

Abstract
Genomic prediction of future phenotypes or genetic merit using dense SNP genotypes can be used for prediction of disease risk, forensics, and genomic selection of livestock and domesticated plant species. The reliability of genomic predictions is their squared correlation with the true genetic merit and indicates the proportion of the genetic variance that is explained. As reliability relies heavily on the number of phenotypes, combining data sets from multiple populations may be attractive as a way to increase reliabilities, particularly when phenotypes are scarce. However, this strategy may also decrease reliabilities if the marker effects are very different between the populations. The effect of combining multiple populations on the reliability of genomic predictions was assessed for two simulated cattle populations, A and B, that had diverged for T = 6, 30, or 300 generations. The training set comprised phenotypes of 1000 individuals from population A and 0, 300, 600, or 1000 individuals from population B, while marker density and trait heritability were varied. Adding individuals from population B to the training set increased the reliability in population A by up to 0.12 when the marker density was high and T = 6, whereas it decreased the reliability in population A by up to 0.07 when the marker density was low and T = 300. Without individuals from population B in the training set, the reliability in population B was up to 0.77 lower than in population A, especially for large T. Adding individuals from population B to the training set increased the reliability in population B to close to the same level as in population A when the marker density was sufficiently high for the marker–QTL linkage disequilibrium to persist across populations. Our results suggest that the most accurate genomic predictions are achieved when phenotypes from all populations are combined in one training set, while for more diverged populations a higher marker density is required.
GENOMIC predictions of future phenotypes or total genetic merit can be used for prediction of disease risk, for forensics, and for selection of livestock and domesticated plant species (Wray et al. 2007; Lee et al. 2008; Goddard and Hayes 2009; Vanraden et al. 2009; Zhong et al. 2009). Genomic predictions rely on linkage disequilibrium (LD) between genetic markers and quantitative trait loci (QTL) in a population. A common approach has been to select significant markers from a genomewide association study as predictors (Lande and Thompson 1990; Morrison et al. 2007; Van Hoek et al. 2008) but this approach is likely to select false positive markers and ignores markers below the significance threshold. Methods that use all markers simultaneously may therefore result in higher reliabilities of predictions of the total genetic merit, indicating that a larger proportion of the genetic variance is explained (Meuwissen et al. 2001; Gianola et al. 2003; Xu 2003; Hoggart et al. 2008). The additive effects of the markers can be estimated from a training set, i.e., a large set of genotyped individuals with phenotypic data. Genomic predictions for other individuals can subsequently be calculated by summing the effects of the markers across all loci (Meuwissen et al. 2001; Gianola et al. 2003; Xu 2003). This procedure assumes that the individuals come from the same population as the training set so the LD between genetic markers and QTL persists from the training set to other individuals. In practice, one may also be interested in individuals from other populations, such as individuals from other population substructures, other selection lines, or other breeds, in which the LD between genetic markers and QTL may be different. The reliability of the genomic predictions for individuals from other populations may therefore be lower, especially when the population is genetically more distant from the training set. Furthermore, combining phenotypes from multiple populations may be a way to increase the size of the training set. Thus far, most studies regarding genomic prediction of total genetic values have considered a single population. Little is known about genomic prediction in a multiple-population context.
In artificial breeding of animals or plants, selection based on genomic predictions, or genomic breeding values (GBVs), is called genomic selection (Meuwissen et al. 2001). A difficulty with genomic selection across multiple populations is the calculation of GBVs because the marker effects may differ across populations. Therefore, GBVs could be predicted with multiple within-population evaluations or with one across-population evaluation in which the training set comprises individuals from all populations. Combining populations in a training set may be advantageous because the effects of the markers can be estimated from a larger number of phenotypes. This is particularly of interest when the training set for one of the populations is too small for a proper within-population evaluation. On the other hand, it is expected that some markers may be in high LD with a QTL in one population but not in the other population, especially when these markers are more distant from the QTL or when the populations have diverged for many generations (Andreescu et al. 2007; Gautier et al. 2007; De Roos et al. 2008). Furthermore, some QTL may be fixed in any one of the populations. The effects of combining populations in a training set on the reliability of GBVs have only recently been studied. Ibán z-Escriche et al. (2009) compared reliabilities of genomic predictions in purebred animals using crossbred descendants in the training set and concluded that across-population evaluations were favorable over within-population evaluations when the populations were closely related, marker density was high, or the number of training records was small.
z-Escriche et al. (2009) compared reliabilities of genomic predictions in purebred animals using crossbred descendants in the training set and concluded that across-population evaluations were favorable over within-population evaluations when the populations were closely related, marker density was high, or the number of training records was small.
