three replicates for each sex and bacterial infection of every line, over three rounds of infections. This yielded nine independent biological replicates of each infection with a total of >21,000 flies infected and 5046 plates counted.
TaqMan RT–PCR:
In addition to bacterial load, expression phenotypes were measured after infection for a subset of 16 lines, chosen from the phenotypic tails of sex difference in load after infection with E. faecalis. For each of these lines, 30 males and 30 females were infected with E. faecalis. Eight hours after infection, three replicates of each line and sex, most with 8–10 flies each, were snap-frozen in liquid nitrogen, along with three replicates of uninfected flies. RNA was extracted using a Trizol:chloroform protocol. cDNA was then synthesized from the isolated nucleic acid and diluted to fulfill TaqMan protocol requirements (Applied Biosystems). Transcripts were quantified using TaqMan RT–PCR, including antimicrobial peptide genes (DiptericinA, Defensin, and Metchnikowin), along with X-linked immune-related genes (Peptidoglycan Recognition Protein-SA and Transferrin1) and ribosomal protein RpL32 as a reference gene (see Table S4 for probe and primer sequence information). We measured the CT value for each sample (number of PCR cycles at which the level of fluorescence for the sample crosses a constant critical threshold value) and used the reciprocal, 1/CT, as a proxy for expression for further calculations.
Statistics and association testing:
Bacterial load was determined for each sample in terms of colony-forming units per fly (cfu/fly). Estimates of bacterial density from Drosophila homogenates (pools of three flies each) range from 1.0 × 100 to 4.0 × 106 cfu (corresponding to 0.3 × 100 to 1.3 × 106 cfu/fly). All empty plates were recorded as true zero counts (on a log scale) rather than as missing data; plates with density calculated above 3.0 × 106 were too dense to be accurately counted, so these were assigned to have densities of 4.0 × 106, which probably underestimates densities in most cases. Residuals from the analysis of variance on the raw cfu counts were distributed nonnormally, and log transformation yielded an adequate fit of residuals to the normal distribution. Statistical analyses were carried out using the R software (R Development Core Team 2007) and SAS/STAT software with the SAS system (SAS Institute, Cary, NC). To test for significant effect of Line, as well as a Line × Sex interaction on variation in bacterial load (for each bacterial infection), we used the mixed models
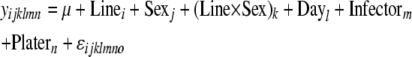 |
(1a) |
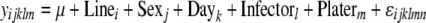 |
(1b) |
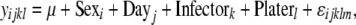 |
(1c) |
where y = ln(cfu/fly) (bacterial count); Line (i = 1 … 168), Sex (j = 1, 2), and the Line × Sex interaction are fixed effects; and Day (l = 1 … 6), Infector (m = 1 … 6), and Plater (n = 1 … 2) are treated as random effects using the R package lme4. ɛ is the error term. The full model (1a) was compared to the partially reduced model (1b) using ANOVAs to test for the effect of a Line × Sex interaction term. Similarly, the partially reduced model (1b) was compared to the reduced model (1c) to determine the effect of Line differences. To test the significance of each effect, load phenotypes were permuted 1000 times in R (for each bacterium), while keeping line, sex, and random effects constant. The coefficients of the model tests from these permutated data provided a null distribution as a basis of comparison for the actual Line and Line × Sex effects estimated from the data, and P-values were calculated for each. The proportions of variance explained by models incorporating just Line effects or Line × Sex interaction effects (r2) were also calculated for each bacterial infection using R.
Mixed models were also employed to test for associations between genotypes and phenotypes. Here, differences in each load phenotype (e.g., total E. faecalis load) between two alleles for each SNP were tested for significance using mixed models
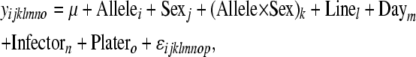 |
(2) |
where y represents the phenotype of interest, Allele (i = 1, 2) corresponds to the genotype at the SNP in question, as a fixed effect, and Sex (j = 1, 2) is also included as a fixed effect. The Allele × Sex interaction term was included as a fixed effect to quantify the effects of sex on SNP associations with bacterial load. Line (l = 1 … 168), Day (m = 1 … 6), Infector (n = 1 … 6), and Plater (o = 1, 2) are all included as random effects. Nearly identical models were used to test allelic effects on bacterial load in either males or females individually, with the exception that these did not include Sex as a fixed effect:
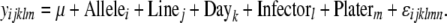 |
(3) |
Because of the potential for linkage disequilibrium among SNPs, tests of association were not all independent, so significance was assessed using permutation tests. Each SNP was tested individually, and genotypes were permuted 1000 times in R, relative to load phenotypes and the line, sex, day, infector, and plater values. The resulting coefficients for Allele or Allele × Sex effects provided a null distribution against which to compare the coefficients from tests with actual values, providing P-values for each. False discovery rate (FDR) was estimated by calculating q-values for each test, using the qvalue R package (Storey 2002). To determine the proportion of variance explained by each SNP, r2 values for models including each SNP alone as a fixed effect were calculated using R.
Associations between SNP genotypes and expression phenotypes were also examined using the mixed model
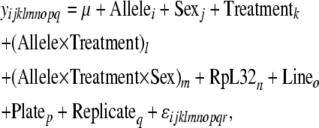 |
(4) |
where y represents the expression level of the gene of interest (1/CT). Here, the fixed effects include Allele (i = 1, 2), corresponding to the genotype at a given SNP; Treatment (j = 1, 2), representing “infected” or “uninfected” state of the flies; Allele × Treatment, the interaction of SNP genotype and infection state to test for the effect of change in expression level after infection; Allele × Treatment × Sex, the influence of sex on this induction effect; and RpL32, the expression of RpL32, as a covariate to normalize the expression phenotype measured. Line (o = 1 … 16), Plate (p = 1, 2), and Replicate (q = 1 … 3) were all included in the model as random effects. For each phenotype, every SNP was tested individually. As above, phenotype–genotype combinations were permuted 1000 times in R, and the coefficients for SNP × Treatment and SNP × Treatment × Sex effects provided null distributions against which to compare the actual coefficients and assign P-values. FDR values were again estimated using q value. r2 values for models including each SNP alone as a fixed effect were calculated using R.
