

Abstract
Codon usage bias (CUB) has been documented across a wide range of taxa and is the subject of numerous studies. While most explanations of CUB invoke some type of natural selection, most measures of CUB adaptation are heuristically defined. In contrast, we present a novel and mechanistic method for defining and contextualizing CUB adaptation to reduce the cost of nonsense errors during protein translation. Using a model of protein translation, we develop a general approach for measuring the protein production cost in the face of nonsense errors of a given allele as well as the mean and variance of these costs across its coding synonyms. We then use these results to define the nonsense error adaptation index (NAI) of the allele or a contiguous subset thereof. Conceptually, the NAI value of an allele is a relative measure of its elevation on a specific and well-defined adaptive landscape. To illustrate its utility, we calculate NAI values for the entire coding sequence and across a set of nonoverlapping windows for each gene in the Saccharomyces cerevisiae S288c genome. Our results provide clear evidence of adaptation to reduce the cost of nonsense errors and increasing adaptation with codon position and expression. The magnitude and nature of this adaptation are also largely consistent with simulation results in which nonsense errors are the only selective force driving CUB evolution. Because NAI is derived from mechanistic models, it is both easier to interpret and more amenable to future refinement than other commonly used measures of codon bias. Further, our approach can also be used as a starting point for developing other mechanistically derived measures of adaptation such as for translational accuracy.
CODON usage bias (CUB) is defined as the nonuniform use of synonymous codons within a gene (Ikemura 1981; Bennetzen and Hall 1982; Sharp and Li 1987). CUB has been extensively documented across a wide range of organisms and varies greatly both within and between genomes (Grantham et al. 1980; Ikemura 1981, 1982, 1985; Bennetzen and Hall 1982; Sharp and Li 1987; Ghosh et al. 2000; Carbone et al. 2003; Mougel et al. 2004; Subramanian 2008). Most explanations of CUB involve a mixture of factors including mutational bias, intron splicing, recombination, gene conversion, DNA packaging, and selection for increased translational efficiency or accuracy (Bernardi and Bernardi 1986; Bulmer 1988, 1991; Shields et al. 1988; Kliman and Hey 1993, 1994; Akashi 1994, 2003; Xia 1996, 1998; Akashi and Eyre-Walker 1998; Musto et al. 1999, 2003; McVean and Charlesworth 1999; Ghosh et al. 2000; Wagner 2000; Birdsell 2002; Comeron and Kreitman 2002; Elf et al. 2003; Chen et al. 2004; Chamary and Hurst 2005a,b; Comeron 2006; Lin et al. 2006; Warnecke and Hurst 2007; Drummond and Wilke 2008; Warnecke et al. 2008). As a result, CUB has played an important role in the neutralist–selectionist debate (e.g., Wolfe and Sharp 1993; Duret and Mouchiroud 1999; Musto et al. 2001; Urrutia and Hurst 2003; Plotkin et al. 2004; Sémon et al. 2005; Chamary et al. 2006; Lynch 2007), interpretations of molecular clocks (e.g., Long and Gillespie 1991; Tamura et al. 2004; Xia 2009), and phylogenetics (e.g., Goldman and Yang 1994; Mooers and Holmes 2000; Nielsen et al. 2007a,b; Anisimova and Kosiol 2009).
Currently, multiple indexes are available for measuring the average CUB of a gene [e.g., Fop, codon bias index (CBI), relative synonymous codon usage (RCSU), codon adaptation index (CAI), Nc, E(g), CodonO, and relative codon bias (RCB) (Ikemura 1981; Bennetzen and Hall 1982; Sharp et al. 1986; Sharp and Li 1987; Wright 1990; Karlin and Mrazek 2000; Wan et al. 2006; Roymondal et al. 2009)]. However, directly relating any of these measures to a specific biological process is difficult. As a result, while certain measures of CUB are more popular in some circles than in others, there is no clear “correct” measure of codon bias. These shortcomings are due, in part, to the fact that these indexes are either heuristic or statistical in origin.
An alternative approach that avoids these shortcomings is to develop mechanistically based indexes that are based on specific biological processes that can drive the evolution of CUB. As an example of such an approach, we present a new CUB index specifically formulated to measure the degree of adaptation an allele exhibits to reduce the cost of nonsense errors during protein translation. We refer to this index as the nonsense error adaptation index (NAI) value of an allele. While NAI is specifically formulated to measure adaptation to reduce the cost of nonsense errors, the approach we present could be altered to measure adaptation in CUB for other aspects like translational efficiency, translational accuracy, or, ideally, a composite measure that evaluates and partitions the importance of these and other selective forces.
Nonsense errors, also referred to as processivity errors, occur when a ribosome terminates the translation of an mRNA transcript prematurely. Nonsense errors have a number of different causes such as ribosome drop-off, improper translation of release factors, frameshifts, and even missense errors (Kurland 1992; Hooper and Berg 2000; Zaher and Green 2009). Direct estimates of the per codon nonsense error rates are rare, but those that do exist for Escherichia coli suggest that they are on the order of 10−4 per codon (Manley 1978; Tsung et al. 1989; Jorgensen and Kurland 1990). For Saccharomyces cerevisiae, no direct estimates of nonsense error rates exist. However, Arava et al. (2003, 2005) provide indirect measures of nonsense error rates for S. cerevisiae that are on the order of 10−3–10−4 per codon. Although there is still great uncertainty, together these data imply that the probability of a nonsense error occurring during the translation of an average length protein of ∼400 amino acids is >20%.
Because most incomplete peptides are expected to be nonfunctional, such peptides impose various costs to the cell. For example, the production of incomplete peptides can tie up essential cell resources such as tRNAs and ribosomes (Dincbas et al. 1999; Cruz-Vera et al. 2004). Further, the recognition and breakdown of incomplete peptides may require additional resources as well and the peptides themselves may be toxic to the cell (Menninger 1978). Another important cost of a nonsense error is the amount of energy invested during protein production into the assembly of the polypeptide chain (Bulmer 1991; Kurland 1992; Eyre-Walker 1996). Because the cell must expend energy during each elongation step of translation, the cost of a nonsense error will increase with codon position at which it occurs. As a result, selection against nonsense errors leads to the unique prediction that CUB should increase intragenically with codon position. Numerous researchers have shown either directly or indirectly that CUB does indeed increase with codon position in E. coli and other microorganisms (Hooper and Berg 2000; Qin et al. 2004; Gilchrist and Wagner 2006; Stoletzki and Eyre-Walker 2007).
In this study we specifically define adaptation as the degree to which an allele reduces the cost of nonsense errors during protein translation. Variation in adaptation between alleles is the result of different synonymous codons having different nonsense error rates. We evaluate an allele's nonsense error cost relative to the coding synonyms of its synonymous genotype space, i.e., the set of alleles that differ in the synonymous codons they use but not the amino acid sequence they encode. It is worth noting that other measures of CUB also restrict their focus to that of coding synonyms. Because we restrict our focus to only synonymous changes in a coding sequence, we avoid the more complex question of how nonsynonymous substitutions affect protein function. Instead, we are able to focus solely on the effect synonymous substitutions have on the expected cost of nonsense errors during protein translation.
To define our NAI we use a simple model of protein elongation to generate a genotype-to-phenotype mapping function. In our mapping function, a genotype is the specific codon usage of an allele and a phenotype is the expected amount of high energy phosphate bonds ∼P that must be broken to generate the benefit equivalent of one functional protein (i.e., a protein production cost–benefit ratio or production cost, for brevity). We then contextualize the production cost of a given allele as a Z-score, using the analytically derived measures of the mean and variance of these costs for its entire set of coding synonyms. The resulting NAI score of an allele is a relative measure of its elevation on a nonsense error cost adaptive landscape for the protein it encodes. More precisely, the NAI score of an allele measures the relative deviation of an allele's nonsense error cost from its expected value scaled by the standard deviation of these costs. Because NAI is well defined from both a statistical and a biological standpoint, it has a number of important advantages over other measures of CUB.