Marker density has an important effect on the reliability of GBVs because a higher number of markers, when equally distributed across the genome, will increase the probability that each QTL is in high LD with at least one marker (Calus et al. 2008; Goddard 2009). Within livestock populations, LD may extend over hundreds of kilobases, as a result of small effective population size (Ne) in recent generations (Heifetz et al. 2005; Du et al. 2007; Mckay et al. 2007). However, only markers that are very close to QTL may have a persistent LD phase with the QTL across breeds or selection lines. For example, between Bos taurus cattle breeds, LD is persistent only for marker pairs that are <10 kb from each other (Gautier et al. 2007; De Roos et al. 2008), and for commercial broiler chicken breeding lines, the correlation between lines of LD measure r ranged from 0.21 to 0.94 for markers <500 kb apart (Andreescu et al. 2007). The number of phenotypes in the training set and the heritability of the phenotypes also have an important effect on the reliability of GBVs (Meuwissen et al. 2001; Calus et al. 2008; Daetwyler et al. 2008; Goddard 2009). For traits with low heritability, many phenotypes are necessary to accurately estimate the marker effects. Therefore, combining populations in a training set may be more advantageous for traits with low heritability than for traits with high heritability.
The objective of this study was to assess the reliability of GBVs across two populations that have diverged for several generations, when the training set comprises individuals from one or both populations. The effects of the heritability of the trait and the marker density on the reliability of GBVs were studied as well. The simulations were intended to model cattle populations, with a similar pattern of LD within and across populations as observed in that species (De Roos et al. 2008). However, our results are relevant to any species where within- and across-population genomic predictions are being considered.
MATERIALS AND METHODS
Simulation of populations:
On the basis of the estimates of past Ne in cattle (De Roos et al. 2008), four phases in cattle population history were distinguished: (1) a predomestication ancestral population with Ne of ∼100,000, (2) a postdomestication population with Ne of a few thousand, (3) a further reduction in Ne to a few hundred, following breed formation, and (4) a modern cattle breed with Ne < 100 because of intensive selection of bulls. To simulate a comparable data set, a base population of 100 individuals was generated with 3 chromosomes of 1 M each and 41,403 equally spaced, monomorphic loci. The cattle genome comprises 30 chromosomes, but simulating 30 chromosomes was computationally too demanding. The number of loci was chosen such that it would generate >20,000 polymorphic loci, on the basis of experience with the simulation software. The individuals were randomly selected as parents and randomly mated for 600 generations, allowing individuals to have multiple progeny. To mimic an effective population size of ∼100,000 (rather than 100) and 600,000 generations of random mating (rather than 600), the mutation rate and recombination rates were increased by 1000, i.e., a mutation rate of 10−5 per locus per generation (Nachman and Crowell 2000) and a recombination rate of 103 per Morgan per generation. In the second step, 200 generations were generated, mimicking the recombination and mutation rate of a population size of ∼1250 for 2000 generations (Figure 1). After that, the number of individuals was increased from 100 to 2000, mutation was stopped, and the recombination rate was 1 per Morgan per generation. In the third step, the number of sires was fixed to 100 and each sire was mated to 20 dams at random, for 50 generations. In step 4, the number of sires was fixed to 25 and each sire was mated to 80 dams at random, for 10 generations.
Figure 1.—

Design of the simulation for populations with T = 6, 30, or 300 generations of divergence and four historical phases with different effective population sizes (Ne). The number of generations in each phase, before and after divergence, is given in each cell.
To simulate two diverged populations, the population was split in two for T generations (T = 6, 30, or 300). After the divergence, each population (A and B) comprised 1000 individuals and within-population random selection and mating was applied, i.e., no exchange of material during T generations (Figure 1). The numbers of sires that were used in the simulation, described above, were those settings that gave the best correspondence with real cattle data in terms of extent of LD within breeds, i.e., average r2 as a function of marker distance (Hill and Robertson 1968). The time of divergence between population A and B was chosen such that it resulted in the same correlation of r values as observed between Dutch and Australian Holstein–Friesian (HF) (T = 6), between Australian HF and New Zealand Friesian (T = 30), and between Australian HF and Australian Angus (T = 300) by De Roos et al. (2008).
Simulation of genotypes and phenotypes:
Of 41,403 loci, 26,294 loci (64%) were polymorphic after step 2, including 6438 loci (16%) that had between three and five alleles. For loci with more than two alleles, one of the mutations was randomly chosen as allele 1 and all other alleles were collapsed into allele 2. Of the polymorphic loci with minor allele frequency >0.10 after step 2 (before divergence), 150 loci were randomly selected as QTL. Their allele substitution effects were randomly drawn from an exponential distribution and randomly given a positive or a negative sign, with equal probability. Four marker sets were generated by randomly selecting M = 300, 1500, 5000, and 18,000 polymorphic loci as genetic markers, without further restrictions on minor allele frequency.
