

Summary
Wnt3a activates the `canonical' signaling pathway, stimulating the nuclear accumulation of β-catenin and activation of Lef/Tcf-sensitive transcription of developmentally important genes. Using totipotent mouse F9 teratocarcinoma cells expressing frizzled-1 (Fz1), we investigated roles of tyrosine kinase activity in Wnt/β-catenin signaling. Treatment with either genistein or Src family kinase inhibitor PP2 attenuates Wnt3a-stimulated Lef/Tcf transcription activation and primitive endoderm formation. siRNA-induced knockdown of Src likewise attenuates Lef/Tcf transcription and primitive endoderm formation in response to Wnt3a, implicating Src as a positive regulator of Wnt/β-catenin signaling. We discovered that Src binds dishevelled-2 (Dvl2), a key phosphoprotein in Wnt signaling, at two positions: an SH3-binding domain and a C-terminal domain. The Y18F mutant of Dvl2 attenuates the Wnt3a-stimulated Lef/Tcf-sensitive transcriptional response. Wnt3a stimulates Src docking to Dvl2 and activation of this tyrosine kinase. Activated Src, in turn, enhances Wnt activation of the canonical pathway. We show that Dvl2 and β-catenin are crucially important substrates for tyrosine phosphorylation in the canonical Wnt/β-catenin pathway.
Keywords: Wnt, Dishevelled, Src, β-catenin
Introduction
Wnt proteins are secreted glycoproteins that bind to members of the frizzled family of G-protein-coupled receptors and to co-receptor low density lipoprotein receptor related protein LRP5 and LRP6 (LRP5/6). Wnt signaling branches into at least three distinct pathways: canonical, non-canonical and planar cell polarity (PCP) (Liu et al., 2001; Willert et al., 2003; He et al., 2004; Wallingford and Habas, 2005; Katanaev et al., 2005; Malbon, 2005). Wnt signaling regulates numerous cellular pathways including embryonic development and adult tissue homeostasis, and when it is dysregulated, promotes cancer (Nusse, 2005; Wodarz and Nusse, 1998; Clevers, 2006; Glass and Karsenty, 2006; Wang and Wynshaw-Boris, 2004). Upon binding of Wnt to frizzled-1 (Fz1) and its co-receptor LRP5/6, a signal is transduced to the cytoplasmic phosphoprotein, dishevelled (Dsh/Dvl) (Yanagawa et al., 1995). Dsh is a key transducer of the Wnt signal that exerts effects at the plasma membrane, in the cytoplasm and in the nucleus (Malbon and Wang, 2006). Fly Dsh and three isoforms of mammalian Dvl proteins (Dvl1-Dvl3) all share several prominent, highly conserved domains: a Dsh-homology domain called DIX; a conserved sequence element with homology to the postsynaptic density protein PSD-95, Discs-large, and ZO-1, termed PDZ; and a Dsh, Egl10, Pleckstrin domain, termed DEP (Wharton, 2003). A proline-rich domain [a putative Src homology 3 (SH3) binding domain], conserved in all three Dvl isoforms, is located between the C-terminus of the PDZ domain and the N-terminus of the DEP domain (Penton et al., 2002). The extreme C-terminus of Dvl proteins is proline-rich and highly conserved, although its function remains unclear (Malbon and Wang, 2006).
Wnt3a binds Fz1 and co-receptor LRP5/6, and activates Dsh/Dvl. Dvl functions via multiprotein complexes that are known to include axin, the gene product of adenomatous polyposis coli (APC), glycogen synthase kinase-3β (GSK3β) and casein kinase-1α (Hart et al., 1998; Liu et al., 2002; Malbon, 2005). Dvl suppresses GSK3β activity, and unphosphorylated β-catenin accumulates in the nucleus where it binds to Lef/Tcf transcription factors and activates genes necessary for early development (Nusse, 1999). Dvl proteins are highly phosphorylated in response to Wnt stimulation and Dvl activity itself is reported to be controlled by multiple phosphorylation events (Willert et al., 1997; McKay et al., 2001; Sun et al., 2001). Thus, Dvl phosphorylation was implicated in the Wnt signaling pathway, especially with regard to serine/threonine protein kinases (Yanagawa et al., 1995; Kishida et al., 2001). We wondered what role, if any, tyrosine kinases, particularly Src family kinases, might play in Dvl biology and Wnt signaling.
Src family tyrosine kinases (TKs) also have important roles in controlling growth, proliferation and differentiation (Brown and Cooper, 1996; Cooper and Howell, 1993). Src family kinases are composed of six major regions/domains: an N-terminal membrane binding region; a `unique' domain; an SH3 domain that binds proline-rich sequences; a phosphotyrosine binding SH2 domain; a tyrosine kinase catalytic domain; and a C-terminal tail region (Brown and Cooper, 1996; Cooper and Howell, 1993). Src activity is regulated by intramolecular interactions. An interaction between the SH2 domain and the C-terminal tail, as well as an interaction between the SH3 domain and a polyproline-type helix in the SH2-kinase linker region, regulates kinase activity (Sicheri et al., 1997; Xu et al., 1997; Williams et al., 1997). Ligands docking to the SH2 and SH3 domains disrupt this autoinhibitory interaction and thereby catalyze TK activation (Moarefi et al., 1997; Briggs et al., 1997; Alexandropoulos and Baltimore, 1996). Many Src family kinase substrates themselves possess SH2 and/or SH3 ligands, which couple enzyme activation to substrate phosphorylation (Brown and Cooper, 1996; Porter et al., 2000; Miller, 2003).
In developmentally relevant signaling, Src is reported to elevate the expression (Karni et al., 2005) and phosphorylation at TyrY654 of β-catenin. The phosphorylation of Tyr654 blocks the E-cadherin–β-catenin interaction (Roura et al., 1999). In addition to Src, Fer, Fyn, transmembrane TK EGFR and c-Met, all downregulate E-cadherin-mediated adhesion via enhanced tyrosine phosphorylation of β-catenin (Lilien and Balsamo, 2005; Piedra et al., 2001). In chronic myeloid leukemia (CML) cells, oncogenic TK Bcr-Abl triggers tyrosine phosphorylation of β-catenin, stabilizes β-catenin levels and enhances nuclear signaling activation (Coluccia et al., 2007). In contrast to its role and actions directly on β-catenin, TK-based phosphorylation of other key elements of the Wnt/β-catenin `canonical' pathway remains poorly understood. In the current study, we demonstrate that Src positively regulates the Wnt/β-catenin pathway at Dvl2, which is a prominent locus for all Wnt signaling.
Results
Tyrosine kinase inhibitors suppress Wnt3a-stimulated Lef/Tcf transcriptional activation and formation of PE
Mouse F9 embryonic teratocarcinoma (F9) cells are totipotent, and can be stimulated to form primitive endoderm (PE) in response to Wnt3a. F9 cells, lacking Fz1 are resistant to Wnt3a, but upon expression of rat Fz1 display Wnt canonical signaling (i.e. Wnt3a-stimulated PE formation) and provide a novel model system (Liu et al., 2001; Yokoyama et al., 2007). Activation of Lef/Tcf-sensitive transcription was adopted as the primary read-out of the Wnt-stimulated canonical pathway (Clevers, 2006). To probe a possible role of TK in the Wnt canonical pathway, Wnt3a-stimulated Lef/Tcf-sensitive transcription was studied in cells pretreated with either genistein (a general TK inhibitor) or the more-selective Src family kinase inhibitor PP2 (Fig. 1A). Wnt3a stimulated activation of Lef/Tcf transcription more than ninefold. Genistein and PP2 treatments individually suppressed Wnt3a-induced Lef/Tcf transcription more than 50% (Fig. 1A). We also tested the effects of Src inhibition on the ability of Wnt3a to promote PE formation. Wnt3a has been shown to stimulate PE formation (Liu et al., 2001; Yokoyama et al., 2007). PE formation was assayed by measuring the expression of the PE marker cytokeratin endo A, using immunoblotting with TROMA-1 antibody (Yang and Petitte, 1994). Wnt3a stimulated the expression of PE marker and Wnt3a-stimulated PE formation was abolished by either genistein or PP2 (Figs 1B,C). Treatment of cells with either genistein or PP2 attenuated the level of activated phosphorylated Src (Src-P) (Fig. 1D), demonstrating the effectiveness of these inhibitors. Direct measurement of Src family TK activity confirms these results (supplementary material Fig. S1A).
Fig. 1.

Tyrosine kinase inhibitors block Wnt/β-signaling pathway. (A) Wnt3a-induced activation of Lef/Tcf-sensitive transcription is attenuated by inhibition of tyrosine kinase activity. Activation of Lcf/Tcf-sensitive transcription was assayed in mouse F9 teratocarcinoma cells co-transfected with rat Fz1 and Super8xTOPFlash (M50) reporter. Before a 30-minute Wnt3a stimulation, cells were preincubated in the presence or absence of the TK inhibitors, genistein or PP2, without or with purified Wnt3a for 8 hours. The luciferase gene reporter was assayed and is displayed relative to the unstimulated cells (set to `1'). The results shown are mean values ± s.e.m. from four independent experiments. (B) Wnt3a-induced formation of PE is attenuated by inhibition of tyrosine kinase activity. The formation of PE was assayed in F9 cells expressing rat Fz1. Cells were stimulated without or with purified Wnt3a for 3 days. Cells were cultured two more days and harvested. TK inhibitor (either genistein or PP2) was added to cultures after rat Fz1 transfection and PE formation was assayed by measuring of expression of PE-marker, cytokeratin endo A, using immunoblotting and staining with anti-TROMA-1 antibody. Blots were quantified using a calibrated scanner and the results are displayed as `fold' compared with control (time = 0, set to `1'). The results shown are mean values ± s.e.m. from ten independent experiments. (C) Representative immunoblots stained with the TROMA-1 antibody, derived from ten independent experiments. (D) Cells were preincubated with or without TK inhibitors for 30 min and then stimulated with Wnt3a for 8 hours in the presence or absence of the same inhibitor. Cell lysates were analyzed with activation-specific, anti-phospho-Src antibody (Tyr418-P, labeled p-Src) or anti-Src antibody (labeled Src).