For example, because it is based on a Z-score, an allele's NAI score is easy to interpret statistically since it can be directly related to the cumulative distribution function (CDF) of a standard normal distribution. As a result, an allele with an NAI score of 0 indicates that it is more adapted to reduce the cost of nonsense errors than half of all of the other alleles in its synonymous genotype space. The fact that the CDF of a standard normal distribution is 95% at 1.645 means that an allele with an NAI score of 1.645 is more adapted than 95% of its coding synonyms and, therefore, could be classified as showing statistically significant signs of adaptation. None of the other commonly used CUB indexes have such a clear statistical interpretation. Further, because the null expectation of NAI is a standard normal distribution, NAI measurements meet the assumptions of most standard statistical approaches such as general linear regression. Taking advantage of this property through the use of a hierarchical regression model, we are able to detect significant signals of adaptation to reduce the cost of nonsense errors across most of the S. cerevisiae genome. Specifically, we find that NAI increases with both codon position and an allele's protein production rate. Using permutation techniques and a simulation model of codon evolution, we show that these observations are consistent with the hypothesis that selection to reduce the cost of nonsense errors plays an important role in driving the evolution of CUB.
MATERIALS AND METHODS
The calculation of an allele's NAI score can be broken down into four steps. The first step is the calculation of the codon-specific elongation and nonsense error probabilities. The second step is to use these probabilities to calculate the nonsense error cost–benefit ratio, η, for the codon sequence of any given allele of a gene. The third step is to calculate the mean and variance in η-values across the synonymous genotype space of that gene. The fourth and final step is to combine a given allele's η-value and the moments of η across its synonymous genotype space to calculate the NAI score for that allele. Because these calculations are based on explicit biological processes, any one of them can be expanded upon or refined in future studies.
Step I. Calculating the per codon elongation and nonsense error probabilities:
Here we use a simple model we developed previously in Gilchrist and Wagner (2006) to calculate codon-specific elongation probabilities. In this model, each elongation step is viewed as an exponential waiting process with two possible outcomes: successful elongation or the occurrence of a nonsense error. Conceptually, we assume that abundances vary between tRNA species and, following the law of mass action, this variation in tRNA abundances leads to variation in elongation rates between codons. We also assume that other factors such as codon wobble can affect the elongation rate of a codon as well. We represent the elongation rate of a particular codon as c(NNN), where N ∈ {A, T, G, C }. While elongation rates can vary between codons, conversely we assume that all codons experience the same universal nonsense error rate b. Given these assumptions, the probability a ribosome will successfully complete an elongation step at some codon NNN is
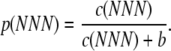 |
(1) |
(See Table 1 for symbols used in this study.) Consequently, the probability a ribosome will experience a nonsense error at the same codon is 1 – p(NNN). For simplicity, we also assume that stop codons always lead to termination of translation; i.e., p(TAA) = p(TAG) = p(TGA) = 0. We emphasize that there is ample room for the development of more complex and biologically accurate relationships between a codon NNN and its elongation probability p(NNN) as defined in Equation 1. However, such elaborations are beyond the scope of this study. As long as the calculation of p(NNN) at each codon is independent of the other codons, any refinement of the model underlying the calculation of p(NNN) will not alter how these values are used in the calculations that follow.
TABLE 1.
List of symbols used in this study
| Symbol | Definition |
|---|---|
| c(NNN) | Elongation rate of codon NNN |
| b | Background nonsense error rate |
| p(NNN) | Elongation probability of codon NNN |
 |
Codon sequence of an allele |
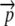 |
Vector of elongation probabilities for codon sequence 
|
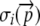 |
Probability a ribosome will translate up to and including the ith codon of the sequence 
|
| βi | Amount of energy expended by the ribosome in translating up to and including the ith codon |
| a1, a2 | Energetic cost of translation initiation and elongation, respectively |
| J | Set of synonymous codons of a given amino acid |
| E(p) | Expected elongation probability of a given amino acid |
| φ | Target production rate of a given protein |
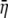 |
Untransformed expected cost–benefit ratio for the coding synonyms of an allele |
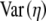 |
Untransformed variance in cost–benefit ratio for the coding synonyms of an allele |
| α, β | Shape and inverse scale parameters of the Gamma distribution, respectively |
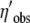 |
Transformed cost–benefit ratio of the observed allele |
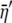 |
Transformed expected cost–benefit ratio for the coding synonyms of an allele |
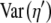 |
Transformed variance in cost–benefit ratio for the coding synonyms of an allele |
| Ai | Intercept of regression of NAI with position |
| Bi | Slope of regression of NAI with position |
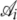 |
Coefficient of hierarchical regression describing how the intercept Ai changes with with ln(φ) |
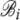 |
Coefficient of hierarchical regression describing how the slope Bi changes with with ln(φ) |
 |
The set of alleles that make up the synonymous genotype space of a gene |
Step II. Calculating the cost of nonsense errors η:
In general, we expect natural selection to favor alleles that produce protein functionality more efficiently than others. Therefore, we define adaptation as the reduction in the expected cost of producing the equivalent of one functional protein, η. More specifically, η describes a cell's expected cost in high energy phosphate bonds ∼P for translating an allele divided by the expected benefit the cell gains from the translation product. The use of a cost–benefit ratio as opposed to the difference between the cost and the benefit of an allele is well justified since the units of cost are different from the units of benefit. More importantly, if we assume that an organism requires a certain amount of protein to be produced at some target rate, the cost–benefit ratio can be used to calculate the expected cost, in ∼P's, for meeting that target. Both Gilchrist (2007) and the simulations we use here provide a clear illustration of this concept.
We explicitly measure peptide utility in relative terms such that one unit of relative utility is equal to the functionality provided by a complete and error-free peptide encoded by a given gene. Measuring the utility of a peptide in this way allows us to focus on how translational errors affect the expected performance of a protein relative to an error-free version as opposed to having to understand the specific function of the encoded protein. Thus, even though the importance of a protein to the organism varies between different genes, because we consider only relative, not absolute, utility, the NAI measure we produce is independent of that importance.
When considering an entire coding sequence, we use subscripts to indicate the position of a codon relative to its start codon. By definition, the start codon is at position 0. Given the fact that the first amino acid of a sequence is part of the ribosome initiation complex, a ribosome cannot experience a nonsense error at position 0. Thus we represent a codon sequence 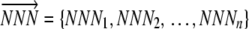 , where n is the number of elongation steps required to make a peptide. Because the start codon is at position 0, n is one less than the number of amino acids in a complete peptide. We use the notation
, where n is the number of elongation steps required to make a peptide. Because the start codon is at position 0, n is one less than the number of amino acids in a complete peptide. We use the notation 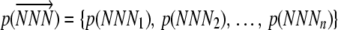 to represent the allele-specific vector of elongation probabilities for a given codon sequence. To make the notation more compact we drop the codon itself from our notation and, instead, simply index elongation probabilities by their position within a sequence, i.e., pi = p(NNNi), and, in a similar vein, leave the codon sequence itself implicit; i.e.,
to represent the allele-specific vector of elongation probabilities for a given codon sequence. To make the notation more compact we drop the codon itself from our notation and, instead, simply index elongation probabilities by their position within a sequence, i.e., pi = p(NNNi), and, in a similar vein, leave the codon sequence itself implicit; i.e., 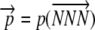 .
.
Using this notation, we now calculate the expected energetic cost per translational initiation event for a given codon sequence based on its corresponding elongation probability vector,  . We begin by noting that to reach the ith codon, a ribosome must first successfully translate the preceding i – 1 codons. Using σi to represent the probability that a ribosome will successfully translate the first i codons of an allele, it follows that
. We begin by noting that to reach the ith codon, a ribosome must first successfully translate the preceding i – 1 codons. Using σi to represent the probability that a ribosome will successfully translate the first i codons of an allele, it follows that
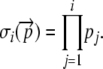 |
(2) |
Successful translation occurs when a ribosome translates all n codons of an allele. The probability of successful translation can therefore be denoted 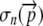 . The probability that a nonsense error will occur somewhere between codon 0 and n is simply
. The probability that a nonsense error will occur somewhere between codon 0 and n is simply 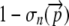 .
.
If a nonsense error occurs at the ith codon, then translation by the ribosome terminates and the amount of energy that has been expended up to this point equals βi. The simplest scenario and the one we employ here is to define βi = a1 + a2(i – 1), where a1 represents the cost of charging the fMet-tRNA (2 ∼ P) and the assembly of the ribosome on the mRNA (2 ∼ P) and a2 represents the cost of tRNA charging (2 ∼ P) and translocation of the ribosome during each elongation step (2 ∼ P) (Bulmer 1991; Wagner 2005). As with calculating p(NNN), more complex cost functions that include additional costs, such as the overhead cost of ribosome usage, could also be used to define βi (see discussion).