Haplotypes of SNPs were assessed for the presence of blocks of high LD across the X chromosome, using the program Haploview (Barrett et al. 2005). Since these lines are homozygous, the comparisons essentially involve counts of gametes. Missing SNP data (Table S3) were imputed using the program fastPHASE version 1.1 (Scheet and Stephens 2006). The 10 haplotype blocks (sets of two to three SNPs within nine different genes) indicated to have significant levels of LD by the Haploview program were tested for associations with both load and expression phenotypes. These association tests were performed in the same manner as listed above for single SNPs, using the mixed models
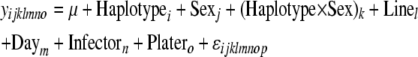 |
(5a) |
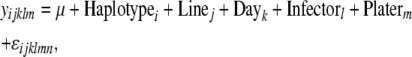 |
(5b) |
where Equation 5a tests for genotypic effect of Haplotype (i = 1 … 10) on bacterial load and Haplotype × Sex effects on bacterial load in all flies; Equation 5b was used to test for effects of Haplotype on bacterial load in males or females individually. As above, P-values were assigned on the basis of null distributions of coefficients of haplotype effects from permuted data sets.
In addition to associations between single SNP genotypes and phenotypes, effects of epistatic interactions were also examined. Here, the effects of interactions between every possible combination of SNP pairs (both within and between genes) were tested. Rigorous inference of pairwise epistasis normally requires consideration of all nine two-locus genotypes, and the usual caveats of fitting linear models with sparse marginal counts apply (Cockerham 1954). Here we have homozygous lines, so there are only four genotypes to contrast, and only 1 d.f. for tests of the single epistatic component and fixed marginal frequencies, so the model is closer to that of Cheverud and Routman (1995). With the low-frequency alleles of some SNPs, not every SNP pair allowed for valid tests of associations with all four genotype combinations, so these pairs were not included. For each valid test, two-way ANOVAs were performed to test associations with each phenotype using models both with and without SNP interaction terms; a significant difference between the fit of the two models to the data indicated an effect of the SNP interaction. The full and reduced models compared here are
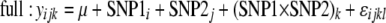 |
(6a) |
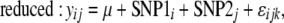 |
(6b) |
where y is the load or expression phenotype, SNP1 (i = 1 … 88) and SNP2 (j = 1 … 88) are the two SNPs of interest, and SNP1 × SNP2 is the interaction term of the allelic effects of these two SNPs. Due to the computational time needed to test all SNP combinations, these simpler linear models were applied, using estimated line means for the load and expression phenotypes. To accommodate the same random effects as above, the phenotypic values used were the least-squares means for each line obtained using mixed models in SAS, based on Equation 1b for load phenotypes and Equation 4 for expression phenotypes. These SNP interaction effects were tested for associations with load in males, females, and both sexes combined, along with the sex difference in load (female load minus male load) for each bacterium. In addition, associations were tested with induction of expression (infected minus uninfected expression levels) in males, females, and both combined. As above, with these ANOVA tests, we calculated P-values by permuting the genotype–phenotype combinations 1000 times and comparing actual F-statistics to the null distributions of F-statistics from tests with the permuted data. Again, r2 values were calculated to quantify the proportion of variance explained by the interaction term; this was determined from the difference in r2 values of the full and reduced models.
Beyond tests of association between the genotypes and phenotypes of these lines, we also tested the ability of expression phenotypes to predict load after infection. More specifically, we tested the effects of uninfected expression levels and induction of expression on E. faecalis levels after infection. These tests used the following models:
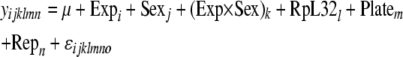 |
(7a) |
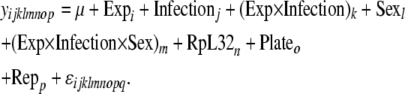 |
(7b) |
Here, y is the load phenotype, ln(cfu/fly), and the model includes Exp (the expression level of the gene assayed, 1/CT) as a fixed effect, along with RpL32 (expression level of RpL32) as a covariate to normalize the expression level of the gene of interest. Sex (j = 1, 2) and sex interaction terms are also included as fixed effects in both (except when each sex is considered individually). Plate (m = 1, 2) and Rep (n = 1 … 3, replicate) were also included as random effects in these models. In Equation 7a, Infection status is not included; here only uninfected or infected samples are considered at one time. In Equation 7b, however, the Expression × Infection term accounts for induction effects (if uninfected and infected flies have significantly different expression levels). Again, significance values for these tests were calculated on the basis of null distributions of coefficients from tests using data permuted 1000 times.
RESULTS
Variation observed in X-linked immune genes:
To quantify effects of naturally occurring X-linked variation in immune genes on immune phenotypes, X chromosomes from a natural population of D. melanogaster were extracted into co-isogenic autosomal backgrounds. To find polymorphic sites in the immune genes in these lines, 21 candidate genes were resequenced in eight sample lines. In the ∼67.5 kb of sequence obtained in these lines (including intronic, exonic, 3′- and 5′-untranslated, and intergenic regions), 947 SNPs were uncovered, 1 SNP about every 71 bases on average. Of the SNPs found in this sample, 172 are in coding regions, and 23 of these (13%) are nonsynonymous. An analysis of the sequence polymorphisms seen here shows nonskewed values of Tajima's D, but somewhat lower levels of variation (Table 1) than have been seen in other population genetic analyses of Drosophila immune genes. Compared to studies of non-African populations of D. melanogaster (Ramos-Onsins and Aguadé 1998; Andolfatto 2001), these X-linked immune genes have much lower values of θW than autosomal immune genes (t-test, P = 0.0002), while still showing significantly higher levels of variation than X-linked nonimmune genes (t-test, P = 0.0127). Most of the autosomal immune genes assayed for polymorphism, though, have been AMPs, and since no AMPs exist on the X chromosome, the disparity in levels of variation between X-linked and autosomal immune genes could be due at least in part to differences among functional groups.
Genetic variation in bacterial load:
We calculated bacterial load means for each D. melanogaster X-extraction line 26–30 hr after infection with E. faecalis or S. marcescens. The line means in load span a range of 9.82–16.73 ln(cfu/fly) for E. faecalis and 7.23–10.29 ln(cfu/fly) for S. marcescens, representing 1007-fold and 21-fold ranges that span 1.6–1.1 average within-line phenotypic standard deviations, respectively (Figure 1). Analyses of variance showed that this variation was significant among lines for both bacteria (P < 0.0001 for each). Furthermore, the line means of load for the two bacterial species are not correlated (correlation coefficient = 0.035, NS, Figure 1C). A lack of correlation of load across bacterial types has been noted in earlier studies (Lazzaro et al. 2004, 2006), and the interpretation has been that bacterial–host interactions are bacterial species specific, which can lead to different immune response dynamics, depending on the virulence mechanisms employed by the bacteria and the host response to this infection.