Src knockdown suppresses Wnt3a-stimulated Lef/Tcf-sensitive transcription and PE formation
Based upon the chemical inhibitor studies, we adopted a knockdown strategy to test the role of one leading candidate, Src, in Wnt canonical signaling. The siRNA-induced knockdown of Src yields a >85% reduction in Src expression; GAPDH expression (as a control) in siRNA-treated cells, by contrast, was unaffected (Fig. 2A). Src knockdown suppressed the Lef/Tcf transcription response by >80% and essentially abolished PE formation in response to Wnt3a (Fig. 2B). Src appears to function as a positive regulator of Wnt/β-catenin signaling, operating to the level of Lef/Tcf transcription and PE formation. Knockdown of another Src family TK, Yes, also attenuated Lef/Tcf-sensitive transcription (supplementary material Fig. S1B). These same chemical TK inhibitors suppressed Wnt3a-stimulated Lef/Tcf transcription in human embryonic kidney 293 (HEK293) cells (supplementary material Fig. S2A). Knockdown of Src in HEK293 cells also reduced Wnt3a-stimulated Lef/Tcf-sensitive transcription (supplementary material Fig. S2A). Finally, TK inhibitors also suppressed activation of the Wnt canonical pathway in human mesenchymal stem cells (supplementary material Fig. S2B). Knockdown of Src stimulated, rather than inhibited, AP-1 transcription [a marker of PCP pathway activation (Bikkavilli et al., 2008; Weber et al., 2008)] in response to Wnt3a, whereas it inhibited NF-AT transcription [a marker of the Ca2+-cGMP pathway (Ahumada et al., 2002; Ma and Wang, 2007)] in response to Wnt5a (supplementary material Fig. S3A,B).
Fig. 2.

Knockdown of Src attenuates whereas overexpression of Src enhances Wnt/β-catenin signaling. (A) siRNA targeting Src inhibits Wnt3a activation of Lef/Tcf-sensitive transcription. F9 cells were treated with siRNAs targeting Src for 1 day before co-transfection of the cells with rat Fz1 and Super8xpTOPFlash plasmids. Cells were stimulated with or without Wnt 3a for 8 hours. The activity of the luciferase reported gene was assayed and the transcriptional activity displayed relative to the unstimulated cells (set to `1'). The results show mean values ± s.e.m. obtained from five separate experiments. Cell lysates were analyzed by immunoblotting; blots were stained with anti-Src antibody (or anti-GAPDH antibody as a loading control). (B) siRNA targeting Src inhibits Wn3a activation of PE formation. F9 cells were transfected with siRNA targeting Src 1 day before rat Fz1 transfection. Cells were treated with or without Wnt3a for 3 days and cultured for a further 2 days. Blots that were stained with the PE marker TROMA-1 antibody were quantified by the calibrated scanner and results were displayed as `fold' (time = 0, set to `1'). The results shown are mean values ± s.e.m. from ten independent experiments. (C) Overexpression of Src enhances Wnt3a-sensitive Lef/Tcf-sensitive transcription. F9 cells were co-transfected with rat Fz1, Super8xTOPFlash reporter, and a Src expression vector one day before cells were stimulated without or with purified Wnt3a for 8 hours. Cell lysates were assayed for Lef/Tcf luciferase transcription activity. Results are displayed relative to the unstimulated cell (set to `1'). The results are shown as mean values ± s.e.m. from six independent experiments. Statistical significance is indicated (*P<0.05; **P<0.01). Right panels display cellular content of Src and of GAPDH (as loading control). Representative immunoblots are shown. (D) Overexpression of Src alone stimulates β-catenin accumulation. Control (–) and Src-overexpressing (Src) F9 cells were lysed with RIPA buffer and cell lysates were analyzed for β-catenin content by immunoblotting with anti-β-catenin antibody or anti-GAPDH antibody (loading control). Blots representative of three separate experiments are displayed.
Src overexpression enhances Wnt/β-catenin signaling
If the hypothesis that Src modulates Wnt canonical signaling is correct, then overexpression of Src might enhance signaling in the Wnt canonical pathway. To test this hypothesis, cells were transiently transfected (Fig. 2C). Overexpression of Src enhances Wnt3a-stimulated Lef/Tcf transcription. Overexpression of Src was confirmed functionally by increased Src family TK activity, i.e. increased Src-P (the active form of Src) levels and enzyme activity (supplementary material Fig. S4A,B). GAPDH expression was used as a control for loading equivalence. Likewise, overexpression of Src stimulated an increase in the abundance of β-catenin (Fig. 2D). Overexpression of the Src family kinase Hck, similarly to Src overexpression, enhanced Wnt/β-catenin signaling (results not shown).
Src docks to the C-terminus of Dvl2
Similarly to their fly homologue Dsh, Dvl1, Dvl2 and Dvl3 display a putative SH3-binding domain (residues 370-376 of Dvl2) (Penton et al., 2002). Since knockdown of Src suppresses, whereas overexpression enhances, Wnt canonical signaling, we investigated whether the most abundant mammalian Dvl isoform (Dvl2) might dock Src (Lee et al., 2008). Dvl2 docking to SH2 and SH3 domains of Src, hematopoietic cell kinase (Hck), as well as to SH3 domains of Nck, Crk and Grb2 was investigated. F9 cell lysates were incubated with GST fusion proteins of each SH3 domain (Src, Hck, Nck, Grb2 and Crk; Fig. 3A). The SH3 GST fusion proteins were immobilized and analyzed for bound Dvl2 by SDS-PAGE and immunoblotting. Blots were probed with antibodies against either Dvl2 or GST. Docking of Dvl2 was prominent to Src SH2/3 domains and Hck SH3 domains. By contrast, Dvl2 showed relatively weak docking to Grb2 SH3, Crk SH3, Hck SH2 or Src SH2 domains. Dvl2 did not dock to GST alone or to Nck SH3 domain. The ability of the SH3 domains of Src or Hck, but not Nck to enable docking to Dvl2 reflects the specificity of the scaffold-kinase interaction.
Fig. 3.

Src docking to Dvl2: analysis of potential docking domains. (A) Src SH2/SH3 and Hck SH3 domains display docking of Dvl2. Docking of Dvl2 to SH3 domains was analyzed by use of immobilized GST-SH3 domain fusion proteins. Cell lysates were incubated with each of the immobilized GST SH3 domains indicated as well as with GST itself (as a control) at 4°C for 1 hour. Bound proteins were analyzed by SDS-PAGE, blotted, and stained with either anti-Dvl2 (top panel) or anti-GST antibodies (bottom panel). Blots representative of three separate experiments are displayed. (B) Dvl2 domains dock Src and Hck tyrosine kinases. Cell lysates were incubated with an immobilized Dvl2 domain: 11-93 (DIX), 433-507 (DEP), 267-309 (PDZ), 356-378 (putative SH3 binding containing region, SH3B), 511-736 (C-terminus) and GST itself, at 4°C for 1 hour. Proteins docking to the Dvl2 domains were resolved by SDS-PAGE and analyzed by immunoblotting and staining with anti-Src, anti-Hck and anti-GST (loading control). Blots representative of four independent experiments are displayed. (C) Mutagenesis of the Dvl2 SH3-binding domain abolishes docking of Src and Hck. Cell lysates were incubated with either GST itself or GST-putative SH3-binding containing region (356-378, SH3B) or GST-mutant SH3 binding containing region (370-376, PxxPxxP to AxxAxxA in 356-378, SH3B-PA mutant). Bound proteins were analyzed by SDS-PAGE and immunoblotting, and stained with anti-Src, anti-Hck and anti-GST antibodies. Blots representative of three independent experiments are displayed.
The Src TKs possess SH2 and SH3, as well as catalytic domains. Intramolecular autoinhibitory interactions between these domains tightly regulate Src activity (Brown and Cooper, 1996; Cooper and Howell, 1993). Analysis of the primary sequence of Dvl2 suggested that the amino acid region 370-376, displaying a consensus sequence for a class I core SH3-protein-binding motif RTEPVRP (Penton et al., 2002), was the most likely binding site for SH3 domains. The ability of Src to dock to well-known domains of Dvl2 (DIX, PDZ, DEP) as well as residues 356-378 was tested. Whole-cell F9 lysates were incubated with GST-fusion proteins to six Dvl2 domains: GST (0); GST-DIX (11-93); GST-PDZ (267-309), GST-putative SH3-binding region (356-378), GST-DEP (433-507), or GST-polyproline-rich C-terminus (511-736). Of interest, the C-terminal 511-736 sequence of Dvl2 displays 26 prolyl residues. Following incubation, GST fusion proteins were immobilized and their bound proteins analyzed by SDS-PAGE and immunoblotting. Src displayed prominent docking to the 356-378 putative SH3-binding region and the C-terminal 511-736 domains of Dvl2 (Fig. 3B). By contrast, DIX, DEP and PDZ domains of Dvl2 displayed little docking of Src. Immobilized GST itself (control) failed to dock TKs at all. Dvl2 region 356-378 and the polyproline-rich C-terminus region constitute dominant sites of docking for Src (as well as Hck). Proline-to-alanine substitutions (370-376, PxxPxxP to AxxAxxA) of the Dvl2 SH3-binding domain resulted in the loss of docking for Src (as well as Hck) (Fig. 3C). We tested whether previous Wnt3a treatment might enable or reveal docking of Dvl2 to the immobilized Src SH2 domain, but no docking to the SH2 domain was observed, not even after Wnt3a treatment (supplementary material Fig. S5).
