

Summary
Mutations in rod opsin, the archetypal G-protein-coupled receptor, cause retinitis pigmentosa. The majority of mutations, e.g. P23H, cause protein misfolding, resulting in ER retention, induction of the unfolded protein response and degradation by ERAD. If misfolded rod opsin escapes degradation, it aggregates and forms intracellular inclusions. Therefore, it is important to identify the chaperones that mediate the folding or degradation of rod opsin. ER degradation enhancing α-mannosidase-like 1 (EDEM1) can enhance the release of terminally misfolded glycoproteins from the calnexin chaperone system. Here, we identify EDEM1 as a novel chaperone of rod opsin. EDEM1 expression promoted the degradation of P23H rod opsin and decreased its aggregation. By contrast, shRNA-mediated knockdown of EDEM1 increased both the amount of P23H rod opsin and its aggregation into inclusions. EDEM1 was detected in rod photoreceptor inner segments and EndoH-sensitive rod opsin co-immunoprecipitated with EDEM1 from retina, suggesting that rod opsin is a physiological EDEM1 client. Unexpectedly, EDEM1 binding to rod opsin was independent of mannose trimming and EDEM1 promoted the cell-surface expression of mutant rod opsin. Collectively, the data suggest that EDEM1 is a chaperone for rod opsin and that expression of EDEM1 can be used to promote correct folding, as well as enhanced degradation, of mutant proteins in the ER to combat protein-misfolding disease.
Keywords: GPCR, Molecular chaperone, Rhodopsin, ERAD, EDEM, Quality control, Neurodegeneration, Retinitis pigmentosa, Traffic
Introduction
Rhodopsin is an archetypal seven-transmembrane G-protein-coupled receptor (GPCR) responsible for scotopic vision under dim light conditions. Rhodopsin is formed from rod opsin and the 11-cis-retinal chromophore. The apoprotein of rhodopsin, rod opsin, is synthesized in the inner segment (IS) of rod photoreceptor cells before vectorial transport to the rod outer segment (OS) photosensory cilia. Mutations in rod opsin cause the neurodegenerative disease retinitis pigmentosa (RP), which leads to blindness (Dryja et al., 1990) (OMIM:180380). Dominant mutations in rod opsin account for ∼25% of all autosomal dominant RP (ADRP) cases and over 200 point mutations have been identified to date (http://www.sph.uth.tmc.edu/Retnet/).
Different amino acid substitutions in rod opsin result in distinct cellular and biochemical defects; hence, a functional classification was suggested (Sung et al., 1993), which was recently extended (Mendes et al., 2005). The distinction between rod opsin mutational consequences is vital for understanding the disease pathogenesis of RP. The majority of rod opsin mutations, such as the most common mutation in North America that changes intradiscal (or lumenal) Pro23 to His (P23H), are class II mutations, which result in the misfolding of the protein. Heterologous expression of class II rod opsin mutants in 293S and COS1 cells revealed that these were retained in the ER, suggesting that the quality control system of this compartment did not permit further traffic of folding-defective polypeptides through the secretory system (Kaushal and Khorana, 1994; Sung et al., 1991).
P23H rod opsin misfolding stimulates the unfolded protein response (UPR) (Lin et al., 2007). The UPR is an adaptive ER stress response designed to enhance the folding, as well as degradation of misfolded proteins, thereby reducing misfolding stress; however, rod opsin misfolding might lead to persistent stress that cannot be overcome. Misfolded P23H mutant rod opsin protein was shown to have less helical structure and tertiary contacts, and more susceptibility to trypsin digestion, suggesting that the misfolded proteins are less compact than the folded forms (Liu et al., 1996). Misfolded P23H mutant rod opsin can spontaneously aggregate and form microscopically visible inclusion bodies, which are considered to be end-stage products of protein aggregation in the cell (Illing et al., 2002; Saliba et al., 2002). The aggregation of P23H rod opsin leads to global impairment of proteasome function, which might lead to reduced degradation of misfolded proteins and other clients of the ubiquitin proteasome system (UPS) (Bennett et al., 2005). In the presence of proteasome inhibitors, P23H rod opsin is ubiquitylated, degradation is inhibited, and export from the ER to the cytosol is delayed (Illing et al., 2002; Saliba et al., 2002), confirming that turnover is mediated by the UPS and underscoring the role of the proteasome in the retrotranslocation and ER-associated degradation (ERAD) of mutant rod opsin.
Rhodopsin is N-glycoslyated at Asn2 and Asn15 in the first intradiscal (or lumenal) domain. These glycan chains of P23H rod opsin are required for efficient recognition by the ERAD machinery in targeting the mutant protein for degradation by the UPS (Saliba et al., 2002), hence a lectin chaperone was suggested to mediate the degradation of rod opsin. A recent study revealed that this key chaperone is unlikely to be calnexin, because cells with dysfunctional calnexin processed P23H rod opsin similarly to controls (Kosmaoglou and Cheetham, 2008).
EDEM1 (ER degradation-enhancing, α-mannosidase-like 1) enhances ERAD of a terminally misfolded α1-antitrypsin variant, null Hong Kong (NHK, HK A1AT) (Hosokawa et al., 2001). The overexpression of recombinant EDEM1 can enhance the release of terminally misfolded glycoproteins from the calnexin chaperone system and their demannosylation (Molinari et al., 2003; Oda et al., 2003), thereby accelerating their elimination from the ER lumen. Furthermore, reduction of endogenous EDEM1 by RNA interference (Molinari et al., 2003) and inactivation of the UPR IRE1-XBP1 pathway have both been shown to decrease the efficiency of glycoprotein ERAD, which eventually compromises cellular secretory capacity (Eriksson et al., 2004; Yoshida et al., 2003). Therefore, EDEM1 is a potential component of the normal response to rod opsin misfolding and manipulation of EDEM1 could affect mutant rod opsin degradation.