Figure 1.—

Line means of bacterial load after infection with (A) Enterococcus faecalis, B) Serratia marcescens, and (C) scatterplot of means of load for the two bacteria. Lines are plotted in rank order for each bacterium in A and B. Bacterial load is measured as the natural log of the count of colony-forming units per fly, ln(cfu/fly), shown with the standard errors of the mean.
In addition to differences among lines in bacterial load after infection, we also find variation among lines in load differences between males and females. Figure 2 shows the sex differences in mean load of both bacteria [in terms of ln(cfu/fly)] across all the lines. These differences (female mean cfu/fly minus male mean cfu/fly) range from 1.9 × 107 to −3.6 × 106 cfu/fly (from 1080-fold higher in females to 31-fold higher in males) for E. faecalis, with a median difference (across the line means) of 6.4 × 106 cfu/fly. No effort was made to control for body size between sexes, but these sex differences on load are much larger than what might be expected from body size differences alone. For S. marcescens, the differences range from 9.3 × 104 to −2.8 × 104 cfu/fly (from 11-fold higher in females to 12-fold higher in males), with a median difference of 5.3 × 103 cfu/fly. Significantly more than half the lines bear mean differences greater than zero for both E. faecalis (χ2, d.f. = 1, P = 2.3 × 10−18) and S. marcescens loads (χ2, d.f. = 1, P = 0.0055). With most lines here displaying higher bacterial load in females than in males after infection, this could imply that males in these lines have more effective immune responses than females. McKean and Nunney (2005) find the opposite effect (higher load in males after infection) with plentiful food and mates, yet this study also highlights the condition-dependent nature of these results. Furthermore, these experiments have included load assays after different types of bacterial infections, which might not be expected to yield the same levels of bacterial load or sex differences in load.
Figure 2.—

Sex differences in mean bacterial load after infection with (A) E. faecalis and (B) S. marcescens, displayed as (female ln(cfu/fly) – male ln(cfu/fly)) ± (standard error of the difference), and (C) histogram of P-values of t-tests of sex difference in all lines after E. faecalis infection (black bars) or S. marcescens infection (gray bars).
Many of these lines show significant differences between male and female load, particularly when infected by E. faecalis. When each line is tested for sex effect on load, the distribution of P-values is highly skewed from an expectation of equal load in both sexes, with an excess of t-tests with P < 0.05 (χ2, d.f. = 1, P = 8.8 × 10−16) in flies infected with E. faecalis; however, in those infected with S. marcescens, P-values from t-tests of sex effects show no significant departure from the expected distribution (χ2, d.f. = 1, P = 0.36) (Figure 2C). Even though these lines show a wide range of differences between sexes, bacterial load after infection in males and that in females are significantly correlated for both E. faecalis and S. marcescens (Spearman's τ, P = 0.0025, P = 3.21 × 10−11). Thus, for most of the lines, higher (or lower) bacterial load remains relatively consistent in both sexes. As expected from the greater sex differences in lines infected with E. faecalis, though, load values in males and females infected with this bacterium are less strongly correlated than are those in flies infected with S. marcescens.
Genotypic variation among the extraction lines was tested for association with variation observed in immune phenotypes. X-linked genes from immune-related pathways (Figure 3) were chosen as candidates and genotyped to determine standing levels of variation. Eighty-eight SNPs in 20 candidate immune genes (Table 1) were individually tested for allelic effects on bacterial load phenotypes after infection with both E. faecalis and S. marcescens. Table 2 lists the q-values, based on P-values calculated from permuted null distributions, for those SNPs that showed at least one phenotypic association with FDR < 10% (q < 0.1). While these tests reveal possible associations with multiple SNPs within different immune genes, any given SNP typically explains <8% of the variance in bacterial load phenotypes.
Figure 3.—

Genes in Drosophila immune-related pathways. Those in black are X-linked genes included in this study, and those outlined in black are X-linked, but were not genotyped here. Pathway genes and interactions are included on the basis of information in previous studies (Wassarman et al. 1995; Stronach and Perrimon 2002; Foley and O'Farrell 2004; Leclerc and Reichhart 2004; Arbouzova and Zeidler 2006; Ferrandon et al. 2007).
TABLE 2.
SNPs associating with load phenotypes
| Functional class | Gene | Location | Change | Ef female | Ef male | Ef all | Sm female | Sm male | Sm all | Ef SNP × Sex | Sm SNP × Sex |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Signal | hopscotch | Exon | V → L | 0.688 | 0.739 | 0.981 | <0.001*** | 0.360 | 0.032* | 0.172 | 0.946 |
| Transduction | 0.7 | 0.3 | |||||||||
| hopscotch | Exon | <0.001*** | 0.025* | 0.981 | <0.001*** | 0.076* | <0.001*** | <0.001*** | 0.022* | ||
| 0.4 | 0.5 | 0.8 | 0.2 | 0.4 | 6.2 | 0.9 | |||||
| hopscotch | Exon | <0.001*** | 0.120 | <0.001*** | <0.001*** | 0.735 | 0.372 | 0.274 | 0.043* | ||
| 0.4 | 0.2 | 0.1 | 0.7 | ||||||||
| hemipterous | Exon | Y → C | 0.476 | <0.001*** | <0.001*** | 0.445 | 0.801 | 0.372 | 0.323 | 0.498 | |
| 1.0 | 0.6 | ||||||||||
| hemipterous | Exon | A → S | 0.476 | 0.215 | 0.981 | 0.847 | 0.520 | 0.758 | 0.029* | 0.997 | |
| 5.9 | |||||||||||
| hemipterous | Exon | 0.258 | 0.973 | 0.981 | 0.732 | 0.123 | 0.168 | 0.029* | 0.997 | ||
| 6.2 | |||||||||||
| Tak1 | Intron | 0.258 | 0.973 | 0.404 | 0.775 | <0.001*** | <0.001*** | 0.304 | 0.417 | ||
| 0.8 | 0.5 | ||||||||||
| TRAF3 | Exon | 0.999 | <0.001*** | <0.001*** | 0.283 | 0.393 | 0.042* | 0.000*** | 0.997 | ||