Time course of Src docking to Dvl2 in response to Wnt3a
We next asked whether docking of Src to Dvl2 was dynamic, that is, could it be regulated by Wnt3a? Protein-protein interactions were analyzed by SDS-PAGE and immunoblotting, making use of antibodies specific for Dvl2, Src and Src-P antibodies (phosphorylated Tyr418) (Fig. 4A). Cellular Src was not influenced by Wnt3a. The docking of Src to Dvl2, by contrast, was found to be Wnt3a dependent (Fig. 4A). Src docking to Dvl2 increased several fold within 30 minutes of Wnt3a treatment. The ratio of Src to Dvl2 in the pull-down samples was quantified; the ratio increased over the first 45 minutes of Wnt3a stimulation, and declined thereafter (Fig. 4B). Does docking of Src to Dvl2 in response to Wnt3a increase TK activity? The amount of phosphorylated, activated (Tyr418-P) Src in the pull-down samples increased about threefold in response to Wnt3a (Fig. 4A,B). Src family TK activity was assayed directly in lysates of Wnt3a-treated and untreated cells, using the optimum peptide substrate. Total Src family TK activity was also shown to increase within 30 minutes of Wnt3a, declining thereafter, in a time course that agrees well with the docking of Src to Dvl2, and phosphorylation or activation of Src in response to Wnt3a (Fig. 4B, supplementary material Fig. S6).
Fig. 4.

Wnt3a stimulates Src docking to Dvl2. (A) time-course of Src docking to Dsh2. F9 cells expressing rat Fz1 were either untreated (time = 0) or treated with purified Wnt3a for 5 to 120 min. Cell lysates (2 mg) were immunoprecipitated with anti-Src antibody. Bound proteins were resolved by SDS-PAGE, immunoblotting, and made visible by staining with either anti-Dvl2 or anti-Src, or activation specific, anti-Src-P antibodies. Blots represent five independent experiments. (B) Quantitative analysis of time course of Src docking to Dvl2 and activation of Src. Src docking to Dvl2 was quantified in the blots by calibrated scanning. The results are displayed as `fold' over zero time (i.e. time = 0, set to `1'). Upper panel shows Src activity associated with Dvl2 isolated by pull-down from extracts prepared from cells stimulated with Wnt3a for 0 to 120 minutes. The tyrosine phosphorylation (Tyr418) of Src reflects activation of Src and is displayed as `fold' over zero time (time = 0, set to `1'). Results are displayed as the mean values ± s.e.m. from five independent experiments. (C) Src activity is increased in response to Wnt3a stimulation. Rat Fz1-expressing F9 cells were stimulated with Wnt3a (0-2 hours). Cells extracts were subjected to pull-down of Dvl2-based complexes. Src docked to Dvl2 was immunoprecipitated along with Dvl2 in the pull-downs. The pull-downs were washed with PBS containing 0.5 % NP-40, and washed again with 50 mM Tris-HCl (pH 7.5). Src kinase activities associated with the Dvl2-based pull-downs were assayed on phosphocellulose paper templates using the Src peptide (AEEEIYGEFEAKKKKG) as a substrate. 32P incorporation into peptide was determined. Results are mean values ± s.e.m. from five independent experiments. (D) Wnt-antagonist DKK1 abolished the activation of Src in response to Wnt3a. F9 cells expressing rat Fz1 were either untreated or pretreated with DKK1 (400 ng/ml) for 1 hour, before stimulation with or without Wnt3a (20 ng/ml) for 30 minutes in the presence or absence of DKK1. Cell extracts were subjected to pull-down of Src, which associated with Dvl2 by anti-Dvl2 antibody. Src kinase activity of Dvl2 pull-downs was assayed using the Src peptide, as described previously. Results shown are mean values ± s.e.m. from seven independent experiments.
Ligands for SH2 or SH3 domains are known to disrupt autoinhibitory interactions and thereby stimulate Src activity (Alexandropoulos and Baltimore, 1996; Moarefi et al., 1997; Briggs et al., 1997). The catalytic activity of Src in Dvl2 pull-down samples from cells treated with or without Wnt3a was measured over 2 hours (Fig. 4C). Samples from untreated cells displayed little TK activity, whereas Wnt3a provoked increased Src activity as early as 5 minutes after stimulation, which peaked at 45 minutes (Fig. 4C). The time courses of Src docking to Dvl2 and of TK activation in response to Wnt3a agree well. We next considered whether TK activation of Dvl2 pull-down samples in response to Wnt3a be blocked by Dickkopf homologue 1 (DKK1), a well-known Wnt antagonist (Nusse, 2001). Cells were pretreated with or without DKK1 and then stimulated with Wnt3a in the continued presence versus absence of DKK1. DKK1 alone did not alter TK activity of whole-cell lysates, whereas Src family TK activity in response to Wnt3a stimulation was reduced ∼50% by DKK1 (results not shown). Src family TK activity associated with the Dvl2 pull-down samples was unaffected by DKK1 alone, increased sevenfold in response to Wnt3a and was blocked by DKK1 when DKK1 was included with Wnt3a (Fig. 4D). The ability of DKK1 to specifically suppress Dvl2-associated TK activity in Wnt3a-stimulated cells precludes the possibility that some growth factor other than the purified Wnt3a is responsible for the activation of Src.
Knockdown of Dvl2 blocks Wnt3a-induced activation of Src
Dvl2 docks and activates Src, so we asked whether knockdown of Dvl2 affects Src family TK activity in response to Wnt3a. Dvl2 expression was suppressed effectively by use of siRNA (Fig. 5A). Although Wnt3a provoked the activation of Src family TK activity (Fig. 4 and Fig. 5A), it failed to stimulate this TK activity in cells first made deficient in Dvl2 (Fig. 5A). The knockdown of Dvl2 provoked a decline, rather than an increase, in Src family TK activity in response to Wnt3a.
Fig. 5.

Knockdown of Dvl2 blocks activation of Src in response to Wnt3a. (A) Dvl2 knockdown eliminates Wnt3a-stimulated activity of Src family TKs. F9 cell were transfected with or without siRNA targeting of Dvl2 1 day before rat Fz1 transfection. Cells were untreated or treated with Wnt3a for 30 minutes. Assay for Src family TK was performed using cell lysates, as described previously. Results are shown as the mean values ± s.e.m. from seven independent experiments. Statistical significance is indicated (*P<0.005). Cell lysates were analyzed by immunoblotting and the blots were stained with either anti-Dvl2 antibody or anti-GAPDH antibody (loading control). (B) Effect of Src knockdown on Wnt3a-stimulated Lef/Tcf-sensitive transcription can be rescued by expression of Src. F9 cells stably expressing rat Fz1 and M50 were transfected with siRNA targeting Src 1 day before transfection with either wild-type (left) or kinase-dead (right) Src. A day later, cells were either unstimulated or stimulated with Wnt3a for 7 hours. Assay for Lef/Tcf-sensitive transcription was performed. Results shown are mean values ± s.e.m. from five independent experiments. Cell lysates were analyzed by immunoblotting and the blots were stained with either anti-Src antibody or anti-GAPDH antibody (loading control).
Kinase-dead Src partially rescues the effects of Src knockdown
Not all pathways involving Src might require its kinase activity. In osteoclasts, expression of kinase-deficient Src partially rescues the Src-knockdown phenotype (Schwartzberg et al., 1997), although Src kinase activity is essential for function of osteoclasts (Miyazaki et al., 2004). We tested whether the effects of Src knockdown could be influenced by expression of kinase-dead Src. First, we probed the ability of wild-type Src to rescue lesions provoked by the Src deficiency; expression of wild-type Src restores the ability of Wnt3a to stimulate Lef/Tcf sensitive transcription (Fig. 5B, left panel). Expression of kinase-dead Src (K295R mutant) was similarly investigated for its ability to rescue Src-deficient cells. As noted in the Src-deficient osteoclasts (Schwartzberg et al., 1997), expression of kinase-dead Src yields some rescue of the Src deficiency (Fig. 5B, right panel). We found that Hck also docks to Dvl2 (Fig. 3B) and that siRNA targeting Hck also partially blocks Wnt3a-induced Lef/Tcf transcription (results not shown). Inhibition of Lef/Tcf transcription by Src knockdown was partially restored by expression of Hck (result not shown). Constitutively active Src (Y527F) was also tested. At the lowest levels of Y527F Src expression tested, basal Lef/Tcf-sensitive transcription activity increased ∼twofold (supplementary material Fig. S7A). Expression of kinase-dead Src modestly enhanced the transcriptional response to Wnt3a (supplementary material Fig. S7B). Knockdown of Src therefore attenuates, whereas constitutively active Src potentiates, the ability of Wnt3a to activate the Wnt canonical pathway.