To calculate the expected protein production cost per initiation event, 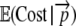 , one simply sums up the cost of each possible outcome weighted by its probability of occurring. Doing so gives
, one simply sums up the cost of each possible outcome weighted by its probability of occurring. Doing so gives
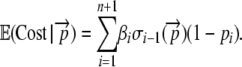 |
(3) |
Note that the summation is taken up to n + 1 to account for the stop codon where, by definition, (1 – pn+1) = 1.
To calculate the expected protein utility per initiation event, we first define the function ui as the utility of a peptide for which translation has terminated at codon i. We then calculate the expected utility of a gene,  , by simply summing up the utility of each peptide given its length weighted by the probability of producing it. Doing so gives
, by simply summing up the utility of each peptide given its length weighted by the probability of producing it. Doing so gives
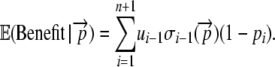 |
(4) |
Here and in our previous work we assume that ui follows a step function where ui = 0 for all i < j; i.e., all nonsense errors prior to codon j lead to a nonfunctional protein. In the case of such a step function it follows that 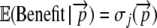 and in the current study we assume that j = n. As with other aspects of this work, our assumptions about ui can easily be relaxed in future studies. For example, ui could be assumed to be a logistic function of i. Alternatively, one could expand the formulation of
and in the current study we assume that j = n. As with other aspects of this work, our assumptions about ui can easily be relaxed in future studies. For example, ui could be assumed to be a logistic function of i. Alternatively, one could expand the formulation of  further to include the possibility of missense errors and their effects.
further to include the possibility of missense errors and their effects.
Combining our results from Equations 3 and 4, the expected cost over expected benefit of a coding sequence is
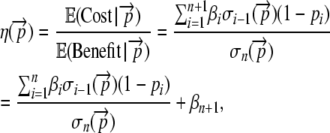 |
(5) |
where the first term on the right-hand side of Equation 5 represents the cost–benefit of the incomplete proteins and the βn+1 term represents the cost–benefit of translating one complete protein (Gilchrist 2007). In summary, the term 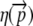 represents the expected amount of ∼P that must be spent to get the benefit of one unit of utility from an allele with a given codon sequence.
represents the expected amount of ∼P that must be spent to get the benefit of one unit of utility from an allele with a given codon sequence.
Step III. Calculating the central moments of η:
We now shift our focus from calculating the protein production cost η for a specific allele to calculating the central moments of η across the entire set of coding synonyms. For simplicity we focus on calculating these moments for the entire length of an allele. In supporting information, File S1, A, we present the details on carrying out similar calculations for the set of coding synonyms that differ from the observed allele only over a restricted window of codons, e.g., from codons 1–20, 21–40, 41–60, and so on. These calculations of NAI over a window explicitly take into account the codon usage outside of the window, thus providing a context-specific measure of adaptation to reduce the cost of nonsense errors within the window. These moments can also be estimated through simulation and we have exploited this fact to verify that our analytic estimates of η's central moments are correct.
Calculating the expected cost–benefit ratio 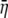 :
:
We begin by computing the expected cost–benefit ratio 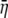 for the coding synonyms of an allele. Beginning with the definition of η in Equation 5 and assuming that the choice of codon at each position is independent of the other, we can calculate the expected value of η for the entire set of coding synonyms
for the coding synonyms of an allele. Beginning with the definition of η in Equation 5 and assuming that the choice of codon at each position is independent of the other, we can calculate the expected value of η for the entire set of coding synonyms 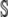 as
as
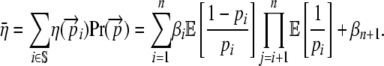 |
(6) |
The expectations over functions of pi, such as 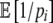 , are taken over the set of synonymous codons
, are taken over the set of synonymous codons 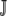 for a given amino acid. Using the amino acid tyrosine (Y) as an example,
for a given amino acid. Using the amino acid tyrosine (Y) as an example, 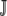 Y = {TAT, TAC }. Similarly, for proline (P),
Y = {TAT, TAC }. Similarly, for proline (P), 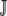 P = {CCT, CCC, CCA, CCG }. The set of synonymous codons for the amino acid serine is unique because they occur in two distinct subsets that cannot be connected via a single-nucleotide substitution. Thus, we treat each subset as a distinct amino acid; i.e.,
P = {CCT, CCC, CCA, CCG }. The set of synonymous codons for the amino acid serine is unique because they occur in two distinct subsets that cannot be connected via a single-nucleotide substitution. Thus, we treat each subset as a distinct amino acid; i.e., 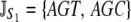 and
and 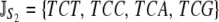 .
.
Generally speaking, the expected inverse elongation probability for an amino acid is 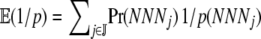 , where Pr(NNNj) is the probability of codon NNNj occurring in a given genome. In the absence of any mutational bias all codons have equal probability of occurring, Pr(NNNj) = 1/|
, where Pr(NNNj) is the probability of codon NNNj occurring in a given genome. In the absence of any mutational bias all codons have equal probability of occurring, Pr(NNNj) = 1/|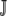 | for all j in
| for all j in 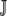 . If mutational bias does occur, then the Pr(NNNj) for each codon is simply the equilibrium frequency of each codon given the biased mutation rate. For example, to include the effect of a genomewide AT bias in our calculation of
. If mutational bias does occur, then the Pr(NNNj) for each codon is simply the equilibrium frequency of each codon given the biased mutation rate. For example, to include the effect of a genomewide AT bias in our calculation of 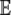 (p), we simply set
(p), we simply set
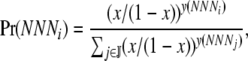 |
where x is the degree of observed AT bias and y(NNNi) is the number of A or T nucleotides in codon NNNi.
Calculating the variance in the cost–benefit ratio Var(η):
Given our assumption of independence between pi values at different positions, the variance in η-values across the synonymous genotype space 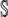 is
is
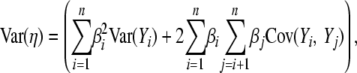 |
(7) |
where
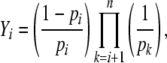 |
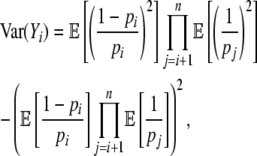 |
(8) |
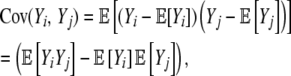 |
(9) |
and
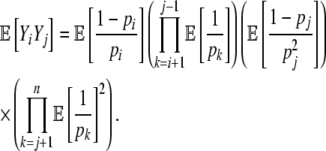 |
(10) |
Substituting the results from Equations 8–10 into Equation 7 allows for the direct calculation of the variance in the η-values around 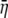 for all
for all 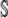 genotypes.
genotypes.
Step IV. Calculating the NAI score of a gene:
We define ηmin as the cost–benefit ratio of producing a protein that uses only the most optimal codons and hence represents the minimum cost of producing that protein. Random samples of synonyms for different genes indicate that, after subtracting off ηmin from each value, the distribution of η-values for the synonyms of an allele is approximately Gamma distributed (see File S1, B for details). Under the assumption of a Gamma distribution, we can solve for the shape and inverse scale parameters, α and β, respectively, on the basis of the 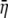 and
and 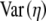 values calculated in step III. Doing so gives
values calculated in step III. Doing so gives 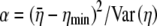 and
and 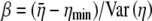 (Rice 1995).
(Rice 1995).
As mentioned earlier, the NAI of a gene is essentially a Z-score, which, in turn, is based on a standard normal distribution. For a given mean and variance, the Gamma distribution has greater skewness than the normal distribution. Because of this skewness, if we calculate a gene's NAI score using the raw, untransformed moments, 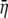 and
and 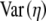 , we will underestimate the true proportion of genotype space with alleles that are less adaptive in their η-values. To reduce this effect, we apply a Box–Cox transformation with h =
, we will underestimate the true proportion of genotype space with alleles that are less adaptive in their η-values. To reduce this effect, we apply a Box–Cox transformation with h = 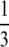 , which is the best skewness-reducing transformation for the Gamma distribution among the Box–Cox family (Pace and Salvan 1997) (see File S1, C for details). Using ′ to denote the Box–Cox-transformed values, we define the NAI score of a gene as
, which is the best skewness-reducing transformation for the Gamma distribution among the Box–Cox family (Pace and Salvan 1997) (see File S1, C for details). Using ′ to denote the Box–Cox-transformed values, we define the NAI score of a gene as
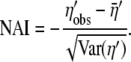 |
(11) |
The negative sign is included in the definition of the NAI score because while natural selection favors a reduction in η-values, adaptation is generally defined as being increased by natural selection. Thus the inclusion of the negative sign means that NAI is an increasing function of adaptation to reduce the cost of nonsense errors.