The breeding value of an individual was equal to the sum of the QTL allele substitution effects, assuming only additive QTL effects and no other genetic variation than the 150 simulated QTL. Phenotypes were generated for 1000 individuals in population A and 1000 individuals in population B, by adding residuals, randomly drawn from a normal distribution with mean equal to zero, to the true breeding values. The variance of the random residuals was chosen such that the heritability in population A (h2) was 0.1, 0.3, 0.7, 0.9, or 1.0. The individuals with phenotypes were the sires and grandsires of the individuals in the last generation, plus additional individuals from the same generations as those sires and grandsires, but excluding dams and granddams of the individuals in the last generation. This may correspond to a situation where the training set comprises average progeny performance records as phenotypes.
GBVs were calculated for the last generation, including 1000 individuals in population A and 1000 individuals in population B. The training set comprised the 1000 individuals with a phenotype from population A plus NB = 0, 300, 600, or 1000 individuals with a phenotype from population B. For scenarios with NB > 0, the sires and grandsires of the individuals in the last generation were always included in the training set.
GBVs were calculated for three sets of populations that diverged for a different number of generations (T = 6, 30, or 300), with four different marker densities (M = 300, 1500, 5000, or 18,000), five different heritabilities (h2 = 0.1, 0.3, 0.7, 0.9, or 1.0), and four different numbers of individuals from population B in the training set (NB = 0, 300, 600, or 1000). Each scenario was replicated 10 times, summing up to 2400 evaluations.
Model:
The genomic prediction model, which was derived from the Bayesian multiple-QTL model of Meuwissen and Goddard (2004), was 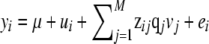 , where yi is the phenotype of individual i, μ is the mean, ui is the polygenic effect of individual i, vj is a scalar to model the allele substitution effect for marker j, qj is a vector of (nonscaled) allele effects for marker j, zij is a design vector for individual i at marker j, which was
, where yi is the phenotype of individual i, μ is the mean, ui is the polygenic effect of individual i, vj is a scalar to model the allele substitution effect for marker j, qj is a vector of (nonscaled) allele effects for marker j, zij is a design vector for individual i at marker j, which was 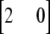 ,
, 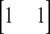 , or
, or 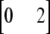 for individuals that are homozygous for allele 1, heterozygous, or homozygous for allele 2, respectively, and ei is the residual corresponding to individual i. The covariance among polygenic effects was modeled as
for individuals that are homozygous for allele 1, heterozygous, or homozygous for allele 2, respectively, and ei is the residual corresponding to individual i. The covariance among polygenic effects was modeled as 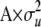 , where A is the numerator relationship matrix based on the full pedigree of the last five generations and
, where A is the numerator relationship matrix based on the full pedigree of the last five generations and 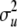 is the polygenic variance. The nonscaled allele effects at each marker (qj) had a variance of 1 and were assumed independent. The variance of the scalar for marker j (vj) was assumed
is the polygenic variance. The nonscaled allele effects at each marker (qj) had a variance of 1 and were assumed independent. The variance of the scalar for marker j (vj) was assumed  or
or 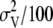 , depending on whether the marker was associated with a QTL or not. An inverted chi-square distribution was assumed for
, depending on whether the marker was associated with a QTL or not. An inverted chi-square distribution was assumed for 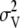 , with a prior variance equal to 1% of the variance of the total breeding values of the individuals in the training set. Whether marker j was associated with a QTL was sampled from a Bernoulli distribution with probability equal to
, with a prior variance equal to 1% of the variance of the total breeding values of the individuals in the training set. Whether marker j was associated with a QTL was sampled from a Bernoulli distribution with probability equal to 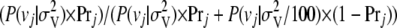 , where
, where 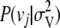 is the probability of sampling vj from
is the probability of sampling vj from 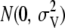 , i.e.,
, i.e., 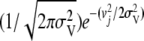 , and Prj is the prior probability of the presence of a QTL at marker j that was equal to 150/M. More details on the prior distributions and the fully conditional distributions can be found in Meuwissen and Goddard (2004). All parameters were estimated with a Markov chain Monte Carlo method using Gibbs sampling with residual updating. The Gibbs sampler was run for 25,000 iterations and 5000 iterations were discarded as burn-in. For comparison, an alternative model that included only the mean and the polygenic effect was also used:
, and Prj is the prior probability of the presence of a QTL at marker j that was equal to 150/M. More details on the prior distributions and the fully conditional distributions can be found in Meuwissen and Goddard (2004). All parameters were estimated with a Markov chain Monte Carlo method using Gibbs sampling with residual updating. The Gibbs sampler was run for 25,000 iterations and 5000 iterations were discarded as burn-in. For comparison, an alternative model that included only the mean and the polygenic effect was also used: 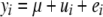 . This scenario is referred to as M = 0.