Src phosphorylates Dvl2
Was Dvl2 itself was a substrate for Src-catalyzed phosphorylation? Recombinant Dvl2 (rDvl2), expressed and purified from insect Sf9 cells, was incubated in vitro in the absence versus presence of purified recombinant Src (rSrc) in a standard phosphorylation reaction buffer containing [γ-32P]ATP. Src catalyzed the phosphorylation of Dvl2; phosphorylation of Dvl2 was demonstrated by autoradiography of the resolved proteins first separated by SDS-PAGE. Addition of purified rHck to the kinase reaction mixture also supported phosphorylation of Dvl2 (supplementary material Fig. S8A). To better define the nature of the site(s) on Dvl2 phosphorylated by Src, GST-fusion proteins of Dvl2 domains were incubated with purified rSrc. Src catalyzed the phosphorylation of GST-fusion proteins containing DIX, PDZ and DEP domains of Dvl2 (Fig. 6A). GST alone and the GST-SH3 fusion protein, by contrast, displayed weak phosphorylation by Src (Fig. 6A). Similar in vitro experiments performed using purified rHck yielded phosphorylation of the same domains as Src (supplementary material Fig. S8B). Mass-spectrometry-based peptide mapping was used to identify the sites of Dvl2 phosphorylation. Phosphorylated rDvl2 was digested with trypsin and subjected to analysis using API QSTAR Pulser liquid chromatography/mass spectrometry (LS/MS). Phosphopeptides were identified and tyrosine phosphorylation established. Five phosphorylation sites on Dvl2 were detected [Y18, Y27 (DIX domain), Y275, Y295 (PDZ domain) and Y463 (DEP domain)], in agreement with the GST-fusion protein analysis. Phosphorylation studies performed with either Src or Hck yielded similar sites of phosphorylation (data not shown).
Fig. 6.

Src phosphorylates Dvl2 in vitro and in vivo. (A) Src phosphorylates Dv2 domains in vitro. GST fusions proteins of Dvl2 domains were purified and incubated with purified rSrc in phosphorylation reaction buffer containing [γ-32P]ATP (500-1000 c.p.m./pmol) at 30°C for 1 hour. Phosphorylation reactions were terminated by addition of SDS sample buffer and then analyzed by SDS-PAGE and autoradiography. Samples were separately analyzed by SDS-PAGE and subsequent immunoblotting with anti-GST antibody to establish loading equivalence (bottom panel). Results shown are representative of four independent experiments. (B) Dvl2 displays tyrosine phosphorylation upon stimulation of F9 cells with Wnt3a. F9 cells expressing rat Fz1 were either untreated or treated with Wnt3a for indicated time. Cell lysates were subjected to immunoprecipitation by anti-Dvl2 antibody. Bound protein was analyzed with either anti-Dvl2 or anti-Tyr-P-specific antibodies. Blots shown are representative of four independent experiments. (C) Time course of tyrosine phosphorylation of β-catenin in response to Wnt3a. F9 cells expressing Rat Fz1were treated with or without Wnt3a for indicated time. Cells were lysed with RIPA buffer and these lysates incubated with ConA-Sepharose at 4°C for 1 hour. Supernatant fractions were further incubated with ConA-Sepharose at 4°C for another hour. Supernatant fractions (2 mg protein) were subjected to immunoprecipitation by anti-β-catenin antibody. Bound proteins were analyzed by SDS-PAGE and subjected to immunoblotting. The blots were stained with either anti-β-catenin antibody (left panel, top) or anti-Tyr-P-specific antibody (left panel, bottom). Right panel shows abundance of β-catenin in the subcellular cytosol-enriched fractions, as well as GAPDH content (as a loading control). Blots displayed are representative of six independent experiments. (D) Alanine-substitution mutagenesis of the Dvl2 SH3-binding domain attenuates Lef/Tcf-sensitive transcription. F9 cell were co-transfected with rat Fz1, Super8xTOPFlash (M50) reporter and either wild-type Dvl2 or a Dvl2 with a mutation of the SH3 binding domain (PxxPxxP to AxxAxxA). 36 hours later, the cells were assayed for Lef/Tcf-sensitive transcription (left panel). Right panel shows the expression of Dvl2 (anti-HA blot) and GAPDH (as loading control), as assayed by immunoblotting. Subcellular cytoplasmic-enriched fractions prepared from cells transfected with or without an expression vector harboring either wild-type Dvl2 or the Dvl2 mutant lacking a proper SH3 domain were subjected to pull-down with anti-β-catenin antibody. The pull-down samples were subjected to SDS-PAGE, immunoblotting, and the blots stained with anti-β-catenin or anti-Tyr-P-specific antibodies (right panel, bottom).
Wnt3a stimulates Dvl2 tyrosine phosphorylation in cells
Was Dvl2 tyrosine phosphorylated in response to Wnt3a? Proteins of pull-down samples of Dvl2 from lysates of untreated and Wnt3a-treated cells were resolved and then stained both with anti-Dvl2 and antibody against phosphorylated tyrosine (Fig. 6B). Wnt3a stimulated Dvl2 phosphorylation in a time-dependent manner (Fig. 6B). Pull-downs using anti-tyrosine-P antibody likewise demonstrated an increase in Dvl2 content (results not shown). Activation of Src alone has been reported to elevate the expression of β-catenin and to result in the phosphorylation of Tyr654 of β-catenin (Karni et al., 2005). Pull-downs of β-catenin from the cytoplasmic subcellular fraction of Wnt3a-treated cells were resolved by SDS-PAGE and stained with anti-tyrosine-P and anti-β-catenin antibodies. The tyrosine phosphorylation content of β-catenin was also increased (for up to 1 hour) after Wnt3a stimulation (Fig. 6C). Cytoplasmic levels of β-catenin continued to increase for several hours; the phosphotyrosine content of the molecule declined after 1 hour of stimulation by Wnt3a.
Expression of mutant of SH3 binding domain of Dvl2 reduces Lef/Tcf transcription
We show that Src docks to the SH3-binding domain of Dvl2, a docking site for Src and its family of TKs. To probe the role of the SH3-binding domain of Dvl2, proline-to-alanine substitutions (370-376, PxxPxxP to AxxAxxA) of Dvl2 were prepared. Basal Lef/Tcf-sensitive transcription was assayed in cells overexpressing wild-type versus mutant Dvl2 (Fig. 6D). Expression of wild-type Dvl2 increases Lef/Tcf sensitive transcription and phosphotyrosine content of β-catenin, whereas expression of the Dvl2 lacking the SH3-binding domain displayed far less of both read-outs at the same level of expression (Fig. 6D).
Y18F substitution in Dvl2, similarly to knockdown of Src, attenuates Lef/Tcf transcription
Dvl2 levels were knocked down and the ability of YF mutants (Dvl2 Y18F, Y27F, Y275F, Y295F and Y463F) versus wild-type Dvl2 to rescue Wnt3a-stimulated Lef/Tcf-sensitive transcription was probed. In the Dvl2-knockdown cells, expression of wild-type Dvl2 restored the Wnt3a-stimulated Lef/Tcf-sensitive transcription, whereas the Y18F Dvl2 mutant, in particular, attenuated the response by more than half (Fig. 7A). The Y27F and Y275F mutants, by contrast, displayed a much-reduced ability to rescue the Wnt3a-stimulated response, and the Y295F and Y463F mutants displayed none. The Y18F Dvl2 was likewise unable to rescue the Wnt3a-stimulated tyrosine phosphorylation of β-catenin (Fig. 7B). It is important to note that knockdown of Src itself attenuated Wnt3a-stimulated tyrosine phosphorylation of Dvl2 (Fig. 7C) as well as of cytosolic β-catenin (Fig. 7D).
Fig. 7.

Y-F mutants of Dvl2 inhibit Lef/Tcf transcription and tyrosine phosphorylation of β-catenin. (A) Tyrosine-to-phenylalanine substitution mutants of Dvl2 inhibit Lef/Tcf transcription. F9 cells stably expressing rat Fz1 and M50 were transfected with siRNA targeting Dvl2 1 day before transfection with either wild-type or a YF mutant of Dvl2 (Y18F, Y27F, Y275F, Y295F and Y463F). On the following day, cells were stimulated with Wnt3a for 7 hours. Assay for Lef/Tcf-sensitive transcription was performed. Results shown are mean values ± s.e.m. from four independent experiments. (B) Tyrosine-to-phenylalanine mutagenesis of Dvl2 inhibits tyrosine phosphorylation of β-catenin. F9 cells were transfected with siRNA targeting Dvl2. On the next day, cells were transfected with rat Fz1 and either wild-type Dvl2 or Y18F mutant of Dvl2. 24 hours later, cells were stimulated with Wnt3a for 1 hour. Cytoplasmic fractions of cells were subjected to immunoprecipitation with β-catenin antibody. Precipitated proteins were analyzed by either anti-tyrosine-P antibody or anti-β-catenin antibody. Blots shown are representative of five independent experiments. (C,D) Src knockdown attenuates tyrosine phosphorylation of Dvl2 and of β-catenin. F9 cells were transfected siRNA targeting Src 1 day before rat Fz1 transfection. On the following day, cells were stimulated with or without Wnt3a for 1 hour. Proteins were immunoprecipitated by either anti-Dvl2 antibody (C) or anti-β-catenin antibody (D). Bound proteins were analyzed by SDS-PAGE and subjected to immunoblotting. The blots were stained with anti-Dvl2 (C) or anti-β-catenin antibody (D) and anti-Tyr-P-specific antibody (C,D). Blots shown are representative of eight independent experiments.