| 1.0 | 0.0 | 0.1 | 6.2 | ||||||||
| Secreted | outstretched | Exon | A → S | 0.404 | 0.973 | 0.521 | 0.117 | <0.001*** | <0.001*** | 0.850 | <0.001*** |
| 0.8 | 0.4 | 1.1 | |||||||||
| upd2 | Exon | 0.999 | 0.356 | 0.536 | 0.067* | 0.633 | 0.140 | 0.925 | 0.731 | ||
| 0.4 | |||||||||||
| upd3 | Intergenic | 0.306 | <0.001*** | <0.001*** | <0.001*** | 0.336 | <0.001*** | 0.123 | <0.001*** | ||
| 0.4 | 0.2 | 0.4 | 0.2 | 0.9 | |||||||
| Iron metabolism | Tsf1 | Exon | 0.999 | <0.001*** | 0.012* | 0.912 | 0.814 | 0.906 | 0.151 | 0.997 | |
| 0.9 | 0.4 | ||||||||||
| Hematopoesis | lozenge | Exon | 0.306 | <0.001*** | 0.688 | 0.092* | <0.001*** | <0.001*** | <0.001*** | 0.997 | |
| 1.0 | 0.6 | 1.2 | 0.8 | 7.7 | |||||||
| Pvf1 | 5′-UTR | 0.999 | 0.973 | 0.981 | 0.851 | 0.135 | 0.443 | 0.987 | 0.037* | ||
| 0.6 | |||||||||||
| Pvf1 | Exon | 0.043* | 0.120 | <0.001*** | 0.832 | 0.633 | 0.890 | 0.987 | 0.731 | ||
| 1.5 | 0.9 | ||||||||||
| Pvf1 | Exon | 0.000*** | 0.221 | <0.001*** | 0.065* | 0.829 | 0.271 | 0.487 | 0.034* | ||
| 0.5 | 0.3 | 0.3 | 0.7 | ||||||||
| Pvf1 | Intron | 0.453 | 0.226 | 0.134 | 0.445 | 0.109 | 0.028* | 0.808 | 0.997 | ||
| 0.0 | |||||||||||
| Rps6 | Exon | 0.999 | 0.951 | 0.981 | 0.820 | 0.000*** | 0.017* | 0.987 | 0.037* | ||
| 0.6 | 0.2 | 1.0 | |||||||||
| Serine protease | Ser7 | Intergenic | 0.999 | <0.001*** | 0.022* | 0.117 | <0.001*** | 0.833 | 0.029* | <0.001*** | |
| 0.2 |
0.1 |
0.4 |
6.2 |
0.9 |
q-values are shown for SNPs that associate with at least one load phenotype (on the basis of having FDR q < 0.1). Percentage of total phenotypic variance explained is shown for each SNP with q < 0.1 (below q-value). Change is the amino acid change associated with SNP, where applicable. Sixty-nine of 88 SNPs show no association with load phenotypes with q < 0.1. *q < 0.1, ***q < 0.001. Ef, E. faecalis; Sm, S. marcescens.
Of the 19 SNPs that associate with one or more of the load phenotypes at this level, 8 associate (at least marginally) with phenotypes for both bacteria; however, 5 of these show opposite effects across bacteria in one or both sexes. Examples of this include 2 SNPs in the gene hopscotch (hop). As depicted in Figure 4, A and B, 1 SNP in exon 7 of the gene has significant allelic effects on load in both males and females, with both bacteria. The effects of the two infections in males, though, appear in opposite directions: a substitution from the “A” allele to the “G” allele of this SNP associates with a lower E. faecalis load, yet a higher S. marcescens load after infection. Similarly, for the second SNP in hop exon 7 (306 bp downstream from the first; Figure 4, C and D), allelic effects are once again significantly associated with load in females infected by either bacterium, yet the load variation in females occurs in opposite directions for the two bacteria.
Figure 4.—

Example effects of SNP on both E. faecalis (A and C) and S. marcescens (B and D) load after infection in females (solid lines) and males (dashed lines). SNPs shown include hop exon 7-01 (synonymous, residue 870) in A and B and hop exon 7-02 (synonymous, residue 968) in C and D.
Besides the distinct phenotypes and associations appearing in response to each of the two bacterial infections, some SNPs also associate with load variation in a sex-specific or even sexually antagonistic manner. Of the 19 SNPs associating with load, 12 show evidence of sex interactions influencing the associations with the load of one or both bacterial infections, and several of these actually appear to have opposite effects in males and females. Most of these SNPs do not show significant associations in both sexes individually, though, lessening our ability to find clear instances of sexually antagonistic associations. Any potentially sexually antagonistic effects appear only with one of the two bacteria in each case. An example of this is seen with the SNP in hop exon 7 in Figure 4A.
In addition to single-SNP tests of association, we consider the possibility that multiple SNPs that fall into particular haplotype configurations might correlate with differences in immune function. We identified haplotypes as collections of SNPs in LD and subsequently tested these for associations with immune phenotypes. Haplotype blocks with significant levels of LD (as defined by the program Haploview, see materials and methods) were identified in 10 sets of two to three SNPs across nine genes. Most of these blocks involved fairly closely located SNPs. Less than 17% of all SNP pairs within 1 kb of each other were found to be in high LD, consistent with previous findings that LD decays quickly along the Drosophila genome (Long et al. 1998; Carbone et al. 2006). These blocks of high LD were tested for associations with load phenotypes. Variation in 6 of these haplotype blocks significantly associates (P < 0.05) with differences in one or more bacterial load phenotypes (Table 3). Twelve individual haplotype–phenotype associations appear with P < 0.05, 10 of which have a FDR < 10% (q < 0.1). Many of these associations appear only with E. faecalis load phenotypes; 3 haplotypes appear to associate with S. marcescens load, but only in sex-by-haplotype interactions. Additionally, for all the haplotype blocks that show significant associations, none of the SNPs included in each haplotype associate individually with the same load phenotype (with FDR < 10%). Furthermore, many of the SNPs included in these haplotypes are noncoding or synonymous; the only nonsynonymous SNPs among these clusters are the two in the Pvf1 haplotype. These two SNPs, both located in exon 1, appear to be outside of the identified PDGF domain and the putative signal peptide of the gene, so no obvious disruption of function is inferred from their amino acid changes. Overall, it appears that the SNPs within the significantly associating haplotypes are most likely in linkage disequilibrium with any variation that could directly lead to phenotypic differences in these lines.
TABLE 3.