. This scenario is referred to as M = 0.
The GBV for individual i was calculated as 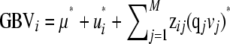 , where the * indicates the posterior mean of the 20,000 samples of μ, ui, and (qjvj) obtained from the stationary phase of the Gibbs chain. The reliability of the GBVs was calculated as the squared correlation between the GBVs and the simulated breeding values for the individuals in the last generation, separately for populations A and B. Standard errors were computed as the standard deviation of the reliabilities across the 10 replicates, divided by
, where the * indicates the posterior mean of the 20,000 samples of μ, ui, and (qjvj) obtained from the stationary phase of the Gibbs chain. The reliability of the GBVs was calculated as the squared correlation between the GBVs and the simulated breeding values for the individuals in the last generation, separately for populations A and B. Standard errors were computed as the standard deviation of the reliabilities across the 10 replicates, divided by 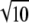 .
.
RESULTS
Linkage disequilibrium and persistence of phase:
The extent of LD in all the simulated populations was very consistent with what has been observed in real cattle data, as was attempted in the simulation. For example, the decay of average r2 with genomic distance is very similar between population A of scenario T = 6 and Australian HF cattle, assuming 1 Mb = 1 cM in cattle (Figure 2). The decay of average r2 with genomic distance did not differ between populations A and B and between the scenarios where the number of generations of divergence was different, T = 6, 30, or 300 (maximum difference 0.007). The average r2 was 0.46, 0.30, 0.18, 0.11, and 0.04 at 0.007, 0.022, 0.051, 0.101, and 1.000 cM, respectively. The average genomic distance between adjacent markers was 0.995, 0.200, 0.060, and 0.017 cM for scenarios with M = 300, 1500, 5000, and 18,000 markers, respectively, whereas the respective average r2 between adjacent markers was 0.06, 0.13, 0.23, and 0.37.
Figure 2.—

Average r2 as a function of genomic distance for one of the simulated data sets (population A in the scenario T = 6) and for Australian Holstein–Friesian bulls, as obtained from De Roos et al. (2008). Each data point was based on ∼8000 or 400 marker pairs for the simulated or real data, respectively.
The correlation of r for the same marker pairs between populations A and B in scenario T = 6 was 0.98 for marker pairs at 0.007 cM distance (Figure 3), which means that the LD between very close markers was very persistent between these two populations. For markers that were more distant from each other the correlation of r was lower, because more recombinations between the markers have occurred during the six generations of divergence, which caused the LD between those markers to differ between the two diverging populations. For scenario T = 6, the correlation of r dropped below 0.80 at ∼0.45 cM. For populations that have diverged for more generations, the correlation of r was lower at small distances and dropped quicker with increasing distance. For scenarios T = 30 and T = 300, the correlation of r dropped below 0.80 at ∼0.055 and ∼0.010 cM, respectively (Figure 3). When compared with real cattle data, the correlations of r for scenario T = 6 were slightly lower than those observed between Australian and Dutch HF bulls. The correlations of r for scenario T = 30 were very consistent with those between Australian HF and New Zealand Friesian bulls across all distances. The correlations of r for scenario T = 300 were consistent with those between Australian HF and Australian Angus, for markers that were <0.1 cM apart. For larger distances, the correlations of r in the simulated data were lower than those between HF and Angus (Figure 3).
Figure 3.—

Correlation of r of the same marker pairs between two populations as a function of genomic distance, in simulations with T = 6, 30, or 300 generations of divergence between populations A and B and in real cattle data (De Roos et al. 2008) between Australian Holstein–Friesians (HF_AUS) on one hand and Dutch Holstein–Friesian (HF_NLD), New Zealand Friesians (HF_NZL), or Australian Angus (ANG_AUS) on the other hand. Each data point was based on ∼8000 or 400 marker pairs for the simulated or real data, respectively.
Simulated QTL:
For each population (T = 6, 30, or 300) and each of 10 replicates, 150 QTL were simulated. The absolute QTL allele substitution effects (a) varied from 0.001 to 7.60, whereas the median and average were 0.68 and 1.02, respectively, which were very close to the expected values for an exponential distribution (0.69 and 1.00, respectively). In the training set of population A, the average QTL minor allele frequency (p) was 0.24 and on average 10 QTL were fixed. The QTL variance (2p(1 − p)a2) was 0.69 on average and 21.8 at maximum. The number of QTL, when ordered by their variance, that was necessary to explain 50 or 90% of the total QTL variance was on average 10 or 44, respectively. For each QTL, the r2 with all syntenic markers was calculated and the maximum value was kept. When averaged across all QTL, this maximum value of marker–QTL r2 was 0.20, 0.41, 0.62, and 0.81 for scenarios with M = 300, 1500, 5000, and 18,000 markers, respectively. The training sets of population B had similar characteristics. The average absolute difference in QTL allele frequency between the training sets of populations A and B was 0.07, 0.12, and 0.20 for T = 6, 30, and 300, respectively.