Src knockdown abolishes Wnt3a-dependent accumulation of nuclear β-catenin
Finally, we investigated whether Src knockdown itself modulated trafficking of β-catenin – a process that we characterized earlier (Yokoyama et al., 2007). In the absence of Wnt3a stimulation, suppression of Src alone significantly reduced the amount of nuclear β-catenin. Wnt3a stimulates the accumulation of β-catenin in the nucleus (Yokoyama et al., 2007), but this increase was abolished in cells in which Src expression was knocked down by siRNA (Fig. 8).
Fig. 8.
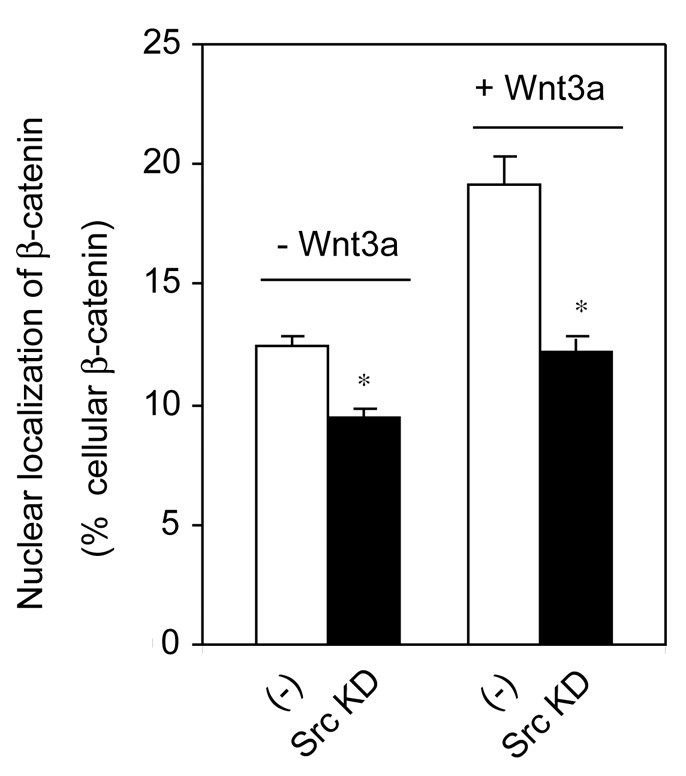
Knockdown of Src attenuates nuclear localization of β-catenin in response to Wnt3a. F9 cells were treated with or without Src-targeting siRNA. 24 hours later, cells were transfected with rat Fz1. Cells were stimulated with or without Wnt3a for 6 hours. Whole-cell lysates were prepared, subfractionated and the nuclear β-catenin analyzed. Fractions were analyzed by SDS-PAGE and immunoblotting with anti-β-catenin antibody. The relative amounts of nuclear β-catenin were established densitometrically by calibrated scanning, as reported earlier. Results shown are mean values ± s.e.m. from four independent experiments. Statistical significance is indicated (*P<0.001).
Discussion
Wnt regulation of gene transcription requires dynamic multiprotein complexes, that include APC, axin and the phosphoprotein Dsh/Dvl, as well as many other proteins (Hart et al., 1998; Liu et al., 2002). Dishevelled proteins have been shown to be downstream of Frizzled/LRP6/G-protein and upstream of GSK3β (Katanaev et al., 2005; Schwarz-Romond et al., 2005), functioning prominently in three Wnt-signaling paradigms: the PDE6-cGMP-Ca2+ pathway, the planar cell polarity pathway and in the Wnt/β-catenin/Lef-Tcf-sensitive transcription (canonical) pathway. All Dvl proteins display three highly conserved domains in their protein sequences: DIX, DEP and PDZ. The DIX domain of Dvl2 is crucial for its ability to recruit axin and for DIX-dependent dynamic self-association of Dvl2 (Schwarz-Romond et al., 2007a; Schwarz-Romond et al., 2007b). The PDZ domain is believed to recruit Dvl2 to Frizzled proteins (Wong et al., 2003). The DEP domain of Dvl has been shown to be essential for Dvl2-mediated signaling (Schwarz-Romond et al., 2007b; Wong et al., 2000). The current study is the first to demonstrate Src as a new docking partner of Dvl2, which is necessary for proper functioning of Wnt canonical signaling. Src is shown to act as a positive regulator of Wnt/β-catenin signaling. Previously, it was reported that tyrosine phosphorylation of β-catenin blocks its recruitment by the axin-GSK3β complex and thereby stimulates Tcf-sensitive transcription (Coluccia et al., 2007). In the current work, overexpression of Src alone is shown to increase the cellular abundance of β-catenin, whereas TK inhibitors or siRNA targeting Src decreases cellular content of β-catenin. Src enhances trafficking of β-catenin from cytoplasm to nuclei: knockdown of Src reduces normal trafficking of β-catenin to the nucleus in response to Wnt3a. Our observations illuminate important aspects of the previous finding that once phosphorylated, β-catenin translocates to nuclei and binds to Tcf transcription factors (Coluccia et al., 2007).
The results presented here shed light on earlier observations on TK and development. In Caenorhabditis elegans, Src family kinases were proposed to collaborate with Wnt pathway elements to specify cell fate development via phosphorylation of downstream molecules (Bei et al., 2002). Dsh-2 has been localized to the borders crucial for asymmetrical cell division in development. Dsh proteins differentially participate in aligning the spindles of EMS and Abar, which vary with respect to their regulation by Src during spindle orientation (Walston et al., 2004). Our data are the first to demonstrate convergence of Wnt signaling and tyrosine kinase signaling at a single well-characterized locus, Dvl2. Src docks to Dvl2 in a dynamic, Wnt3a-dependent manner that provokes activation of Src TK activity. Dvl2 knockdown suppresses the activation of Src family TK activity in response to Wnt3a. Treating cells with the Wnt antagonist DKK1 abolished this TK activation that was stimulated in response to Wnt3a.
At least two prominent components of the multiprotein complexes – Dvl2 and β-catenin – are phosphorylated on tyrosine residues in response to activation of the Wnt canonical pathway. The five tyrosine residues (Y18, Y27, Y275, Y295 and Y463) phosphorylated were identified by mass spectrometry. Src family TK can phosphorylate in vitro two tyrosine residues (Y18 and Y27) of the DIX domain, two residues (Y275 and Y295) of the PDZ domain, and a single residue (Y463) in the DEP domain. Tyrosine phosphorylation of Y18, Y27 and Y275 appears to contribute, in some complex manner, to the ability of Src to enhance Wnt3a/β-catenin signaling.
Src is expressed in osteoblasts as well as in osteoclasts; expression in both cell types is required for normal bone resorption. In Src-knockout mice, the osteoclast lineage is intact, but the cells are functionally impaired and unable to resorb bone, provoking osteopetrosis (Soriano et al., 1991; Lowe et al., 1993). In osteoclasts, expression of kinase-deficient Src rescued the Src–/– phenotype (Schwartzberg et al., 1997). For osteoclasts, by contrast, Src kinase activity is essential for their normal function (Miyazaki et al., 2004). Our data suggest that TK activity is not dispensable for the normal Lef/Tcf-sensitive transcriptional response to Wnt3a because inhibition of Src activity sharply attenuates Wnt/β-catenin signaling. Furthermore, overexpression of stabilized β-catenin (S33Y β-catenin) does not completely rescue the inhibition of Lef/Tcf transcription stimulated in response to either knockdown of Src or Src family TK inhibitor (PP2). Taken together, these observations suggest that Src targets Dvl2 in the canonical Wnt pathway (results not shown).
Dvl2 binds Src in a Wnt3a-dependent manner. Dvl2 docks primarily to the SH3 domain of Src. The prominent region of Dvl2 through which Src appears to dock in response to Wnt3a is localized between the PDZ domain and the DEP domain, a region that is proline-rich, and possesses a consensus sequence for a class I core SH3-binding motif, RTEPVRP (Penton et al., 2002). Dvl alleles in Drosophila containing mutations in this same motif strongly disrupt Wnt (Wingless) signaling (Penton et al., 2002). Our data demonstrate that residues 370-376 of Dvl2 function as a bona fide SH3-binding domain in canonical Wnt signaling. Docking of Src to Dvl2 through this SH3-binding domain and C-terminus proline-rich domain increased in response to stimulation by Wnt3a. Indeed, disruption of this Dvl2 SH3-binding domain inhibits Lef/Tcf-sensitive transcription. We propose that, in the absence of Wnt, Src is found in its standard autoinhibited, inactive conformation (Fig. 9). Proteomic analysis reveals Dvl2 to be a constituent of large multiprotein complexes (Yokoyama et al., 2007). Docking of Src to Dvl2 SH3-binding domains disrupts Src autoinhibition (Fig. 9), resulting from SH3 domain interaction with a linker region of SH2/SH3 domains (Alexandropoulos and Baltimore, 1996; Moarefi et al., 1997; Briggs et al., 1997). Docking to Dvl2 relieves Src autoinhibition, enabling phosphorylation of Src substrates (Brown and Cooper, 1996; Porter et al., 2000; Pellicena and Miller, 2001; Miller, 2003), including Dvl2. Src docks to Dvl2, is catalytically activated through docking, uses both Dvl2 as well as β-catenin as substrates, and acts as a positive regulator of Wnt canonical signaling. All facets of these Src-based functions are provoked in response to Wnt3a. Thus, Dvl2 both recruits a tyrosine kinase to its multiprotein complexes and activates it in a well-defined spatiotemporal environment intimate to Wnt signaling.
Fig. 9.