Multiple SNP clusters associating with load phenotypes
| Gene (SNPs) | Location | Ef female | Ef male | Ef all | Sm female | Sm male | Sm all | Ef Sex × Hap | Sm Sex × Hap |
|---|---|---|---|---|---|---|---|---|---|
| Dredd (2, 3, 4) | Exon 2, intron 1, 5′ intergenic | 0.139 | 0.226 | 0.489 | 0.195 | 0.931 | 0.443 | 0.191 | 0.021* |
| phl (6, 7) | Exon 4 | 0.187 | 0.250 | 0.017* | 0.650 | 0.096 | 0.473 | 0.722 | 0.328 |
| mxc (1, 2) | 5′ intergenic and exon 2 | 0.013* | 0.037* | 0.101 | 0.325 | 0.150 | 0.123 | 0.028* | 0.285 |
| hep (9, 10, 11) | Exon 5, exon 4, exon 3 | 0.043* | 0.084 | 0.121 | 0.311 | 0.336 | 0.209 | <0.001** | 0.026* |
| Pvf1 (3, 4) | Exon 1 | <0.001** | 0.001** | 0.019* | 0.095 | 0.274 | 0.104 | 0.348 | 0.625 |
|
Tak1 (1, 2) |
Intron 4 and exon 3 |
0.956 |
0.945 |
0.974 |
0.575 |
0.486 |
0.484 |
0.241 |
0.033* |
*P < 0.05, **P < 0.01. Ef, E. faecalis; Sm, S. marcescens; Hap, haplotype.
For each possible pair among the 88 SNPs, we tested for pairwise epistasis on the basis of the significance of the interaction term in two-way ANOVAs. Figure 5 depicts those pairs of genes within the Toll, imd, JAK/STAT, and JNK pathways that bear SNPs with interactions associating with load phenotypes with q-values <0.1 for tests of models using SNP pair interactions to explain variation in bacterial load after infection. Most genes—11 of 12 tested in these pathways—contain SNPs involved in interactions associating with one or more of the load phenotypes at the q < 0.1 level. Additional interactions were found to be significant at this threshold involving genes outside these pathways (thus not depicted on these diagrams), including lozenge, Pvf1, Ser7, and Tsf1. One of the genes, upd2, also had an interaction between a pair of SNPs within the same gene that associate with a load phenotype (q < 0.1). The interaction terms of the models explain different amounts of phenotypic variance, even among interactions showing significant effects (at the q < 0.1 level). Some interactions account for <0.1% of the variance, while others explain up to 20.6% (Pvf1 × Traf2 interaction term with female load after S. marcescens infection). This amount of variance is substantially higher than that explained by each SNP individually; the sum of the percentage of variance explained by the two included SNPs is <0.1% in this instance.
Figure 5.—

Epistatic interactions associating with bacterial load after infection with (A) E. faecalis and (B) S. marcescens. Lines between a pair of genes correspond to at least one interaction between SNPs in those genes having a significant effect on the load phenotype of the corresponding pattern (ANOVA, q < 0.1). Interactions shown associating with male load were only those that did not also associate with load in both sexes combined. (Ef, E. faecalis; Sm, S. marcescens.)
Genetic variation in immune gene induction:
To evaluate the effects of X-linked genetic variation on immune gene induction, a subset of 16 lines was selected from the collection of X-extraction lines, and males and females of these lines were assayed for expression of immune-related genes before and after infection with E. faecalis. We used the differences in these levels to quantify the induction of gene expression in response to infection. The genes examined include those encoding the antimicrobial peptides Defensin (Def), DiptericinA (DptA), and Metchnikowin (Mtk), as well as Peptidoglycan recognition protein-SA (PGRP-SA) and Transferrin1 (Tsf1), involved in iron transport. The subset of lines was selected from the tails of the distribution of mean sex differences of E. faecalis loads, allowing for tests of associations of immune gene induction with sex differences in load.
The 88 immune-related SNPs were tested for association with induction phenotypes for each of these genes, in males, females, and both sexes combined, as well as in phenotypic associations with sex-by-SNP interactions. Table 4 lists the SNPs that showed association (with FDR < 10%) with one or more of the induction phenotypes. Only 10 of the 88 SNPs appear with significant associations, yet these represent variation in seven separate genes (six of which also have SNPs or haplotypes associating with load phenotypes). While some SNPs show associations with more than one phenotype, most show isolated effects: five of the nine associate with only one of the induction phenotypes. Most of these associations appear with induction phenotypes in only one sex, yet only one SNP associates significantly with sex difference in induction. While most of these associations explain <8% of the variance observed (some <1%), the Ser7 exonic SNP appears to explain >14% of the observed variance in Mtk induction in females (Table 4).
TABLE 4.
SNPs associating with immune gene induction phenotypes
| Female induction |
Male induction |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Functional class | Gene | Location | Change | Def | DptA | Mtk | PGRP-SA | Tsf1 | Def | DptA | Mtk | PGRP-SA | Tsf1 |
| Signal | pole hole | Exon | 0.997 | 0.820 | 0.506 | 0.230 | 0.170 | 0.551 | 0.518 | 0.954 | 0.976 | <0.001*** | |
| Transduction | 3.0 | ||||||||||||
| pole hole | Exon | 0.997 | 0.820 | 0.887 | 0.230 | 0.170 | 0.368 | 0.197 | 0.954 | 0.144 | 0.056* | ||
| 1.8 | |||||||||||||
| TRAF2 | Intergenic | 0.023* | 0.870 | 0.813 | 0.161 | 0.247 | 0.551 | 0.922 | NA | 0.976 | 0.806 | ||
| 6.7 | |||||||||||||
| TRAF3 | Exon | NA | <0.001*** | NA | 0.228 | NA | 0.382 | <0.001*** | 0.954 | NA | NA | ||
| 7.1 | 4.8 | ||||||||||||
| Secreted | upd2 | Exon | <0.001*** | 0.820 | 1.000 | 0.248 | 0.170 | 0.551 | 0.943 | 0.681 | 0.976 | 0.042* | |
| 0.3 | 7.9 | ||||||||||||
| Iron metabolism | Tsf1 | Exon | <0.001*** | NA | 0.017* | 0.228 | NA | NA | NA | NA | NA | NA | |
| 4.5 | 1.6 | ||||||||||||
| Hematopoesis | Pvf1 | Exon | A → T | NA | 0.381 | NA | NA | NA | 0.551 | 0.518 | NA | NA | NA |
| Pvf1 | Exon | 0.997 | 0.820 | 0.992 | 0.161 | 0.075* | 0.551 | 0.546 | NA | 0.976 | 0.806 | ||
| 1.7 | |||||||||||||
| Serine protease | Ser7 | Intergenic | <0.001*** | NA | <0.001*** | 0.228 | NA | NA | NA | NA | NA | NA | |
| 4.6 | 1.2 | ||||||||||||