Reliability of genomic breeding values:
Reliabilities of GBVs were calculated for the youngest generations of populations A and B in all scenarios. The number of markers (M) and the heritability (h2) had a large effect on the reliability in population A, in contrast with the time since divergence from population B (T) and the number of individuals from population B in the training set (NB). For T = 6, NB = 0, and h2 = 0.1, the reliability increased from 0.07 (M = 0) to 0.11 (M = 300) to 0.18 (M = 18,000) by using more marker information, whereas for h2 = 1.0, the reliability increased from 0.31 (M = 0) to 0.54 (M = 300) to 0.97 (M = 18,000) (Figure 4). Adding 1000 individuals of population B to the training set (NB = 1000 vs. NB = 0) had some effect on the reliabilities in population A, with a maximum increase of 0.12 (reliability 0.30 vs. 0.18) for scenario T = 6, M = 18,000, and h2 = 0.10 and a maximum decrease of −0.07 (reliability 0.50 vs. 0.57) for scenario T = 300, M = 300, and h2 = 1.0 (Figure 5).
Figure 4.—

Reliability of genomic breeding values for 1000 individuals from the last generation of population A in simulations with six generations of divergence between populations A and B (T = 6), no individuals from population B in the training set (NB = 0), and different numbers of markers and heritabilities (h2). SEs were <0.015 when reliabilities were >0.80 and ∼0.025 otherwise.
Figure 5.—

Effect of adding 1000 individuals from population B to the training set (NB = 1000 vs. NB = 0) on the reliability of genomic breeding values for 1000 individuals from the last generation of population A, in simulations with T = 6 or 300 generations of divergence between populations A and B and different numbers of markers and heritabilities (h2). SEs were <0.005.
Without individuals from population B in the training set (NB = 0), the reliabilities in population B were always lower than in population A, but this difference was more pronounced when populations A and B had diverged for many generations or when the marker density was low (Figure 6). For example, for M = 18,000 and h2 = 1.0, the reliability in population A was 0.97, whereas the reliability in population B was 0.88, 0.65, and 0.36 for T = 6, 30, and 300, respectively. Furthermore, for M = 300 and h2 = 1.0, the reliability in population A was 0.55, whereas the reliability in population B was 0.16, 0.03, and 0.02 for T = 6, 30, and 300, respectively (Figure 6). For scenarios with lower heritability, reliabilities were generally lower in both populations, but the effects of marker density and time since divergence on reliabilities were consistent with Figure 6.
Figure 6.—

Reliability of genomic breeding values for 1000 individuals from the last generation of populations A and B, after T = 6, 30, or 300 generations of divergence between populations A and B, with no individuals from population B in the training set (NB = 0), a heritability of h2 = 1.0, and different numbers of markers. SEs were <0.015 when reliabilities were >0.80 and ∼0.025 otherwise.
When 300 individuals from population B were added to the training set (NB = 300), the reliability in population B increased substantially, irrespective of the marker density, heritability, or time since divergence. This increase in reliability was also observed for scenarios without marker information (M = 0), because the 300 individuals that were added to the training set also comprised the sires and grandsires of the youngest generation in population B. For scenario T = 6 and h2 = 1.0, the reliability in population B reached the same level as in population A when NB = 1000, when NB = 600 and M ≥ 5000, or when NB = 300 and M = 18,000 (Figure 7). For scenario T = 300 and h2 = 1.0, however, NB = 1000 individuals from population B were required in the training set to reach the same reliability as in population A (Figure 8). Figure 8 also shows that the reliability in population A decreases when individuals from population B were added to the training set and the marker density is low. For scenarios with lower heritability, the effects of adding individuals from population B to the training set were similar, except that the reliability in population A decreased to a lesser extent or even increased (Figure 5).
Figure 7.—

Reliability of genomic breeding values for 1000 individuals from the last generation of populations A and B, after T = 6 generations of divergence between populations A and B, with a heritability of h2 = 1.0, NB = 0, 300, 600, or 1000 individuals from population B added to the training set, and different numbers of markers. SEs were <0.015 when reliabilities were >0.80 and ∼0.025 otherwise.
Figure 8.—

Reliability of genomic breeding values for 1000 individuals from the last generation of populations A and B, after T = 300 generations of divergence between populations A and B, with a heritability of h2 = 1.0, NB = 0, 300, 600, or 1000 individuals from population B added to the training set, and different numbers of markers. SEs were <0.015 when reliabilities were >0.80 and ∼0.025 otherwise.