Wnt3a stimulates Src docking to, activation by, and phosphorylation of Dvl2. In this working model, at resting state (in the absence of Wnt stimulation), Src family TKs are in an autoinhibitory `inactive' confirmation. For Src family kinases, two intramolecular interactions tightly regulate enzymatic activity: an interaction between the SH2 domain and the C-terminal tail and an interaction between the SH3 domain and a polyproline type II helix in the SH2-kinase linker region. Wnt3a stimulation of Frizzled-1 and LRP5/6 activates the `canonical' pathway in which multi-protein complexes that include Dvl2 docking via PDZ ligand and PDZ-binding domains. Our results indicate that the SH3 domain of Src family kinase binds to Dvl2 SH3-binding domain (residues 370-376) as well as to a larger, proline-rich C-terminal domain (511-736; proline-rich binding domain, PRO-RD) of Dvl2, or both. Through docking to Dvl2, autoinhibitory conformation of Src is released, disrupted by the SH3 domain and a polyproline type II helix in the SH2-kinase linker region. Wnt3a not only induces the docking of Src to Dvl2, but also release of Src from autoinhibition (i.e. activation), as well as Src-catalyzed phosphorylation of Src substrates (including Dvl2 in the DIX, PDZ and DEP domains) found in the Dvl2-based multiprotein complexes. Thus, Src is a positive regulator of Wnt canonical signaling, which acts by docking to and being activated by Dvl2 in response to Wnt stimulation. Red circles represent tyrosine phosphorylation sites. The schematic of the multiprotein complexes has been simplified. Axin, GSK3β, APC and many other components of the multiprotein complexes are not identified individually in this schematic.
Materials and Methods
Materials
The following reagents were purchased from the indicated commercial supplier(s): anti-Hck, anti-tyrosine-P (4G10), and anti-Src (GD11) from Upstate Biotechnology (Lake Placid, NY); anti-Dvl2 (10B5), anti-Src (SC-18), anti-β-catenin (12F7) and anti-c-Yes (F7) antibodies from Santa Cruz Biotechnology (Santa Cruz, CA); anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody from Abcam (Cambridge, MA); anti-β-catenin antibody from Sigma (St Louis, MO); anti-cytokeratin endoA antibody TROMA-1 from the University of Iowa Developmental Studies Hybridoma Bank (Iowa City, IA); anti-glutathione S-transferase (GST) and glutathione-coupled agarose beads from Molecular Probes/Invitrogen (Eugene, OR); ConA-Sepharose from GE Healthcare (Piscataway, NJ); Immobilon membrane from Millipore (Bedford, MA); purified, biologically active Wnt3a and DKK1 from R&D Systems (Minneapolis, MN). siRNAs targeting Src, Hck, Dvl2 and a scrambled sequence `control' siRNA were purchased from Ambion (Austin, TX). siRNA targeting Yes was designed and purchased from Santa Cruz Biotechnology. The expressing vector harboring the Src (FLAG-tagged, wild-type, kinase-dead K295R-Src, and constitutively active Y527F-Src) and Hck were kind gifts from W. Todd Miller (Department of Physiology and Biophysics, SUNY at Stony Brook, Stony Brook, NY).
Cell culture and transfection
The mouse F9 teratocarcinoma cells (F9) cells were obtained from ATCC collection (Manassas, VA) and grown in Dulbecco's modified Eagle's medium (DMEM) as described previously (Yokoyama et al., 2007). Cells were transfected transiently with an expression vector (pCDNA3) harboring the rat Fz1 using Lipofectamine (Invitrogen, Carlsbad, CA), according to the commercial instructions. HEK293 cells were grown in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin at 37°C in 5% CO2. The human mesenchymal stem cells (hMSCs) were cultured in mesenchymal stem cell growth medium (MSCGM) supplemented with L-glutamine, penicillin, streptomycin and serum according to the protocol provided by Lonza Biosciences (Walkersville, MD). hMSCs were transfected by using the Nucleofection technique (Haleem-Smith et al., 2005). For some experiments, hemagglutinin (HA)- and green fluorescence protein (GFP)-tagged mouse Dvl2 and mutant of SH3 binding domain (residues 370-376, proline to alanine mutation) in Dvl2 were transfected to F9 cells. Site-directed mutagenesis of mouse Dvl2 (residues 370-376, PxxPxxP to AxxAxxA) was carried and mutations were confirmed by automated DNA sequencing.
Lef/Tcf-sensitive transcriptional reporter gene assay
Luciferase assay was carried out as previously described (Yokoyama et al., 2007). F9 cells were grown on 12-well plates and co-transfected with rat Fz1 and Super8xTOPFlash (M50) (Seto and Bellen, 2006). After 1-2 days of transfection, cells were treated with Wnt3a (20 ng/ml) for up to 8 hours. For some experiments, F9 cells coexpressing rat Fz1 and M50 were stimulated with Wnt3a for 8 hours in the presence of genistein (25 μM) or PP2 (1 μM), respectively. For overexpression of Src or Hck, F9 cells were co-transfected with expression vector harboring rat Fz1, Src or Hck and Super8xTOPFlash (M50). After 32 hours of transfection, cells were lysed and further analyzed. Lef/Tcf transcription activity was determined using cell lysates and displayed to relative to unstimulated cells (set to `1').
Formation of primitive endoderm (PE)
Cells were stimulated with purified Wnt3a (20 ng/ml) for 3 days with or without the TK inhibitors, genistein (25 μM) or PP2 (1 μM). Cells were cultured for a further 2 days and then were lysed in RIPA buffer. PE formation (expression of the PE marker protein cytokeratin endoA) was determined as described previously, using TROMA-1 monoclonal antibody (Yokoyama et al., 2007). Formation of PE was displayed as fold to relative to unstimulated cell (set to `1'). For knockout of Src, siRNA targeting Src was transfected 1 day before transfection of rat Fz1.
Immunoblotting and immunoprecipitation
Protein concentration was determined by use of the Bradford assay. Proteins were analyzed by SDS-PAGE and immunoblotting (Yokoyama et al., 2007). Immune complexes were made visible using a horseradish-peroxidase-conjugated, secondary antibody in tandem with ECL chemiluminescence. For immunoprecipitation experiments, cells were washed with phosphate-buffered saline (PBS) twice, and were lysed in RIPA buffer. Cell lysates were immunoprecipitated with primary antibody overnight at 4°C. After the beads were thoroughly washed with PBS containing 0.5% NP-40, bound proteins were eluted by SDS sample buffer, collected from the beads, and then analyzed by SDS-PAGE and immunoblotting. For ease of discussion, throughout this article, we make use of the term `pull-down' to describe a generic approach in which a target molecule is isolated from a mixture by binding to an antibody (immunoprecipitation), fusion protein (binding partner such as GST), or by adsorption of a larger complex in which the target molecule may combine (co-immunoprecipitation).
Separation of cytoplasmic β-catenin
Cell lysates from distinct Wnt3a stimulation were incubated with ConA-Sepharose at 4°C for 1 hour, as described previously (Aghib and McCrea, 1995). After brief centrifugation, supernatant fractions were re-incubated with ConA-Sepharose for another hour. Supernatant fractions were separated by centrifugation and protein concentrations were determined. For some experiments, these membrane-free fractions were subjected to immunoprecipitation using anti-β-catenin antibody.
Knockdown of Src, Hck, Yes and Dvl2 by siRNA
siRNA sequences designed to suppress Src and Hck are: 5′-GGCACCAAACUCAGCCUUAtt-3′ and 5′-UAAGGCUGAGUUUGGUGCCtg-3′; 5′-GGAUACCAUUGUGGUCGCAtt-3′ and 5′-UGCGACCACAAUGGUAUCCtc-3′, respectively. After treatment for 1 day with siRNA targeting Src, Hck or Yes, cells were transfected with M50 and an expression vector harboring rat Fz1. On the following day, F9 cells were stimulated with or without Wnt3a for 8 hours. Cells were washed with PBS and lysed. A scrambled sequence siRNA designed by Ambion was used as a control. Rescue experiments were performed using F9 cells stably expressing rat Fz1 and M50. 24 hours after treatment with siRNA targeting of Src, cells were transfected with either FLAG-tag wild-type Src, kinase-dead Src or Hck, and continuously cultured for another 24 hours. Cells were stimulated with Wnt3a for 7 hours. Cells were lysed and Lef/Tcf transcription activity was determined. For the AP-1 transcription assay, F9 cells were transfected with rat Fz1 and AP-1 reporter gene (Ap-1-Luc). 1 day after transfection, cells were stimulated with Wnt3a for 7 hours. For the NF-AT assay, F9 cells were transfected with rat Fz2 and NF-AT reporter gene [p-NF-AT-Luc (Ma and Wang, 2007)]. 24 hours later, these cells were stimulated with Wnt5a for 7 hours, optimal conditions for assay of the Wnt5a-Ca2+ non-canonical pathway. siRNA sequences targeting Dvl2 are: 5′-GGAAGAGAUCUCCGAUGACtt-3′ and 5′-GUCAUCGGAGAUCUCUUCCtt-3′.