| Ser7 | Exon | 0.261 | 0.820 | 0.046* | 0.272 | 0.075* | 0.551 | 0.197 | NA | 0.976 | 0.806 | ||
| 14.3 | 4.5 | ||||||||||||
| Combined induction |
Effect of Sex × SNP interaction on induction |
||||||||||||
| Functional class |
Gene |
Location |
Change |
Def |
DptA |
Mtk |
PGRP-SA |
Tsf1 |
Def |
DptA |
Mtk |
PGRP-SA |
Tsf1 |
| Signal | pole hole | Exon | 0.582 | 0.682 | 0.425 | 0.481 | 0.277 | 0.923 | 0.620 | NA | 0.759 | 0.539 | |
| Transduction | |||||||||||||
| pole hole | Exon | 0.599 | 0.645 | 0.620 | 0.412 | 0.387 | 0.906 | 0.318 | 0.760 | 0.694 | 0.592 | ||
| TRAF2 | Intergenic | 0.594 | 0.874 | 0.425 | 0.549 | 0.912 | 0.788 | 0.832 | NA | 0.657 | 0.763 | ||
| TRAF3 | Exon | 0.448 | <0.001*** | 0.620 | 0.515 | NA | NA | NA | NA | NA | NA | ||
| 5.9 | |||||||||||||
| Secreted | upd2 | Exon | 0.149 | 0.874 | 0.620 | 0.549 | 0.387 | 0.788 | 0.832 | 0.760 | 0.657 | 0.488 | |
| Iron metabolism | Tsf1 | Exon | 0.119 | 0.899 | <0.001*** | 0.515 | 0.273 | NA | NA | NA | NA | NA | |
| 1.7 | |||||||||||||
| Hematopoesis | Pvf1 | Exon | A → T | 0.849 | 0.450 | NA | 0.515 | 0.657 | NA | 0.620 | NA | <0.001*** | NA |
| 0.3 | |||||||||||||
| Pvf1 | Exon | 0.691 | 0.450 | 0.620 | 0.515 | 0.912 | 0.906 | 0.832 | NA | 0.657 | 0.539 | ||
| Serine protease | Ser7 | Intergenic | <0.001*** | 0.902 | <0.001*** | 0.515 | <0.001*** | NA | NA | NA | NA | NA | |
| 0.2 | 0.5 | 0.0 | |||||||||||
|
Ser7 |
Exon |
0.590 |
0.450 |
0.104 |
0.549 |
0.908 |
NA |
0.741 |
NA |
0.750 |
0.539 |
||
q-values shown for SNPs that associate with at least one induction phenotype at an FDR ≤10% (q ≤ 0.1) are shown; 79 of the 88 SNPs tested showed no association with any induction phenotype. Percentage of phenotypic variance explained by association is shown for each test with q < 0.1 (below q-value). Change, amino acid change associated with SNP, where applicable. *q < 0.1, ***q < 0.001.
Correlations between induction and load phenotypes:
In addition to testing associations between genetic variation and immune gene phenotypes, we also tested whether any of the variation observed in induction of immune genes correlated with variation in bacterial load after infection with E. faecalis. Here, we tested the ability of models incorporating expression levels (before and after infection, as well as levels of induction) to explain levels of bacterial load in these lines of flies. One putative association was found, where the induction of Tsf1 correlates negatively with bacterial load after E. faecalis infection (P = 0.008, on the basis of permuted null distribution) in males. Figure 6 displays bacterial load line means plotted against induction line means of Tsf1 (normalized by RpL32 expression) in both males and females after infection with E. faecalis. Increased induction levels of Tsf1 associate with lower levels of bacterial load after infection in males, while female values show no significant correlation between these traits.
Figure 6.—

Line means for bacterial load vs. Tsf1 induction levels (infected minus uninfected expression, normalized by RpL32 expression) after infection with E. faecalis in males (solid diamonds, solid regression line) and females (open diamonds, dashed regression line).
DISCUSSION
To examine the effects of genomic location on genotypic variation and phenotype, we measured associations between polymorphisms in X-linked immune genes and response to bacterial infection in lines of D. melanogaster. These lines, bearing naturally varying X chromosomes in a co-isogenic autosomal background, were genotyped for SNPs in 20 immune genes. These X-linked immune genes include members of numerous immune-related pathways. The Toll and imd pathways, key to the humoral antimicrobial response in Drosophila, have representatives on the X chromosome, and the JAK/STAT pathway, also involved in response to bacterial infection, has a significant excess of its genes on the X (χ2, d.f. = 1, P = 7.7 × 10−5). Interestingly, while the X-linked genes from these pathways include those with roles in recognition and signaling, there are no antimicrobial peptide genes yet identified on the X chromosome. The absence of antimicrobial peptide genes on the X chromosome is highly significant (Fisher's exact test, P = 0.0036), and the cause for this remains a puzzle.
Other than the genic content of the X compared to the autosomes, this chromosome also provides a unique environment that may allow different levels and types of genetic variation to exist compared to that on the autosomes. Furthermore, since genes on the X chromosome spend one-third of their time in hemizygous males, they are exposed to different selective pressures; hemizygosity may expose recessive alleles, purging deleterious genotypes and fixing beneficial ones. This is expected to result in lower levels of variation on the X chromosome relative to autosomes with the exception of alleles showing antagonistic phenotypes either between sexes or in different environments or genetic backgrounds (Charlesworth et al. 1987).
For immune-related genes on the X chromosome, we expect that variation may be maintained in the population more readily if different alleles provide beneficial effects in diverse environments, such as with different bacterial infections, or in distinct genetic backgrounds, including in males vs. females. The results found in this study agree with this expectation. Genetic variation is observed in X-linked immune genes, frequently associating with phenotypic variation in immune response. Many of these associations, though, appear with one bacterial infection and not the other or act in a sex-specific or sexually antagonistic manner. Alleles such as these, associating with phenotypic variation in a condition-specific manner, presumably would not be selected for or against as rapidly as those with universally beneficial or deleterious effects, even on the X chromosome. A few SNPs tested here do show more general associations with the immune phenotypes examined; presumably alleles that appear relatively detrimental in tests observed here could have been maintained in the population because of beneficial effects in other circumstances (or for other phenotypes).