DISCUSSION
A number of authors have demonstrated that within a population the number of phenotypic records, the heritability of the trait, the number of genetic markers, and the effective population size determine the reliability of genomic predictions (Meuwissen et al. 2001; Muir 2007; Calus et al. 2008; Daetwyler et al. 2008; Goddard 2009). Our results demonstrate the reliability of genomic predictions across populations is, in addition to the above parameters, determined by the extent of marker–QTL allelic phase between the populations, which in turn is at least in part determined by the time of divergence between the populations. The more diverged the populations are, the denser the markers must be to ensure preservation of marker–QTL phase across the populations.
Our results within a population agree with previous results from simulation studies. For example, Meuwissen et al. (2001) found a reliability of 0.62 in a simulation with a training set of 1000 phenotypes with a heritability of 0.5 and one multiallelic marker per centimorgan. To achieve the same LD between adjacent markers as in Meuwissen et al. (2001) (r2 = 0.20), their reliabilities should be compared to scenario M = 5000, which had r2 = 0.23 between adjacent markers and reliabilities of 0.46 (h2 = 0.3) and 0.73 (h2 = 0.7). These results are more consistent with the 0.62 (h2 = 0.5) reported by Meuwissen et al. (2001).
The agreement of our reliabilities for within-population genomic predictions with those from real data is not as clear. For example, VanRaden et al. (2009) predicted GBVs for North American HF bulls using 38,416 SNPs and observed reliabilities of GBVs that were ∼0.23 higher than reliabilities of traditional parent averages, which is substantially lower than the reliabilities found in this simulation study with comparable marker density (M = 5000). One reason may be that in real data the number of QTL is ≫150 and the distribution of their effects is not exponential but normal. In that case, much more phenotypic information is needed to accurately estimate the marker effects (Goddard 2009). This was confirmed by VanRaden et al. (2009) by demonstrating that reliability of GBV increased more by doubling the number of phenotypes than by doubling the number of markers. Second, here we have simulated only three chromosomes and therefore fewer marker effects needed to be estimated than may be required in real data. Again, this would mean that the results of this simulation study are still relevant, but that in experiments in cattle, for example, the number of phenotypes must be much larger to obtain similar reliabilities. Third, in this simulation a random selection of 150 markers was used as QTL, whereas in real data causative mutations underlying genetic variation may have a different allele frequency spectrum because of different mutation rates, allele frequency distribution, and selection pressure. Nonadditive genetic effects were not simulated in our study but they may play a role in quantitative traits in dairy cattle. VanRaden et al. (2009), however, used deregressed estimated breeding values of progeny-tested bulls as phenotypes, which are assumed to be additive, and still achieved reliable predictions.
For multipopulation genomic predictions, our results demonstrate that adding individuals from a second population (population B) to the training set had some effect on the reliability of genomic breeding values in the first population (population A) and was most beneficial when the heritability was low, because then more phenotypic records were needed to estimate the marker effects (Figure 5). Furthermore, the phenotypes from population B were of highest value when the two populations had diverged for only few generations and the marker density was high, because then the marker–QTL LD phase persists across the two populations. On the other hand, when the populations have diverged for many generations and the marker density is low, the marker–QTL LD is different between the two populations, which leads to suboptimal marker effect estimates for each population. As a result, the reliability in population A was decreased by up to 0.07 (Figure 5) when there was a long period of divergence between the populations. These results are consistent with those of Ibán z-Escriche et al. (2009) who concluded that across-population evaluations were favorable over within-population evaluations when the populations were closely related, marker density was high, or the number of training records was small.
z-Escriche et al. (2009) who concluded that across-population evaluations were favorable over within-population evaluations when the populations were closely related, marker density was high, or the number of training records was small.
Without individuals from population B in the training set, the reliability of GBVs in population B was substantially lower than in population A, especially when the populations had diverged for 300 generations and the marker density was low (Figure 6). Again, this is caused by differences in marker–QTL LD phase between the populations. Adding only a limited number of individuals from population B to the training set increased the reliability in population B substantially (Figures 7 and 8). This was partly because the phenotypes of the sires and grandsires of the youngest generation were added to the training set, but additionally that the marker effects were optimized for both populations. It may be possible that without individuals from population B in the training set, the QTL are explained by markers that are quite distant from the QTL, but in LD with the QTL in population A. These markers may not be in LD with the same QTL in population B and therefore result in poorer predictions for population B. By adding individuals from population B to the training set, only markers that are close to QTL are used to explain their variation, and therefore the predictions are much better for population B, without decreasing the reliability in population A. This makes combining populations in a training set of particular interest for QTL fine mapping, as long as the marker density is sufficient to find markers in LD with the QTL across populations (Barendse et al. 2007). It may also be expected that genomic predictions in a third population (C) are better when the training set includes individuals from populations A and B. In association studies, however, population subdivision or recent admixture is often regarded as negative, because if the underlying populations differ in their average phenotype, then any marker that has a different allele frequency between the populations will be associated with the phenotype, leading to many spurious associations (Pritchard and Rosenberg 1999). This problem is specific for association studies; in fact, genomic prediction models may use unlinked markers to estimate coancestry among individuals (Habier et al. 2007; Goddard 2009).