NAI calculations and simulations:
We have developed a computer program called NAI that can be used to calculate the NAI score of an allele as well as the NAI score for a series of moving windows within an allele. This program allows NAI to be calculated in two different ways, either using Equations 6–10 to calculate the exact moments analytically or through random sampling of synonymous genotype space weighted by a given AT bias. The sample population is then used to estimate the moments of η for the entire set of coding synonyms. The results we present here were generated by calculating the moments analytically, but we have used the random sampling approach to verify our results.
We have also developed an additional stochastic, simulation program called codon evolution simulation (CES). CES simulates the evolution of a locus where the resident allele is allowed to evolve across its synonymous genotype space following the allele substitution model described in Sella and Hirsh (2005). The same substitution probabilities were also independently derived by Iwasa (1988) and Berg and Lässig (2003). For further details refer to File S1, D. Both NAI and CES are written in ANSI standard C and released under the GNU Public License 2.0. Both precompiled *nix binaries and source code are available at www.tiem.utk.edu\∼mikeg\Software.
Application to the S. cerevisiae genome:
To illustrate the utility and behavior of NAI, we applied it to the S. cerevisiae strain S288c genome based on the Saccharomyces Genome Database's June 6, 2008 release (Dolinski et al. 2008). Default parameter values for the simulation of the S. cerevisiae genome using both CES and NAI calculations are given in Table 2. We restricted our analysis of gene level NAI scores to the 4674 verified nuclear genes that lack internal stops and our analysis of how NAI changes with window position to the 4377 genes within that set with at least 100 codons. The per codon elongation rates were generated as in Gilchrist and Wagner (2006). These rates are proportional to the tRNA abundance and take into account a set of wobble penalties based on Curran and Yarus (1989). Exact values used are given in the Table S1. Since no reliable empirical measurements of the nonsense error rate in S. cerevisiae exist, on the basis of experiments with E. coli by Jorgensen and Kurland (1990) we used a nonsense error rate of b = 0.0012/sec. Given an average per codon elongation rate of 10/sec, this value is consistent with indirect empirical estimates of b in S. cerevisiae by Arava et al. (2005). Because of the uncertainty in the model parameters and the fact that such parameters are largely unknown for most organisms, we evaluated the sensitivity of NAI to changes in the key parameters: the background nonsense error rate b, the cost of translation initiation a1, the cost of each elongation step a2, and the elongation probability for each codon pi after Hamby (1994).
TABLE 2.
List of default parameter values used for all NAI calculations and the CES simulation
| Parameter | Value |
|---|---|
| b | 0.0012/sec |
| a1 | 4 ∼ P |
| a2 | 4 ∼ P |
| q | 4.19 × 10−7 |
| Ne | 1.36 × 107 |
| μ | 10−9/generation |
| AT bias |
0.62 |
Because selection pressure against nonsense errors increases with codon position along a sequence, we also evaluated how NAI changes with codon position in both the S288c and the simulated genomes. Since we are more interested in general trends across the entire S. cerevisiae genome than in the behavior of specific genes, we used a hierarchical regression model approach when analyzing the data. In this approach, we began by calculating the NAI values for successive, nonoverlapping windows of 20 codons. We then fitted a linear regression model to these intragenic NAI values of an allele. The regression model allows us to estimate the initial degree of adaptation to reduce the cost of nonsense errors through the regression intercept Ai. The model also allows us to estimate how such adaptation changes with position through the regression slope Bi of a given allele i. That is,
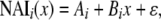 |
(12) |
where ɛ here and below represents a noise term, x is the codon position at the center of the window, i.e., x = {10, 30, 50, … , xmax – 10}, and xmax is the largest multiple of 20 less than or equal to the length n of the coding sequence.
To quantify the general behavior of the output generated by the regression model of Equation 12 across the S. cerevisiae genome, we then fitted second-order regression models to the intercept Ai and slope Bi values for each gene as a function of its log protein production rate, ln(φi). In other words, we fitted the following models
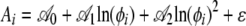 |
(13) |
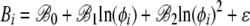 |
(14) |
to the set of maximum-likelihood estimates (MLEs) for the model parameters Ai and Bi in Equation 12 weighted by their standard error. Thus, the hierarchical analysis measures how NAI changes with the two dependent variables: codon position, x and log protein production rate, ln(φ). Although higher-order functions could be fitted to the MLE data, our goal is to capture the general behavior of the system as simply as possible. The gene-specific protein production rates φi were based on a combination of mRNA measurements from AffyMetrix data sets as presented in Beyer et al. (2004) and translation rates per mRNA based on ribosome occupancy data given in Arava et al. (2003) and Mackay et al. (2004). See Gilchrist (2007) for further details.
To better contextualize our results and control for the fact that other selective forces could be driving some or all of the adaptation we observe, we repeated the above analysis on multiple artificial data sets that were generated either by randomizing the codon order of each gene in the S. cerevisiae genome or by randomly assembled genes. In terms of randomly reordered data sets, two different types of data sets were generated: partial and complete reorderings. Partial reordering involved randomly reordering codons on a per amino acid basis such that the codon order of a gene was randomized but the amino acid order of the sequence encoded was not altered. In contrast, complete reordering involved the random reordering of codons independent of the amino acids encoded. While both partial and complete reordering change the codon order of an allele, neither approach alters the set of codons used. Since nonsense errors are the only selective force that is expected to result in increasing CUB with position, these reordered data sets serve as a control. We also generate a random population of alleles for each gene by randomly sampling the population of coding synonyms independent of their specific cost–benefit values η but weighted by an AT bias of 0.62. These random samples serve as controls for when there is no selection on CUB evolution.
To understand how the NAI behaves when CUB evolves solely under selection to reduce the cost of nonsense errors, we simulated the evolution of each gene using CES. The simulation was run assuming that the protein production rate of a gene was equal to its empirically estimated value φi multiplied by a log-normal random variable centered around 1 and with a standard deviation equal to the standard error of the log-transformed mRNA abundance values given in Beyer et al. (2004). The use of this additional noise factor mimics the uncertainty in the estimates of φi inherent in the analysis of the S288c and reordered genomes. The simulation was run for 20/μ generations (i.e., sufficiently long such that we expect 20 substitutions per nucleotide under a pure mutation–drift process) so that the simulation results should represent samples from the stationary distribution of allele fixation. We applied the same hierarchical regression analysis to our CES simulated genome as we used with the S. cerevisiae 288c genome.
RESULTS
General behavior of NAI:
We begin by analyzing the behavior of the NAI for entire coding sequences. First and as we expected, we find that the NAI scores for random samples of coding synonyms are normally distributed around a mean of 0 and a standard deviation of 1 (cf. Figure 1) (Shapiro-Wilk's test, P-value = 0.916). We also see that the distributions of S288c and simulated NAI scores across the entire genome behave in a similar and predictable manner, where genes with greater protein production rates have higher NAI scores (Figure 2). The S288c NAI scores for genes were also well correlated with the NAI scores in our CES-generated genome (ρ ∼ 0.74), which provides further evidence that selection against nonsense errors can explain much of the CUB observed in S. cerevisiae.
Figure 1.—

Distribution of NAI values for 1000 randomly assembled alleles (open bars) and 1000 CES simulated alleles (shaded bars) for the gene YBL068W. CES simulations were done using the empirically estimated protein production rate of φ = 0.21s−1. The dashed line at 4.015 indicates the NAI value for the YBL068W allele in S. cerevisiae S288c, which corresponds to the 99.997th percentile of a standard normal distribution, which is represented by the solid curve. Note that the mean of the NAI scores of the random population does not significantly differ from zero (t = 0.1917, P-value >0.8) while the mean of the NAI scores of the simulated population does (t = 172.2, P-value <10−15).
Figure 2.—

Correlation of NAI values with log protein production rates ln(φ) for the S288c and CES simulated genomes. ρ represents the correlation coefficient between the two variables.
Indeed, if there was no adaptation to reduce the cost of nonsense errors, the distribution of NAI scores across the S. cerevisiae genome would follow a standard normal distribution. Looking at the S. cerevisiae genome, we find that the distribution of these NAI values does not mimic the standard normal distribution, but instead is highly skewed toward higher values (Figure 3). This distribution of NAI scores as summarized in Table 3 indicates that most genes show some degree of adaptation to reduce the cost of nonsense errors.