Previous investigations (Lazzaro et al. 2004, 2006) have involved similar tests of association between genotypic variation in immune genes on the second and third chromosomes of D. melanogaster and differences in immune response phenotypes. It is difficult to make direct comparisons between these studies, involving fly lines from separate populations, different bacterial infections, and distinct experimental setups, including different levels of replication. We do find, though, that similar to those studies, variation in numerous genes throughout immune pathways associates significantly with phenotypic variation. Interestingly, genetic variation on the second chromosome can explain 47.2% of the total variance in bacterial load (after infection with S. marcescens; Lazzaro et al. 2004), and variation on the third chromosome can explain 22.1% of the total observed variance in bacterial load (after infection with Providencia rettgeri; T. Sackton, personal communication), yet X-linked genetic variation in these lines explains only 15.5% of the total variance in bacterial load (after infection with S. marcescens). This suggests a lower level of naturally occurring variation in X-linked immune genes and/or less influence of that variation on immune phenotypes than is observed with autosomal genes. Additionally, this could be due to the fact that the X chromosome, as a shorter portion of the genome compared to the autosomes, may simply contain fewer loci affecting the observed immune phenotypes. Individual polymorphisms within immune genes on the second chromosome, though, appear to explain a larger proportion of the phenotypic variance than was observed with the SNPs here; numerous autosomal variations explained >5% of the phenotypic variance, whereas the most significant explained up to 22.7% (Lazzaro et al. 2004, 2006). Thus, variation in X-linked immune genes could have a lesser influence on phenotypic variance than polymorphism in autosomal genes, relative to environmental and experimental factors influencing differences in bacterial load.
The associations found between autosomal genes and immune phenotypes were strongly biased with respect to the functional class of immunity genes. There was a preponderance of associations between bacterial load and SNPs in recognition molecules and a deficit of associations with SNPs in antimicrobial peptides (Lazzaro et al. 2004; T. Sackton, personal communication). The X chromosome had a markedly different distribution of functional classes of immune genes, including an absence of any antimicrobial peptides, and this may contribute to the observation that there was no departure from a random representation of recognition and signaling functional classes among X-linked genes that associated with bacterial defense (χ2, d.f. = 1, P = 0.327). Furthermore, since variation in autosomal antimicrobial peptide genes appears to lack the phenotypic associations that variation in other autosomal immune genes bears, it seems unlikely that genic makeup alone would lead to different patterns of association between the X-linked and the autosomal immune genes.
While polymorphisms in X-linked genes appear to act mostly in sex-specific or sexually antagonistic associations with phenotypic variation, associations involving autosomal variation were much more likely to be sex independent. An investigation of associations with variation in immune genes on the second chromosomes uncovered no significant sex or sex × line effects on load (Lazzaro et al. 2004). When the specific tests of SNP interactions with sex were done, 8 of the 127 SNPs had a sex × SNP interaction that was significant at the nominal 5% level (Lazzaro et al. 2004), a number that is consistent with the expectation under the null hypothesis. Similarly, SNPs on the third chromosome associating with immune phenotypes show only marginal effects of sex × SNP interaction (T. Sackton, personal communication). The inflated magnitude of sexual dimorphism of X-linked immunity genes over autosomal genes is consistent with several mechanisms that might produce different regulatory responses of X-linked genes, including unique patterns of sex-biased expression (Parisi et al. 2003) or imprecision of dosage compensation.
In addition to dimorphic effects on correlations between genotype and load, we also observe sex-specific associations between induction levels and load in these lines. This suggests that activation of parts of the immune response may be regulated in a sex-specific way, perhaps in response to different physiological demands. Because Drosophila males and females have different fitness impacts of immune system activation (McKean and Nunney 2001, 2005), it is reasonable to expect some alleles to display different sex-specific phenotypic effects.
As well as the sexual dimorphism that appears in the associations between X-linked genetic variation and immune response phenotypes, distinct responses to different species of bacterial infections were also observed here. Four of the 19 SNPs (21.1%) that associate with bacterial load phenotypes in these lines (in both sexes combined, with P < 0.05), though, show associations with variation in response to both E. faecalis and S. marcescens, while only 1 of 36 (2.8%) of all of the autosomal SNPs associating with load differences after infections with one of these two bacteria is commonly found between the two. This excess of overlapping associations among X-linked polymorphisms over those on the autosomes is marginally significant (Fisher's exact test, P = 0.048), indicating a higher level of generality in the X-linked associations with response to different bacterial infections. Most of the variation tested in these lines (both X-linked and autosomal), though, appears to have the same effect across infections: flies bearing an allele that associates with lower load after infection with one bacterium tend to have lower load after the infection with the other bacterium as well. Thus, while the variation among X-linked and autosomal immune genes may vary in generality of response to different bacteria, there is not much evidence for antagonistic variation between bacterial infections.
The widespread presence of sex differences in associations in this study underscores the complexity of the association between immune response and polymorphisms in X-linked immune genes. These effects influencing genotype–phenotype correlations appear to be more striking with X-linked variation than with that on the autosomes; this is not unexpected, though, given the genomic environments of genes on these respective chromosomes. If the X-linked variation existing in natural populations includes alleles detrimental in one sex but not the other, these alleles are less likely to be selected against and may remain in the population in spite of negative phenotypic effects. Thus, even though we may expect genotypic variation associating with phenotypic effects to be relatively uncommon on the X chromosome, phenotypic differences observed here do correlate with polymorphisms in these lines. The complex patterns of association seen, however, show that these segregating polymorphisms bear characteristics consistent with predicted effects of natural selection on X-linked variation.
Acknowledgments
We thank Tracy Mak, Anna Beavis, Ryan Cummings, Kurt McKean, and Phil Olsen for assistance with infections and bacterial load quantifications. We also thank Xiaoyun Wang for help with sequencing, Peter Schweitzer of the Cornell Life Sciences Core Laboratories Center for assistance with genotyping, and Tim Sackton and Brian Lazzaro for helpful discussion and comments on this manuscript, as well as the reviewers for helpful suggestions. This work was supported by National Institutes of Health grant R01 AI064890 to A.G.C. and B. P. Lazzaro.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.108.093971/DC1.