In this study, the reliability was studied for only one generation after the training set. In dairy cattle, it may be expected that new phenotypes will be added to the training set every generation, and selection candidates are only one or two generations younger than the training set. However, in other species it may be more common to establish a training set once and use it for genomic predictions for several generations. Habier et al. (2007) showed that over subsequent generations the reliability of GBVs decreased substantially because of the decay of pedigree relationship with animals with phenotypes. The reliability due to LD, however, was more persistent over generations. It may be expected that if combining populations in a training set leads to associations between QTL and very close markers only, the reliability of GBVs may persist better over generations as well. This is consistent with Muir (2007), who observed that a training set of 4 generations × 512 individuals gave less decay of reliability over generations than a training set of 2 generations × 1024 individuals, and also with Zhong et al. (2009), who observed that genomic prediction models that relied mostly on LD between markers and QTL had more persistent accuracy over generations than models that relied on marker-based coancestry.
An important assumption in this study was that QTL effects were the same across both populations. This assumption is violated when there is QTL-by-environment interaction or QTL-by-genetic background interaction. With substantial QTL-by-environment interaction or QTL-by-genetic background interaction, it may be less advantageous to combine populations in a training set, because marker effects may differ across populations. Alternative methods may help in this respect by allowing population-specific estimation of the allele substitution effects (Ibán z-Escriche et al. 2009), while searching for marker–QTL association across populations.
z-Escriche et al. 2009), while searching for marker–QTL association across populations.
The results of this study imply that for genomic predictions in multiple populations, the highest reliabilities are achieved when the populations are combined in the training set rather then analyzing the populations separately, especially for populations that have diverged for few generations and when the marker density is high. Practically this may mean combining phenotypes from multiple breeds or selection lines and including crossbred animals as well. Combining populations may be very advantageous when one of the populations it too small for a population-specific analysis: for example, in dairy or beef cattle breeding when not enough bulls have been progeny tested for a certain breed; when phenotypes are obtained from crossbred animals, as in pig and poultry breeding (Ibán z-Escriche et al. 2009); in plant breeding, where training sets may be derived from multiple inbred lines (Zhong et al. 2009); and in human genetics, where disease susceptibility may be predicted from marker data using a case–control training set comprising various subpopulations (Wray et al. 2007).
z-Escriche et al. 2009); in plant breeding, where training sets may be derived from multiple inbred lines (Zhong et al. 2009); and in human genetics, where disease susceptibility may be predicted from marker data using a case–control training set comprising various subpopulations (Wray et al. 2007).
Provided marker density is sufficient, combining populations in a training set will increase the reliability of GBVs in plant and livestock breeding, which will result in higher rates of genetic improvement and more efficient breeding programs (Goddard 2009). Similarly, genomic predictions of disease risk will be closer to the true genetic susceptibility to the disease, which may lead to better prevention and treatment.
In conclusion, heritability and marker density have a large effect on the reliability of GBVs. When the training set comprised individuals from only one population, the reliability of the GBVs in another population was substantially lower than in the population of the training set. When relatively few individuals from the other population were added to the training set, however, the reliability in the other population increased substantially, irrespective of the heritability or marker density. The benefits of combining populations in a training set were highest when the populations have diverged for only few generations, when the marker density was high, and when the heritability was low. Our results suggest that the most accurate genomic predictions are achieved when phenotypes from all populations are combined in one training set as opposed to analyzing the data separately within populations. For more diverged populations a high marker density is necessary to ensure markers and QTL are in the same LD phase.