GST-fusion proteins and pull-down experiments using GST-SH2, SH3 domain or GST-Dvl2 domains
GST-fusion proteins of various SH2 or SH3 domains were individually immobilized on glutathione-derivatized agarose matrix. F9 cell extracts (2 mg protein) were incubated with GST-Src SH2, GST-Src SH2/SH3, GST-Hck SH2, GST-Hck SH3, GST-Nck SH3, GST-Grb2 SH3, GST-Crk SH3 or GST alone for 1 hour. The gels were washed with PBS containing 0.5% NP-40. The bound proteins were resolved by SDS-PAGE and analyzed by immunoblotting with anti-GST and anti-Dvl2 antibodies. The following Dvl2 domains were expressed as fusion proteins in E. coli BL21 cells, as described elsewhere (Yokoyama and Malbon, 2007): GST-DIX, residues 11-93 of Dvl2; GST-PDZ, residues 267-309 of Dvl2; GST-putative SH3-binding domain region, residues 356-378 of Dvl2; GST-DEP, residues 433-507 of Dvl2; and GST-C-terminus, residues 511-736 of Dvl2. Mutation of GST-putative SH3 binding domain (residues 370-376, PxxPxxP to AxxAxxA) was carried out using Quikchange Mutagenesis System (Stratagene, La Jolla, CA). Mutations were confirmed by automated DNA sequencing. Cell lysates (2 mg protein) were incubated for 1 hour at 4°C, alone, or in combination with GST-fusion proteins of the DIX, PDZ, DEP, residues 356-378 (putative SH3-binding domain), or residues 511-736 (C-terminus domain) of Dvl2 that were immobilized to glutathione-coupled agarose beads. The bound proteins were collected and analyzed by SDS-PAGE, blotting and immunostaining with anti-GST and anti-Src (or anti-Hck) antibodies.
Expression and purification of Dvl2 in Sf9 cells
Recombinant Dvl2 (rDvl2) with both an N- and C-terminal 6×His tags was generated and purified, as previously described (Yokoyama and Malbon, 2007). Briefly, rDvl2 was expressed in Spodoptera frugiperda (Sf9) cells using Bac-to-Bac Baculovirus system (Invitrogen). Sf9 cells were infected with recombinant Dvl2 baculovirus. 3 days after infection, cells were lysed using a French pressure cell twice in 20 mM Tris-HCl buffer (pH 8.0) containing 1% deoxycholate, 2 mM Na3VO4, 20 mM NaF, 5 mM 2-mercaptoethanol, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 1 mM phenylmethylsulfonyl fluoride. Lysates were centrifuged at 40,000 g for 30 minutes. rDvl2 was purified by Ni-NTA column (Qiagen).
Assay of Src family kinase activity
Cell lysates were subjected to immunoprecipitation with either anti-Src antibody or anti-Dvl2 antibody. Activity of Src family kinases was determined, as described (Yokoyama and Miller, 1999). For some experiments, whole-cell lysates were assayed directly. Reaction mixture contained 20 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 2.5 mM Na3VO4, 0.5 mM dithiothreitol, 0.25 mM ATP, 0.5 mM Src peptide substrate (AEEEIYGEFEAKKKKG), 50 μg BSA and [γ-32P]ATP (∼500 c.p.m./pmol). The reactions were incubated at 30°C for 40 minutes and terminated by addition of 50% acetic acid. These samples then were spotted on p81 phosphocellulose paper (Casnellie, 1991). Incorporation of 32P into peptide was determined by liquid scintillation counting.
Phosphorylation of Dvl2 by tyrosine kinases
Phosphorylation of either rDvl2 or GST fusion proteins of Dvl2 domains was performed in a kinase buffer (10 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 10 mM MnCl2, 1 mM Na3VO4, 50 mM NaF, 0.5 mM [γ-32P]ATP (500 c.p.m./pmol) supplemented either without or with purified rSrc (or rHck) for 1 hour. Reactions were stopped by addition of SDS sample buffer and analyzed by SDS-PAGE and gel autoradiography. rSrc and rHck were expressed in Sf9 cells and purified as described earlier. (Yokoyama and Miller, 1999; Porter et al., 2000).
Subcellular fractionation of cells
Subcellular fractionation was carried out as described previously (Yokoyama et al., 2007). A sample of the whole-cell suspension was lysed directly in RIPA buffer for the determination of marker protein and of protein yields, as reported earlier (Yokoyama et al., 2007).
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/24/4439/DC1
We thank the members of the laboratory for careful discussions and critical review of the manuscript. We thank the staff of the Proteomics Center (State University of New York at Stony Brook) for mass spectrometry analysis. This work was supported generously by United States Public Health Services grants (to C.C.M.) from the National Institute of Diabetes, Digestive and Kidney Diseases, NIH. Deposited in PMC for release after 12 months.
References
- Aghib, D. F. and McCrea, P. D. (1995). The E-cadherin complex contains the src substrate p120. Exp. Cell Res. 218, 359-369. [DOI] [PubMed] [Google Scholar]
- Ahumada, A., Slusarski, D. C., Liu, X., Moon, R. T., Malbon, C. C. and Wang, H. Y. (2002). Signaling of rat Frizzled-2 through phosphodiesterase and cyclic GMP. Science 298, 2006-2010. [DOI] [PubMed] [Google Scholar]
- Alexandropoulos, K. and Baltimore, D. (1996). Coordinate activation of c-Src by SH3- and SH2-binding sites on a novel p130Cas-related protein, Sin. Genes Dev. 10, 1341-1355. [DOI] [PubMed] [Google Scholar]
- Bei, Y., Hogan, J., Berkowitz, L. A., Soto, M., Rocheleau, C. E., Pang, K. M., Collins, J. and Mello, C. C. (2002). SRC-1 and Wnt signaling act together to specify endoderm and to control cleavage orientation in early C. elegans embryos. Dev. Cell 3, 113-125. [DOI] [PubMed] [Google Scholar]
- Bikkavilli, R. K., Feigin, M. E. and Malbon, C. C. (2008). G alpha o mediates WNT-JNK signaling through dishevelled 1 and 3, RhoA family members, and MEKK 1 and 4 in mammalian cells. J. Cell Sci. 121, 234-245. [DOI] [PubMed] [Google Scholar]
- Briggs, S. D., Sharkey, M., Stevenson, M. and Smithgall, T. E. (1997). SH3-mediated Hck tyrosine kinase activation and fibroblast transformation by the Nef protein of HIV-1. J. Biol. Chem. 272, 17899-17902. [DOI] [PubMed] [Google Scholar]
- Brown, M. T. and Cooper, J. A. (1996). Regulation, substrates and functions of src. Biochim. Biophys. Acta 1287, 121-149. [DOI] [PubMed] [Google Scholar]
- Casnellie, J. E. (1991). Assay of protein kinases using peptides with basic residues for phosphocellulose binding. Methods Enzymol. 200, 115-120. [DOI] [PubMed] [Google Scholar]
- Clevers, H. (2006). Wnt/beta-catenin signaling in development and disease. Cell 127, 469-480. [DOI] [PubMed] [Google Scholar]
- Coluccia, A. M., Vacca, A., Dunach, M., Mologni, L., Redaelli, S., Bustos, V. H., Benati, D., Pinna, L. A. and Gambacorti-Passerini, C. (2007). Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J. 26, 1456-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, J. A. and Howell, B. (1993). The when and how of Src regulation. Cell 73, 1051-1054. [DOI] [PubMed] [Google Scholar]
- Glass, D. A., 2nd and Karsenty, G. (2006). Molecular bases of the regulation of bone remodeling by the canonical Wnt signaling pathway. Curr. Top Dev. Biol. 73, 43-84. [DOI] [PubMed] [Google Scholar]
- Haleem-Smith, H., Derfoul, A., Okafor, C., Tuli, R., Olsen, D., Hall, D. J. and Tuan, R. S. (2005). Optimization of high-efficiency transfection of adult human mesenchymal stem cells in vitro. Mol. Biotechnol. 30, 9-20. [DOI] [PubMed] [Google Scholar]
- Hart, M. J., de los Santos, R., Albert, I. N., Rubinfeld, B. and Polakis, P. (1998). Downregulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr. Biol. 8, 573-581. [DOI] [PubMed] [Google Scholar]
- He, X., Semenov, M., Tamai, K. and Zeng, X. (2004). LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development 131, 1663-1677. [DOI] [PubMed] [Google Scholar]
- Karni, R., Gus, Y., Dor, Y., Meyuhas, O. and Levitzki, A. (2005). Active Src elevates the expression of beta-catenin by enhancement of cap-dependent translation. Mol. Cell. Biol. 25, 5031-5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katanaev, V. L., Ponzielli, R., Semeriva, M. and Tomlinson, A. (2005). Trimeric G protein-dependent frizzled signaling in Drosophila. Cell 120, 111-122. [DOI] [PubMed] [Google Scholar]
- Kishida, M., Hino, S., Michiue, T., Yamamoto, H., Kishida, S., Fukui, A., Asashima, M. and Kikuchi, A. (2001). Synergistic activation of the Wnt signaling pathway by Dvl and casein kinase Iepsilon. J. Biol. Chem. 276, 33147-33155. [DOI] [PubMed] [Google Scholar]
- Lee, Y. N., Gao, Y. and Wang, H. Y. (2008). Differential mediation of the Wnt canonical pathway by mammalian Dishevelleds-1, -2, and -3. Cell Signal 20, 443-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilien, J. and Balsamo, J. (2005). The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr. Opin. Cell Biol. 17, 459-465. [DOI] [PubMed] [Google Scholar]