Figure 3.—

Distribution of NAI values of S288c and CES simulated genomes. Shaded bars represent the distribution of NAI scores across all genes in the S288c genome and hatched bars represent NAI scores of CES simulated genes. The solid line represents the standard normal distribution, which is the expected distribution of NAI scores in the absence of any selection.
TABLE 3.
Distribution of NAI scores and their corresponding percentile in a standard normal distribution
| % genome above NAI score |
|||
|---|---|---|---|
| NAI score | % CDF, standard normal | S. cerevisiae | CES simulation |
| 0 | 50 | 92.14 | 85.95 |
| 1.282 | 90 | 74.59 | 61.57 |
| 1.645 | 95 | 67.92 | 55.54 |
| 2.326 | 99 | 54.63 | 45.67 |
| 2.576 | 99.5 | 50.42 | 42.45 |
| 3.090 | 99.9 | 43.27 | 37.38 |
| 3.291 | 99.95 | 39.84 | 35.43 |
| 3.719 |
99.99 |
33.79 |
32.30 |
For example, we find that 92.1% of the S. cerevisiae genes have NAI values >0 . Under a pure mutation–drift process only 50% of the genes would be expected to have NAI scores >0. In fact, ∼68% of S. cerevisiae's genes have NAI scores >1.645. Again, under a pure mutation–drift process we would expect to only see 5% of the genes with NAI scores in this range. These results clearly demonstrate that most genes are significantly more adapted to reducing their cost–benefit ratio η than their coding synonyms. In fact, over half of all S. cerevisiae genes have an NAI score >2.326, indicating that they are more adapted than 99% of their coding synonyms. More striking, a full 33% are more adapted than 99.99% of their coding synonyms (NAI > 3.719). We observed similar levels of adaptation in our simulated sequences and less adaptation in our reordered data sets (Table 3). For example, in a typical simulated data set we found that 86% had NAI values >0 and 55.5% had NAI values >1.645.
The sensitivity of NAI to changes in parameter values is presented in File S1, E, Figure S3 and Table S3. 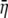 , Var(η), and NAI values for each gene are presented in Table S4, Table S5, Table S6, and Table S7, E. To summarize, NAI scores are relatively insensitive to changes in almost all of the parameters underlying its formulation. This insensitivity is especially strong for the initiation cost, the elongation cost, and the nonsense error rate, a1, a2, and b, respectively.
, Var(η), and NAI values for each gene are presented in Table S4, Table S5, Table S6, and Table S7, E. To summarize, NAI scores are relatively insensitive to changes in almost all of the parameters underlying its formulation. This insensitivity is especially strong for the initiation cost, the elongation cost, and the nonsense error rate, a1, a2, and b, respectively.
Changes in NAI with codon position:
One specific prediction about CUB driven by the cost of nonsense errors is that the strength of selection and, therefore, the degree of adaptation should increase with codon position.
To test this prediction, we calculated the NAI value for each successive, nonoverlapping window of 20 codons for each gene and then fit the first-order regression model in Equation 12. We then fit the weighted second-order regression model in Equations 13 and 14 to the maximum-likelihood estimates of the Ai and Bi parameters in Equation 12. The overall behavior of how NAI changes with codon position and log protein production rate ln(φ) is summarized by the curves of the hierarchical model. Specifically, the parameters given in Table 4 and plotted in Figure 4 describe how the intercept Ai and slope Bi of the NAI vs. position regression model in Equation 12 change with ln(φ). In general, all hierarchical model parameters differed significantly from zero. We also find that the initial NAI value Ai and the rate at which NAI increases with codon position Bi both increase in an accelerating manner with ln(φ).
TABLE 4.
Hierarchical regression analysis results
| MLE | SE | t | P-value | |
|---|---|---|---|---|
| S288c genome: parameters for regression intercept A | ||||
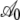 |
2.102 | 0.029 | 71.97 | <2 × 10−16 |
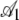 |
0.793 | 0.016 | 50.12 | <2 × 10−16 |
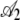 |
0.084 | 2.3 × 10−3 | 36.68 | <2 × 10−16 |
| S288c genome: parameters for regression slope B | ||||
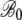 |
2.72 × 10−2 | 1.635 × 10−3 | 16.63 | <2 × 10−16 |
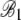 |
8.74 × 10−3 | 7.92 × 10−4 | 11.03 | <2 × 10−16 |
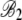 |
7.94 × 10−4 | 9.6 × 10−5 | 8.27 | <2 × 10−16 |
| CES simulated genome: parameters for regression intercept A | ||||
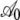 |
1.021 | 0.025 | 40.48 | <2 × 10−16 |
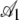 |
0.447 | 0.0145 | 30.89 | <2 × 10−16 |
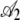 |
0.047 | 2.05 × 10−3 | 22.94 | <2 × 10−16 |
| CES simulated genome: parameters for regression slope B | ||||
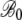 |
0.135 | 1.82 × 10−3 | 74.5 | <2 × 10−16 |
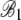 |
4.48 × 10−2 | 8.97 × 10−4 | 49.96 | <2 × 10−16 |
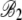 |
3.74 × 10−3 |
1.08 × 10−4 |
34.66 |
<2 × 10−16 |
Figure 4.—


Hierarchical analysis results. Each data point represents the maximum-likelihood estimates of the regression parameters of NAI vs. position (Equation 12) for each individual gene. Curves represent the weighted second-order hierarchical regression curves of A and B given in Equations 13 and 14. Weighting is based on the standard error of each data point.
Quantitatively, looking at the estimates of 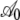 in Table 4 we see that the S. cerevisiae S288c genome shows greater adaptation or higher NAI values at the start of low expression genes than the genome simulated using CES. However, inspection of
in Table 4 we see that the S. cerevisiae S288c genome shows greater adaptation or higher NAI values at the start of low expression genes than the genome simulated using CES. However, inspection of 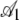 and
and 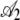 values indicates that for both the observed and the simulated sequences, the NAI values at the beginning of a gene increase with ln(φ) in a similar manner. Given the fact that the 288c alleles tend to have greater than expected NAI values, it is then perhaps not surprising that the rate at which NAI increases with position is lower in the S288c sequences when compared to the simulated sequences. This is illustrated by the fact that
values indicates that for both the observed and the simulated sequences, the NAI values at the beginning of a gene increase with ln(φ) in a similar manner. Given the fact that the 288c alleles tend to have greater than expected NAI values, it is then perhaps not surprising that the rate at which NAI increases with position is lower in the S288c sequences when compared to the simulated sequences. This is illustrated by the fact that 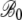 ,
, 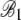 , and
, and 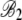 are lower in the observed sequences than in the genome simulated using CES. Although upon first glance the magnitude of the regression slopes for the NAI vs. position Bi may appear slight, these values are substantial as the slope increases in units of standard deviation and the alleles generally encode hundreds of codons. Figure 5 allows us to see these differences in behavior between the S288c and simulated and data sets for six genes from across a wide range of protein production rates.
are lower in the observed sequences than in the genome simulated using CES. Although upon first glance the magnitude of the regression slopes for the NAI vs. position Bi may appear slight, these values are substantial as the slope increases in units of standard deviation and the alleles generally encode hundreds of codons. Figure 5 allows us to see these differences in behavior between the S288c and simulated and data sets for six genes from across a wide range of protein production rates.
Figure 5.—

Change in NAI with position and protein production rate, φ. Shown is how NAI changes with position for six genes chosen from a wide range of empirically estimated protein production rates, φ. Solid circles (•) represent NAI values for the S288c coding sequence of the gene while open circles (○) represent NAI values for a simulated coding sequence. The regression lines through the NAI values are given by – – – for the S288c sequence and by · · · for the simulated sequence.
Stepping back, we note that qualitatively the distributions of Ai and Bi values around the hierarchical regression curves are quite noisy in both observed and simulated data sets (Figure 4). This results from the fact that while the selective forces against nonsense errors are consistent and increase with the protein production rate of a gene, mutation and drift clearly play important roles in the evolution of CUB.
To test our rather strong assumption that only completely translated proteins have any functionality, we repeated the hierarchical analyses where the final 10 or 20 amino acids were excluded from our NAI. This is roughly equivalent to assuming a (0, 1) functionality threshold at n – 10 or n – 20. The results of these analyses showed no statistically significant change in the parameter estimates of our hierarchical model (Table S2).