References
- Andreescu, C., S. Avendano, S. R. Brown, A. Hassen, S. J. Lamont et al., 2007. Linkage disequilibrium in related breeding lines of chickens. Genetics 177 2161–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barendse, W., A. Reverter, R. J. Bunch, B. E. Harrison, W. Barris et al., 2007. A validated whole genome association study of efficient food conversion in cattle. Genetics 176 1893–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calus, M. P. L., T. H. E. Meuwissen, A. P. W. De Roos and R. F. Veerkamp, 2008. Accuracy of genomic selection using different methods to define haplotypes. Genetics 178 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daetwyler, H. D., B. Villanueva and J. A. Woolliams, 2008. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS ONE 3(10): e3395. [DOI] [PMC free article] [PubMed]
- De Roos, A. P. W., B. J. Hayes, R. Spelman and M. E. Goddard, 2008. Linkage disequilibrium and persistence of phase in Holstein-Friesian, Jersey and Angus cattle. Genetics 179 1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, F.-X., A. C. Clutter and M. M. Lohuis, 2007. Characterizing linkage disequilibrium in pig populations. Int. J. Biol. Sci. 3 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier, M., T. Faraut, K. Moazami-Goudarzi, V. Navratil, M. Foglio et al., 2007. Genetic and haplotypic structure in 14 European and African cattle breeds. Genetics 177 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianola, D., M. Perez-Enciso and M. Toro, 2003. On marker-assisted prediction of genetic value: beyond the ridge. Genetics 163 347–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard, M. E., 2009. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica 136 245–257. [DOI] [PubMed] [Google Scholar]
- Goddard, M. E., and B. J. Hayes, 2009. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10 381–391. [DOI] [PubMed] [Google Scholar]
- Habier, D., R. L. Fernando and J. C. M. Dekkers, 2007. The impact of genetic relationship information on genome-assisted breeding values. Genetics 177 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifetz, E. M., J. E. Fulton, N. O'Sullivan, H. Zhao, J. C. M. Dekkers et al., 2005. Extent and consistency across generations of linkage disequilibrium in commercial layer chicken breeding populations. Genetics 171 1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., and A. Robertson, 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38 226–231. [DOI] [PubMed] [Google Scholar]
- Hoggart, C. J., J. C. Whittaker, M. De Iorio and D. J. Balding, 2008. Simultaneous analysis of all SNPs in genome-wide and re-sequencing association studies. PLoS Genet. 4(7): e1000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Ibán
 z-Escriche, N., R. L. Fernando, A. Toosi and J. C. M. Dekkers, 2009. Genomic selection of purebreds for crossbred performance. Genet. Sel. Evol. 41 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
z-Escriche, N., R. L. Fernando, A. Toosi and J. C. M. Dekkers, 2009. Genomic selection of purebreds for crossbred performance. Genet. Sel. Evol. 41 12. [DOI] [PMC free article] [PubMed] [Google Scholar] - Lande, R., and R. Thompson, 1990. Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics 124 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. H., J. H. J. van der Werf, B. J. Hayes, M. E. Goddard and P. M. Visscher, 2008. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. PLoS Genet. 4(10): e1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay, S. D., R. D. Schnabel, B. M. Murdoch, L. K. Matukumalli, J. Aerts et al., 2007. Whole genome linkage disequilibrium maps in cattle. BMC Genet. 8 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2004. Mapping multiple QTL using linkage disequilibrium and linkage analysis information and multitrait data. Genet. Sel. Evol. 36 261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., B. J. Hayes and M. E. Goddard, 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison, A. C., L. A. Bare, L. E. Chambless, S. G. Ellis, M. Malloy et al., 2007. Prediction of coronary heart disease risk using a genetic risk score: the atherosclerosis risk in communities study. Am. J. Epidemiol. 166(1): 28–35. [DOI] [PubMed] [Google Scholar]
- Muir, W. M., 2007. Comparison of genomic and traditional BLUP-estimated breeding value accuracy and selection response under alternative trait and genomic parameters. J. Anim. Breed. Genet. 124 342–355. [DOI] [PubMed] [Google Scholar]
- Nachman, M. W., and S. L. Crowell, 2000. Estimate of the mutation rate per nucleotide in humans. Genetics 156 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., and N. A. Rosenberg, 1999. Use of unlinked genetic markers to detect population stratification in association studies. Am. J. Hum. Genet. 65 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hoek, M., A. Dehghan, J. C. Witteman, C. M. van Duijn, A. G. Uitterlinden et al., 2008. Predicting type 2 diabetes based on polymorphisms from genome-wide association studies: a population-based study. Diabetes 57(11): 3122–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRaden, P. M., C. P. VanTassell, G. R. Wiggans, T. S. Sonstegard, R. D. Schnabel et al., 2009. Invited review: reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 92 16–24. [DOI] [PubMed] [Google Scholar]
- Wray, N. R., M. E. Goddard and P. M. Visscher, 2007. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 17 1520–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., 2003. Estimating polygenic effects using markers of the entire genome. Genetics 163 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, S., J. C. M. Dekkers, R. L. Fernando and J.-L. Jannink, 2009. Factors affecting accuracy from genomic selection in populations derived from multiple inbred lines: a barley case study. Genetics 182 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]