- Liu, C., Li, Y., Semenov, M., Han, C., Baeg, G. H., Tan, Y., Zhang, Z., Lin, X. and He, X. (2002). Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837-847. [DOI] [PubMed] [Google Scholar]
- Liu, T., DeCostanzo, A. J., Liu, X., Wang, H., Hallagan, S., Moon, R. T. and Malbon, C. C. (2001). G protein signaling from activated rat frizzled-1 to the beta-catenin-Lef-Tcf pathway. Science 292, 1718-1722. [DOI] [PubMed] [Google Scholar]
- Lowe, C., Yoneda, T., Boyce, B. F., Chen, H., Mundy, G. R. and Soriano, P. (1993). Osteopetrosis in Src-deficient mice is due to an autonomous defect of osteoclasts. Proc. Natl. Acad. Sci. USA 90, 4485-4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L. and Wang, H. Y. (2007). Mitogen-activated protein kinase p38 regulates the Wnt/cyclic GMP/Ca2+ non-canonical pathway. J. Biol. Chem. 282, 28980-28990. [DOI] [PubMed] [Google Scholar]
- Malbon, C. C. (2005). G proteins in development. Nat. Rev. Mol. Cell. Biol. 6, 689-701. [DOI] [PubMed] [Google Scholar]
- Malbon, C. C. and Wang, H. Y. (2006). Dishevelled: a mobile scaffold catalyzing development. Curr. Top Dev. Biol. 72, 153-166. [DOI] [PubMed] [Google Scholar]
- McKay, R. M., Peters, J. M. and Graff, J. M. (2001). The casein kinase I family in Wnt signaling. Dev. Biol. 235, 388-396. [DOI] [PubMed] [Google Scholar]
- Miller, W. T. (2003). Determinants of substrate recognition in nonreceptor tyrosine kinases. Acc. Chem. Res. 36, 393-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki, T., Sanjay, A., Neff, L., Tanaka, S., Horne, W. C. and Baron, R. (2004). Src kinase activity is essential for osteoclast function. J. Biol. Chem. 279, 17660-17666. [DOI] [PubMed] [Google Scholar]
- Moarefi, I., LaFevre-Bernt, M., Sicheri, F., Huse, M., Lee, C. H., Kuriyan, J. and Miller, W. T. (1997). Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 385, 650-653. [DOI] [PubMed] [Google Scholar]
- Nusse, R. (1999). WNT targets. Repression and activation. Trends Genet. 15, 1-3. [DOI] [PubMed] [Google Scholar]
- Nusse, R. (2001). Developmental biology. Making head or tail of Dickkopf. Nature 411, 255-256. [DOI] [PubMed] [Google Scholar]
- Nusse, R. (2005). Wnt signaling in disease and in development. Cell Res. 15, 28-32. [DOI] [PubMed] [Google Scholar]
- Pellicena, P. and Miller, W. T. (2001). Processive phosphorylation of p130Cas by Src depends on SH3-polyproline interactions. J. Biol. Chem. 276, 28190-28196. [DOI] [PubMed] [Google Scholar]
- Penton, A., Wodarz, A. and Nusse, R. (2002). A mutational analysis of dishevelled in Drosophila defines novel domains in the dishevelled protein as well as novel suppressing alleles of axin. Genetics 161, 747-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedra, J., Martinez, D., Castano, J., Miravet, S., Dunach, M. and de Herreros, A. G. (2001). Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J. Biol. Chem. 276, 20436-20443. [DOI] [PubMed] [Google Scholar]
- Porter, M., Schindler, T., Kuriyan, J. and Miller, W. T. (2000). Reciprocal regulation of Hck activity by phosphorylation of Tyr(527) and Tyr(416). Effect of introducing a high affinity intramolecular SH2 ligand. J. Biol. Chem. 275, 2721-2726. [DOI] [PubMed] [Google Scholar]
- Roura, S., Miravet, S., Piedra, J., Garcia de Herreros, A. and Dunach, M. (1999). Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J. Biol. Chem. 274, 36734-36740. [DOI] [PubMed] [Google Scholar]
- Schwartzberg, P. L., Xing, L., Hoffmann, O., Lowell, C. A., Garrett, L., Boyce, B. F. and Varmus, H. E. (1997). Rescue of osteoclast function by transgenic expression of kinase-deficient Src in src–/– mutant mice. Genes Dev. 11, 2835-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz-Romond, T., Merrifield, C., Nichols, B. J. and Bienz, M. (2005). The Wnt signalling effector Dishevelled forms dynamic protein assemblies rather than stable associations with cytoplasmic vesicles. J. Cell Sci. 118, 5269-5277. [DOI] [PubMed] [Google Scholar]
- Schwarz-Romond, T., Fiedler, M., Shibata, N., Butler, P. J., Kikuchi, A., Higuchi, Y. and Bienz, M. (2007a). The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat. Struct. Mol. Biol. 14, 484-492. [DOI] [PubMed] [Google Scholar]
- Schwarz-Romond, T., Metcalfe, C. and Bienz, M. (2007b). Dynamic recruitment of axin by Dishevelled protein assemblies. J. Cell Sci. 120, 2402-2412. [DOI] [PubMed] [Google Scholar]
- Seto, E. S. and Bellen, H. J. (2006). Internalization is required for proper Wingless signaling in Drosophila melanogaster. J. Cell Biol. 173, 95-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicheri, F., Moarefi, I. and Kuriyan, J. (1997). Crystal structure of the Src family tyrosine kinase Hck. Nature 385, 602-609. [DOI] [PubMed] [Google Scholar]
- Soriano, P., Montgomery, C., Geske, R. and Bradley, A. (1991). Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 64, 693-702. [DOI] [PubMed] [Google Scholar]
- Sun, T. Q., Lu, B., Feng, J. J., Reinhard, C., Jan, Y. N., Fantl, W. J. and Williams, L. T. (2001). PAR-1 is a Dishevelled-associated kinase and a positive regulator of Wnt signalling. Nat. Cell Biol. 3, 628-636. [DOI] [PubMed] [Google Scholar]
- Wallingford, J. B. and Habas, R. (2005). The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development 132, 4421-4436. [DOI] [PubMed] [Google Scholar]
- Walston, T., Tuskey, C., Edgar, L., Hawkins, N., Ellis, G., Bowerman, B., Wood, W. and Hardin, J. (2004). Multiple Wnt signaling pathways converge to orient the mitotic spindle in early C. elegans embryos. Dev. Cell 7, 831-841. [DOI] [PubMed] [Google Scholar]
- Wang, J. and Wynshaw-Boris, A. (2004). The canonical Wnt pathway in early mammalian embryogenesis and stem cell maintenance/differentiation. Curr. Opin. Genet. Dev. 14, 533-539. [DOI] [PubMed] [Google Scholar]
- Weber, U., Pataki, C., Mihaly, J. and Mlodzik, M. (2008). Combinatorial signaling by the Frizzled/PCP and Egfr pathways during planar cell polarity establishment in the Drosophila eye. Dev. Biol. 316, 110-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton, K. A., Jr (2003). Runnin' with the Dvl: proteins that associate with Dsh/Dvl and their significance to Wnt signal transduction. Dev. Biol. 253, 1-17. [DOI] [PubMed] [Google Scholar]
- Willert, K., Brink, M., Wodarz, A., Varmus, H. and Nusse, R. (1997). Casein kinase 2 associates with and phosphorylates dishevelled. EMBO J. 16, 3089-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert, K., Brown, J. D., Danenberg, E., Duncan, A. W., Weissman, I. L., Reya, T., Yates, J. R., 3rd and Nusse, R. (2003). Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423, 448-452. [DOI] [PubMed] [Google Scholar]
- Williams, J. C., Weijland, A., Gonfloni, S., Thompson, A., Courtneidge, S. A., Superti-Furga, G. and Wierenga, R. K. (1997). The 2.35 A crystal structure of the inactivated form of chicken Src: a dynamic molecule with multiple regulatory interactions. J. Mol. Biol. 274, 757-775. [DOI] [PubMed] [Google Scholar]
- Wodarz, A. and Nusse, R. (1998). Mechanisms of Wnt signaling in development. Annu. Rev. Cell Dev. Biol. 14, 59-88. [DOI] [PubMed] [Google Scholar]
- Wong, H. C., Mao, J., Nguyen, J. T., Srinivas, S., Zhang, W., Liu, B., Li, L., Wu, D. and Zheng, J. (2000). Structural basis of the recognition of the dishevelled DEP domain in the Wnt signaling pathway. Nat. Struct. Biol. 7, 1178-1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, H. C., Bourdelas, A., Krauss, A., Lee, H. J., Shao, Y., Wu, D., Mlodzik, M., Shi, D. L. and Zheng, J. (2003). Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol. Cell 12, 1251-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W., Harrison, S. C. and Eck, M. J. (1997). Three-dimensional structure of the tyrosine kinase c-Src. Nature 385, 595-602. [DOI] [PubMed] [Google Scholar]
- Yanagawa, S., van Leeuwen, F., Wodarz, A., Klingensmith, J. and Nusse, R. (1995). The dishevelled protein is modified by wingless signaling in Drosophila. Genes Dev. 9, 1087-1097. [DOI] [PubMed] [Google Scholar]
- Yang, Z. and Petitte, J. N. (1994). Use of avian cytokines in mammalian embryonic stem cell culture. Poult Sci. 73, 965-974. [DOI] [PubMed] [Google Scholar]
- Yokoyama, N. and Miller, W. T. (1999). Identification of residues involved in v-Src substrate recognition by site-directed mutagenesis. FEBS Lett. 456, 403-408. [DOI] [PubMed] [Google Scholar]
- Yokoyama, N. and Malbon, C. C. (2007). Phosphoprotein phosphatase-2A docks to Dishevelled and counterregulates Wnt3a/beta-catenin signaling. J. Mol. Signal 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama, N., Yin, D. and Malbon, C. C. (2007). Abundance, complexation, and trafficking of Wnt/beta-catenin signaling elements in response to Wnt3a. J. Mol. Signal 2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.