Given the complexity of our NAI calculations, it is possible that the observed changes in NAI values with codon position and log protein production rate, ln(φ), in the S. cerevisiae genome are actually an artifact of selection for translational accuracy or translational efficiency irrespective of nonsense errors. If this were the actual case, then we would expect to see no difference between how NAI changes with position in the observed and reordered genomes. Instead we find that 66% of the genes in the S. cerevisiae genome have slope parameters Bi > 0, a frequency that is significantly greater than what was observed in each set of partially reordered, completely reordered, and randomly assembled genomes that are all distributed around 0.5 (see Table 5). The difference in behavior between the observed and the reordered genomes can also be observed in the hierarchical regression curves, a typical example of which is illustrated in Figure S2. Note that both partially and completely reordered genomes do show a very slight upward bias in their individual regression slopes Bi and this bias is unexpected, but increases with protein production rate. The cause of this bias and its relationship to ln(φ) is not understood at this point. For example, it may be caused by subtle compositional effects or the fact that the true distribution of η-values calculated over a window is not actually Gamma distributed as we assume. Whatever the cause, the effect is small when compared with the fraction of genes with positive regression slopes Bi and the magnitude of the parameters of our hierarchical model.
TABLE 5.
Frequency of individual regression slopes Bi > 0
| % mean frequency |
vs. S288c genome |
vs. CES simulation |
||||
|---|---|---|---|---|---|---|
| SD | t-statistic | P-value | t-statistic | P-value | ||
| Random | 49.9 | 0.0086 | −604.630 | <2 × 10−16 | −1021.83 | <2 × 10−16 |
| Partially reordered | 51.6 | 0.0083 | −560.143 | <2 × 10−16 | −990.58 | <2 × 10−16 |
| Completely reordered |
50.2 |
0.0087 |
−586.051 |
<2 × 10−16 |
−996.92 |
<2 × 10−16 |
Shown is the frequency of genes with positive regression slopes based on a sample of 1000 genomes for each category as well as a comparison of these populations to the frequency for the S288c genome (77%) and the CES simulation genome (66%).
DISCUSSION
In this study we developed a method for quantifying the adaptation an allele displays to reduce the cost of nonsense errors during protein translation η relative to its set of alternative coding synonyms. The approach presented here is a generalization of the definition of η derived in Gilchrist (2007) and provides analytic expressions for the mean and variance of η-values in a given synonymous genotype space. These values are used to contextualize the η-value for any given point in synonymous genotype space into a single NAI value. To our knowledge, our work is the first to provide a general means of surveying a biologically meaningful adaptive landscape and then use these results to contextualize adaptation at the molecular level.
NAI scores are based on a mechanistic model of protein translation and functionality. As a result, the nature of adaption of NAI measures is clearly defined. In addition, because of its mechanistic derivation, our approach is flexible enough to allow for future refinement or expansion of the NAI as our understanding of the processes involved in protein translation and other processes progresses. This flexibility is best illustrated in the seamless manner with which we are able to incorporate mutation bias into the calculation of the NAI. The fact that we can easily incorporate codon-specific nonsense error rates into our model by simply using codon-specific b values in Equation 1 also illustrates this point. Another example of this flexibility is in the cost of function β(i) used in Equation 3.
In our current formulation β(i) includes only the direct assembly cost of a protein. However, β(i) could be expanded to include other costs as well. For example, given our assumption about exponential waiting times during each elongation step, the combined assembly and ribosome overhead costs at a given codon i could be calculated as 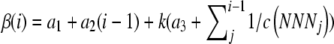 , where k is a scaling constant representing the per second overhead cost of the ribosome cost in ∼P's and a3 is the expected time it takes for a ribosome to intercept an mRNA and initiate translation. Expanding our cost function β(i) in this way highlights the potentially large effect nonsense errors can have on the overall translational efficiency of a ribosome, a point overlooked in most discussions of CUB.
, where k is a scaling constant representing the per second overhead cost of the ribosome cost in ∼P's and a3 is the expected time it takes for a ribosome to intercept an mRNA and initiate translation. Expanding our cost function β(i) in this way highlights the potentially large effect nonsense errors can have on the overall translational efficiency of a ribosome, a point overlooked in most discussions of CUB.
In a similar manner, the utility function ui could be defined by a continuous, sigmoidal function rather than the step function we use here. Doing this would allow calculations of η to include the contribution of incomplete, but partially functional peptides. More generally, ui could take on negative values, allowing it to describe any toxic or interference effects some short, incomplete peptide may have. A final example of how our approach can be extended can be found by examining one of the strongest assumptions we make, the lack of interactions between ribosomes on the same mRNA. Because the presence of a ribosome at one position can interfere with the behavior of another ribosome upstream from it, this assumption is clearly violated. Relaxing this assumption would make the computation of NAI substantially more challenging (e.g., see Chou 2003; Basu and Chowdhury 2007). While we believe that our current approach is a reasonable first approximation, other research suggests relaxing this assumption might be useful. For example, Bulmer (1991) showed this type of interribosomal interference can lead to additional selection for increased translational efficiency at the start of a coding sequence. This idea of interribosomal interference is consistent with our own observation of greater observed NAI values at the start of a sequence than expected on the basis of our simulation.
It is important to recognize that one of the most common uses of CUB is to predict relative gene expression levels from the coding sequence of a gene. While the primary purpose of NAI is not to predict gene expression, the concepts underlying it can be used for that purpose (Gilchrist 2007). Nevertheless, NAI values are well correlated with the most commonly used CUB indexes (ρ = 0.793–0.822, Figure S1).
Most CUB indexes are based on some sort of distance measure in synonymous genotype space. For example, consider Sharp and Li's (1987) CAI, which is probably the most commonly used measure of CUB. The CAI value of an allele is based on the geometric mean frequency of codons it uses relative to the usage within a subset of highly expressed genes. Conceptually, CAI is a multiplicative distance measurement of an observed allele from an allele whose CUB mirrors that of a subset of highly expressed genes. Another, more recent example can be found in Roymondal et al.'s (2009) RCB index. RCB measures the CUB of an allele relative to its expected position on the basis of mutation and drift alone. Similarly, the CUB indexes E(g) (Karlin and Mrazek 2000), Fop (Ikemura 1981), and Nc (Wright 1990) can be thought of as providing similar types of distance measures.
While the NAI is similar to these other indexes in that it is a measure of relative distance, the distance measurement is not in terms of positioning within an allele's synonymous genotype space, but is based on the relative altitude of an allele on an adaptive landscape. The fact that the NAI is based on the phenotypic adaptation of an allele rather than its position in genotype space makes it fundamentally different from these other measures. Unlike most other commonly used indexes, the NAI explicitly takes into account the effect of genomewide AT bias in shaping the codon usage of a gene. In addition, because the NAI is based on a Z-score, its easier to interpret and has desirable statistical properties that are absent in most CUB indexes.
Despite the fact that selection against codon usage bias is generally thought to be a rather weak selective force (Stoletzki and Eyre-Walker 2007; Hershberg and Petrov 2008), the NAI scores for the S. cerevisiae genome indicate that most (>92%) of its coding sequences are more adapted to reduce the cost of nonsense errors than expected. Indeed, >67% of the S. cerevisiae alleles are at a higher point on the adaptive landscape than 95% of their coding synonyms. Perhaps more striking is the finding that >33% of the S. cerevisiae alleles are found above the 99.99th percentile of the adaptive landscape. Thus, almost a third of all alleles can be found in the far upper reaches of the nonsense error adaptive landscape. In addition, we also observe that NAI values generally increase with codon position of a gene, a unique pattern expected to result only from selection against nonsense errors. Indeed, the increases we see are consistent with the increases observed in our simulations where nonsense errors are the sole selective force. Taken together, our results provide additional evidence that nonsense errors play an important role in CUB evolution of S. cerevisiae.
One shortcoming of our study is that we consider only one source of selection: nonsense errors. In reality, many other selective forces contribute to the evolution of CUB. Indeed, the emerging picture from the field clearly indicates that any synonymous change in the coding region of a gene is likely to be pleiotropic, possibly affecting mRNA folding, translational accuracy, translational efficiency, protein folding, etc. This suggests that there are many places for these sources of selection to either conflict with or reinforce one another. Even though some of the adaptation we observe is likely due to these other forces, the fact that we do not consider these other forces when calculating an NAI value does not invalidate its meaning. NAI is simply a measure of adaptation to the cost of nonsense errors and does not depend on the forces ultimately responsible for that adaptation. While the tendency of NAI to increase with codon position can currently be explained only by selection against nonsense errors, the exact degree to which the overall adaptation of an allele can be directly attributed to this selective force is still open to debate. One way of resolving this debate would be to expand the approach developed here to include other potential selective forces. Indeed, we hope that the approach we develop here will serve as a starting point for generating other measures of adaptation. Only when we have a combination of such measures will researchers be able to evaluate the importance of the different selective forces driving the evolution of CUB.