References
- Andolfatto, P., 2001. Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Mol. Biol. Evol. 18 279–290. [DOI] [PubMed] [Google Scholar]
- Arbouzova, N. I., and M. P. Zeidler, 2006. JAK/STAT signalling in Drosophila: insights into conserved regulatory and cellular functions. Development 133 2605–2616. [DOI] [PubMed] [Google Scholar]
- Barrett, J. C., B. Fry, J. Maller and M. J. Daly, 2005. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 263–265. [DOI] [PubMed] [Google Scholar]
- Brennan, C. A., and K. V. Anderson, 2004. Drosophila: the genetics of innate immune recognition and response. Annu. Rev. Immunol. 22 457–483. [DOI] [PubMed] [Google Scholar]
- Carbone, M. A., K. W. Jordan, R. F. Lyman, S. T. Harbison, J. Leips et al., 2006. Phenotypic variation and natural selection at catsup, a pleiotropic quantitative trait gene in Drosophila. Curr. Biol. 16 912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B., J. A. Coyne and N. H. Barton, 1987. The relative rates of evolution of sex chromosomes and autosomes. Am. Nat. 130 113–146. [Google Scholar]
- Cheverud, J. M., and E. J. Routman, 1995. Epistasis and its contribution to genetic variance components. Genetics 139 1455–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A. G., and L. Wang, 1997. Molecular population genetics of Drosophila immune system genes. Genetics 147 713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerham, C. C., 1954. An extension of the concept of partitioning hereditary variance for analysis of covariances among relatives when epistasis is present. Genetics 29 859–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date, A., Y. Satta, N. Takahata and S. I. Chigusa, 1998. Evolutionary history and mechanism of the Drosophila Cecropin gene family. Immunogenetics 47 417–429. [DOI] [PubMed] [Google Scholar]
- Ferrandon, D., J.-L. Imler, C. Hetru and J. A. Hoffmann, 2007. The Drosophila systemic immune response: sensing and signaling during bacterial and fungal infections. Nat. Rev. Immunol. 7 862–874. [DOI] [PubMed] [Google Scholar]
- Foley, E., and P. H. O'Farrell, 2004. Functional dissection of an innate immune response by a genome-wide RNAi screen. PLoS Biol. 2 E203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, J. R., A. K. Chippendale and W. R. Rice, 2002. The X chromosome is a hot spot for sexually antagonistic fitness variation. Proc. R. Soc. Lond. Ser. B 269 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving, P., L. Troxler and C. Hetru, 2004. Is innate enough? The innate immune response in Drosophila. C. R. Biol. 327 557–570. [DOI] [PubMed] [Google Scholar]
- Jiggins, F. M., and G. D. D. Hurst, 2003. The evolution of parasite recognition genes in the innate immune system: purifying selection on Drosophila melanogaster peptidoglycan recognition proteins. J. Mol. Evol. 57 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrell, D. A., and B. Beutler, 2001. The evolution and genetics of innate immunity. Nat. Rev. Genet. 2 256–267. [DOI] [PubMed] [Google Scholar]
- Lazzaro, B. P., 2005. Elevated polymorphism and divergence in the class C scavenger receptors of Drosophila melanogaster and D. simulans. Genetics 169 2023–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro, B. P., and A. G. Clark, 2003. Molecular population genetics of inducible antibacterial peptide genes in Drosophila melanogaster. Mol. Biol. Evol. 20 914–923. [DOI] [PubMed] [Google Scholar]
- Lazzaro, B. P., B. K. Sceurman and A. G. Clark, 2004. Genetic basis of natural variation in D. melanogaster antibacterial immunity. Science 303 1873–1876. [DOI] [PubMed] [Google Scholar]
- Lazzaro, B. P., T. B. Sackton and A. G. Clark, 2006. Genetic variation in Drosophila melanogaster resistance to infection: a comparison across bacteria. Genetics 174 1539–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc, V., and J. M. Reichhart, 2004. The immune response of Drosophila melanogaster. Immunol. Rev. 198 59–71. [DOI] [PubMed] [Google Scholar]
- Long, A. D., R. F. Lyman, C. H. Langley and T. F. Mackay, 1998. Two sites in the Delta region contribute to naturally occurring variation in bristle number in Drosophila melanogaster. Genetics 149 999–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov, R., and C. A. Janeway, Jr., 1997. Innate immunity: impact on the adaptive immune response. Curr. Opin. Immunol. 9 4–9. [DOI] [PubMed] [Google Scholar]
- McKean, K. A., and L. Nunney, 2001. Increased sexual activity reduces male immune function in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 98 7904–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKean, K. A., and L. Nunney, 2005. Bateman's principle and immunity: phenotypically plastic reproductive strategies predict changes in immunological sex differences. Evolution 59 1510–1517. [PubMed] [Google Scholar]
- Parisi, M., R. Nuttall, D. Naiman, G. Bouffard, J. Malley et al., 2003. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 299 697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team, 2007. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. (http://www.R-project.org).
- Ramos-Onsins, S., and M. Aguadé, 1998. Molecular evolution of the Cecropin family in Drosophila: functional genes vs. pseudogenes. Genetics 150 157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, W. R., 1984. Sex chromosomes and the evolution of sexual dimorphism. Evolution 35 735–742. [DOI] [PubMed] [Google Scholar]
- Rozas, J., and R. Rozas, 1995. DnaSP, DNA sequence polymorphism: an interactive program for estimating population genetics parameters from DNA sequence data. Comput. Appl. Biosci. 11 621–625. [DOI] [PubMed] [Google Scholar]
- Sackton, T. B., B. P. Lazzaro, T. A. Schlenke, J. D. Evans, D. Hultmark et al., 2007. Dynamic evolution of the innate immune system in Drosophila. Nat. Genet. 39 1461–1468. [DOI] [PubMed] [Google Scholar]
- Scheet, P., and M. Stephens, 2006. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenke, T. A., and D. J. Begun, 2003. Natural selection drives Drosophila immune system evolution. Genetics 164 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, N. D., A. M. Larracuente and A. G. Clark, 2008. Contrasting the efficacy of selection on the X and autosomes in Drosophila. Mol. Biol. Evol. 25 454–467. [DOI] [PubMed] [Google Scholar]
- Storey, J. D., 2002. A direct approach to false discovery rates. J. R. Stat. Soc. Ser. B 64 479–498. [Google Scholar]
- Stronach, B., and N. Perrimon, 2002. Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev. 16 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella, A. J., A. Blanco-Garcia, S. Hutter and J. Rozas, 2005. VariScan: analysis of evolutionary patterns from large-scale DNA sequence polymorphism data. Bioinformatics 21 2791–2793. [DOI] [PubMed] [Google Scholar]
- Wassarman, D. A., M. Therrien and G. M. Rubin, 1995. The Ras signaling pathway in Drosophila. Curr. Opin. Genet. Dev. 5 44–50. [DOI] [PubMed] [Google Scholar]