Acknowledgments
We thank Sergey Gavrilets, Michael Saum, and Hong Qin for providing helpful suggestions and comments on this manuscript. We also thank two anonymous reviewers for their constructive criticisms and suggestions that have greatly improved this manuscript.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.108209/DC1.
References
- Akashi, H., 1994. Synonymous codon usage in Drosophila melanogaster: natural-selection and translational accuracy. Genetics 136 927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., 2003. Translational selection and yeast proteome evolution. Genetics 164 1291–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., and A. Eyre-Walker, 1998. Translational selection and molecular evolution. Curr. Opin. Genet. Dev. 8 688–693. [DOI] [PubMed] [Google Scholar]
- Anisimova, M., and C. Kosiol, 2009. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol. Biol. Evol. 26 255–271. [DOI] [PubMed] [Google Scholar]
- Arava, Y., Y. L. Wang, J. D. Storey, C. L. Liu, P. O. Brown et al., 2003. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 100 3889–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arava, Y., F. E. Boas, P. O. Brown and D. Herschlag, 2005. Dissecting eukaryotic translation and its control by ribosome density mapping. Nucleic Acids Res. 33 2421–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu, A., and D. Chowdhury, 2007. Traffic of interacting ribosomes: effects of single-machine mechanochemistry on protein synthesis. Phys. Rev. E 75 021902-1–021902-11. [DOI] [PubMed] [Google Scholar]
- Bennetzen, J. L., and B. D. Hall, 1982. Codon selection in yeast. J. Biol. Chem. 257 3026–3031. [PubMed] [Google Scholar]
- Berg, J., and M. Lässig, 2003. Stochastic evolution and transcription factor binding sites. Biophysics 48 S36–S44. [Google Scholar]
- Bernardi, G., and G. Bernardi, 1986. Compositional constraints and genome evolution. J. Mol. Evol. 24 1–11. [DOI] [PubMed] [Google Scholar]
- Beyer, A., J. Hollunder, H.-P. Nasheuer and T. Wilhelm, 2004. Post-transcriptional expression regulation in the yeast Saccharomyces cerevisiae on a genomic scale. Mol. Cell. Proteomics 3 1083–1092. [DOI] [PubMed] [Google Scholar]
- Birdsell, J. A., 2002. Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol. Biol. Evol. 19 1181–1197. [DOI] [PubMed] [Google Scholar]
- Bulmer, M., 1988. Are codon usage patterns in unicellular organisms determined by selection-mutation balance. J. Evol. Biol. 1 15–26. [Google Scholar]
- Bulmer, M., 1991. The selection-mutation-drift theory of synonymous codon usage. Genetics 129 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone, A., A. Zinovyev and F. Kepes, 2003. Codon adaptation index as a measure of dominating codon bias. Bioinformatics 19 2005–2015. [DOI] [PubMed] [Google Scholar]
- Chamary, J. V., and L. D. Hurst, 2005. a Biased codon usage near intron-exon junctions: Selection on splicing enhancers, splice-site recognition or something else? Trends Genet. 21 256–259. [DOI] [PubMed] [Google Scholar]
- Chamary, J. V., and L. D. Hurst, 2005. b Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol. 6 R75.1–R75.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary, J. V., J. L. Parmley and L. D. Hurst, 2006. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 7 98–108. [DOI] [PubMed] [Google Scholar]
- Chen, S. L., W. Lee, A. K. Hottes, L. Shapiro and H. H. McAdams, 2004. Codon usage between genomes is constrained by genome-wide mutational processes. Proc. Natl. Acad. Sci. USA 101 3480–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, T., 2003. Ribosome recycling, diffusion, and mRNA loop formation in translational regulation. Biophys. J. 85 755–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron, J. M., 2006. Weak selection and recent mutational changes influence polymorphic synonymous mutations in humans. Proc. Natl. Acad. Sci. USA 103 6940–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron, J. M., and M. Kreitman, 2002. Population, evolutionary and genomic consequences of interference selection. Genetics 161 389–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Vera, L. R., M. A. Magos-Castro, E. Zamora-Romo and G. Guarneros, 2004. Ribosome stalling and peptidyl-tRNA drop-off during translational delay at aga codons. Nucleic Acids Res. 32 4462–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran, J. F., and M. Yarus, 1989. Rates of aminoacyl-trans-RNA selection at 29 sense codons in vivo. J. Mol. Biol. 209 65–77. [DOI] [PubMed] [Google Scholar]
- Dincbas, V., V. Heurgue-Hamard, R. H. Buckingham, R. Karimi and M. Ehrenberg, 1999. Shutdown in protein synthesis due to the expression of mini-genes in bacteria. J. Mol. Biol. 291 745–759. [DOI] [PubMed] [Google Scholar]
- Dolinski, K., R. Balakrishnan, K. R. Christie, M. C. Costanzo and S. S. Dwight, 2008. Saccharomyces Genome Database. ftp://ftp.yeastgenome.org/yeast/. [DOI] [PubMed]
- Drummond, D. A., and C. O. Wilke, 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret, L., and D. Mouchiroud, 1999. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, Arabidopsis. Proc. Natl. Acad. Sci. USA 96 4482–4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elf, J., D. Nilsson, T. Tenson and M. Ehrenberg, 2003. Selective charging of tRNA isoacceptors explains patterns of codon usage. Science 300 1718–1722. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A., 1996. Synonymous codon bias is related to gene length in Escherichia coli: Selection for translational accuracy? Mol. Biol. Evol. 13 864–872. [DOI] [PubMed] [Google Scholar]
- Ghosh, T. C., S. K. Gupta and S. Majumdar, 2000. Studies on codon usage in Entamoeba histolytica. Int. J. Parasitol. 30 715–722. [DOI] [PubMed] [Google Scholar]
- Gilchrist, M. A., 2007. Combining models of protein translation and population genetics to predict protein production rates from codon usage patterns. Mol. Biol. Evol. 24 2362–2373. [DOI] [PubMed] [Google Scholar]
- Gilchrist, M. A., and A. Wagner, 2006. A model of protein translation including codon bias, nonsense errors, and ribosome recycling. J. Theor. Biol. 239 417–434. [DOI] [PubMed] [Google Scholar]
- Goldman, N., and Z. H. Yang, 1994. Codon-based model of nucleotide substitution for protein-coding DNA-sequences. Mol. Biol. Evol. 11 725–736. [DOI] [PubMed] [Google Scholar]
- Grantham, R., C. Gautier and M. Gouy, 1980. Codon frequencies in 119 individual genes confirm consistent choices of degenerate bases according to genome type. Nucleic Acids Res. 8 1893–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamby, D. M., 1994. A review of techniques for parameter sensitivity analysis of environmental models. Environ. Monit. Assess. 32 135–154. [DOI] [PubMed] [Google Scholar]
- Hershberg, R., and D. A. Petrov, 2008. Selection on codon bias. Annu. Rev. Genet. 42 287–299. [DOI] [PubMed] [Google Scholar]
- Hooper, S. D., and O. G. Berg, 2000. Gradients in nucleotide and codon usage along Escherichia coli genes. Nucleic Acids Res. 28 3517–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura, T., 1981. Correlation between the abundance of Escherichia-coli transfer-RNAs and the occurrence of the respective codons in its protein genes—a proposal for a synonymous codon choice that is optimal for the Escherichia-coli translational system. J. Mol. Biol. 151 389–409. [DOI] [PubMed] [Google Scholar]
- Ikemura, T., 1982. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. Differences in synonymous codon choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting transfer RNAs. J. Mol. Biol. 158 573–597. [DOI] [PubMed] [Google Scholar]
- Ikemura, T., 1985. Codon usage and transfer-RNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 2 13–34. [DOI] [PubMed] [Google Scholar]
- Iwasa, Y., 1988. Free fitness that always increases in evolution. J. Theor. Biol. 135 265–281. [DOI] [PubMed] [Google Scholar]
- Jorgensen, F., and C. G. Kurland, 1990. Processivity errors of gene-expression in Escherichia coli. J. Mol. Biol. 215 511–521. [DOI] [PubMed] [Google Scholar]
- Karlin, S., and J. Mrazek, 2000. Predicted highly expressed genes of diverse prokaryotic genomes. J. Bacteriol. 182 5238–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliman, R. M., and J. Hey, 1993. Reduced natural-selection associated with low recombination in Drosophila melanogaster. Mol. Biol. Evol. 10 1239–1258. [DOI] [PubMed] [Google Scholar]
- Kliman, R. M., and J. Hey, 1994. The effects of mutation and natural-selection on codon bias in the genes of Drosophila. Genetics 137 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland, C. G., 1992. Translational accuracy and the fitness of bacteria. Annu. Rev. Genet. 26 29–50. [DOI] [PubMed] [Google Scholar]
- Lin, Y. S., J. K. Byrnes, J. K. Hwang and W. H. Li, 2006. Codon-usage bias versus gene conversion in the evolution of yeast duplicate genes. Proc. Natl. Acad. Sci. USA 103 14412–14416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, M., and J. H. Gillespie, 1991. Codon usage divergence of homologous vertebrate genes and codon usage clock. J. Mol. Evol. 32 6–15. [DOI] [PubMed] [Google Scholar]
- Lynch, M., 2007. The Origins of Genome Architecture. Sinauer Associates, Sunderland, MA.
- MacKay, V. L., X. H. Li, M. R. Flory, E. Turcott and G. L. Law, 2004. Gene expression analyzed by high-resolution state array analysis and quantitative proteomics—response of yeast to mating pheromone. Mol. Cell. Proteomics 3 478–489. [DOI] [PubMed] [Google Scholar]
- Manley, J. L., 1978. Synthesis and degradation of termination and premature-termination fragments of beta-galactosidase in vitro and in vivo. J. Mol. Biol. 125 407–432. [DOI] [PubMed] [Google Scholar]
- McVean, G. A. T., and B. Charlesworth, 1999. A population genetic model for the evolution of synonymous codon usage: patterns and predictions. Genet. Res. 74 145–158. [Google Scholar]
- Menninger, J. R., 1978. Accumulation as peptidyl-transfer RNA of isoaccepting transfer-RNA families in Escherichia-coli with temperature-sensitive peptidyl-transfer RNA hydrolase. J. Biol. Chem. 253 6808–6813. [PubMed] [Google Scholar]
- Mooers, A., and E. Holmes, 2000. The evolution of base composition and phylogenetic inference. Trends Ecol. Evol. 15 365–369. [DOI] [PubMed] [Google Scholar]
- Mougel, F., C. Manichanh, G. D. N'Guyen and M. Termier, 2004. Genomic choice of codons in 16 microbial species. J. Biomol. Struct. Dyn. 22 315–329. [DOI] [PubMed] [Google Scholar]
- Musto, H., H. Romero, A. Zavala, K. Jabbari and G. Bernardi, 1999. Synonymous codon choices in the extremely GC-poor genome of Plasmodium falciparum: compositional constraints and translational selection. J. Mol. Evol. 49 27–35. [DOI] [PubMed] [Google Scholar]
- Musto, H., S. Cruveiller, G. D'Onofrio, H. Romero and G. Bernardi, 2001. Translational selection on codon usage in Xenopus laevis. Mol. Biol. Evol. 18 1703–1707. [DOI] [PubMed] [Google Scholar]
- Musto, H., H. Romero and A. Zavala, 2003. Translational selection is operative for synonymous codon usage in Clostridium perfringens and Clostridium acetobutylicum. Microbiology 149 855–863. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., B. DuMont, L. Vanessa and M. Hubisz, 2007. a Maximum likelihood estimation of ancestral codon usage bias parameters in Drosophila. Mol. Biol. Evol 24 228–235. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., I. Hellmann, M. Hubisz, C. Bustamante and A. G. Clark, 2007. b Recent and ongoing selection in the human genome. Nat. Rev. Genet. 8 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, L., and A. Salvan, 1997. Principles of Statistical Inference (Advanced Series on Statistical Science & Probability, Vol. 4, Ed. 1). World Scientific, Singapore.
- Plotkin, J. B., H. Robins and A. J. Levine, 2004. Tissue-specific codon usage and the expression of human genes. Proc. Natl. Acad. Sci. USA 101 12588–12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, H., W. B. Wu, J. M. Comeron, M. Kreitman and W. H. Li, 2004. Intragenic spatial patterns of codon usage bias in prokaryotic and eukaryotic genomes. Genetics 168 2245–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, J. A., 1995. Mathematical Statistics and Data Analysis, Ed. 2. Duxbury Press, Belmont, CA.
- Roymondal, U., S. Das and S. Sahoo, 2009. Predicting gene expression level from relative codon usage bias: an application to Escherichia coli genome. DNA Res. 16 13–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sella, G., and A. E. Hirsh, 2005. The application of statistical physics to evolutionary biology. Proc. Natl. Acad. Sci. USA 102 9541–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, P. M., and W. H. Li, 1987. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, P. M., T. M. F. Tuohy and K. R. Mosurski, 1986. Codon usage in yeast—cluster-analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 14 5125–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields, D. C., P. M. Sharp, D. G. Higgins and F. Wright, 1988. Silent sites in Drosophila genes are not neutral—evidence of selection among synonymous codons. Mol. Biol. Evol. 5 704–716. [DOI] [PubMed] [Google Scholar]
- Stoletzki, N., and A. Eyre-Walker, 2007. Synonymous codon usage in Escherichia coli: selection for translational accuracy. Mol. Biol. Evol. 24 374–381. [DOI] [PubMed] [Google Scholar]
- Subramanian, S., 2008. Nearly neutrality and the evolution of codon usage bias in eukaryotic genomes. Genetics 178 2429–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sémon, M., D. Mouchiroud and L. Duret, 2005. Relationship between gene expression and gc-content in mammals: statistical significance and biological relevance. Hum. Mol. Genet. 14 421–427. [DOI] [PubMed] [Google Scholar]
- Tamura, K., S. Subramanian and S. Kumar, 2004. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol. Biol. Evol. 21 36–44. [DOI] [PubMed] [Google Scholar]
- Tsung, K., S. Inouye and M. Inouye, 1989. Factors affecting the efficiency of protein-synthesis in Escherichia coli—production of a polypeptide of more than 6000 amino-acid residues. J. Biol. Chem. 264 4428–4433. [PubMed] [Google Scholar]
- Urrutia, A. O., and L. D. Hurst, 2003. The signature of selection mediated by expression on human genes. Genome Res. 13 2260–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, A., 2000. Inferring lifestyle from gene expression patterns. Mol. Biol. Evol. 17 1985–1987. [DOI] [PubMed] [Google Scholar]
- Wagner, A., 2005. Energy constraints on the evolution of gene expression. Mol. Biol. Evol. 22 1365–1374. [DOI] [PubMed] [Google Scholar]
- Wan, X. F., J. Zhou and D. Xu, 2006. Codono: a new informatics method for measuring synonymous codon usage bias within and across genomes. Int. J. Gen. Syst. 35 109–125. [Google Scholar]
- Warnecke, T., and L. D. Hurst, 2007. Evidence for a trade-off between translational efficiency and splicing regulation in determining synonymous codon usage in Drosophila melanogaster. Mol. Biol. Evol. 24 2755–2762. [DOI] [PubMed] [Google Scholar]
- Warnecke, T., N. N. Batada and L. D. Hurst, 2008. The impact of the nucleosome code on protein-coding sequence evolution in yeast. PLoS Genet. 4 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, K. H., and P. M. Sharp, 1993. Mammalian gene evolution: nucleotide sequence divergence between mouse and rat. J. Mol. Evol. 37 441–456. [DOI] [PubMed] [Google Scholar]
- Wright, F., 1990. The effective number of codons used in a gene. Gene 87 23–29. [DOI] [PubMed] [Google Scholar]
- Xia, X., 2009. Information-theoretic indices and an approximate significance test for testing the molecular clock hypothesis with genetic distances. Mol. Phylogenet. Evol. 52 665–676. [DOI] [PubMed] [Google Scholar]
- Xia, X. H., 1996. Maximizing transcription efficiency causes codon usage bias. Genetics 144 1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, X. H., 1998. How optimized is the translational machinery in Escherichia coli, Salmonella typhimurium and Saccharomyces cerevisiae? Genetics 149 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaher, H. S., and R. Green, 2009. Fidelity at the molecular level: lessons from protein synthesis. Cell 136 746–762. [DOI] [PMC free article] [PubMed] [Google Scholar]