Here, we investigated the potential role of EDEM1 in the modulation of rod opsin processing. EDEM1 overexpression and knockdown were analysed on P23H mutant rod opsin degradation, aggregation and traffic. Formation of EDEM1–rod-opsin complexes was probed in cells and in the retina. Collectively, the data suggest that rod opsin is an EDEM1 client and that manipulation of EDEM1 could be a useful therapeutic target in rhodopsin RP and other protein-misfolding diseases.
Results
EDEM1 and rod opsin localisation
The localisation of EDEM1 relative to rod opsin was investigated in SK-N-SH cells (Fig. 1). SK-N-SH cells were transfected with wild-type (WT) and P23H rod opsin tagged at the cytoplasmic C-terminus with GFP (WT-GFP, P23H-GFP) (Saliba et al., 2002). At 24 hours after transfection, the cells were stained with antibodies against EDEM1 and were analysed by confocal microscopy. EDEM1 localised in punctate structures that were distinct from WT-GFP localisation (Fig. 1A-D). This subcellular localisation is consistent with previous data showing EDEM1 in vesicles and tubular structures budding from the ER (Zuber et al., 2007). EDEM1 was found to partially overlap with P23H-GFP (Fig. 1E-G), as shown by the yellow and orange areas of punctate staining, which were interspersed with GFP staining alone, suggesting that the colocalisation was not perfect (Fig. 1H). This pattern is similar to that previously observed for the EDEM1 and its client HK A1AT (Zuber et al., 2007), with overlap in punctuate and finger-like structures, because EDEM1 is not detected throughout the ER, unlike P23H-GFP.
Fig. 1.
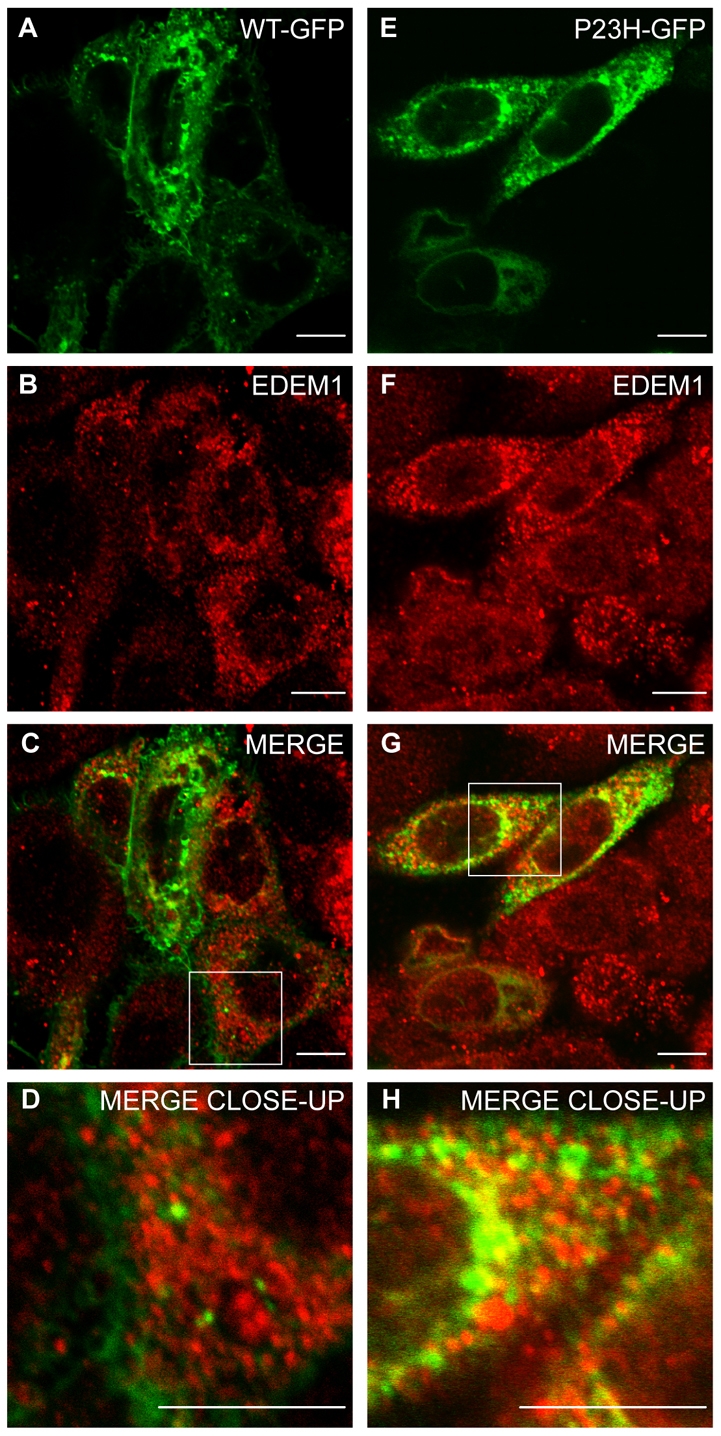
Endogenous EDEM1 partially colocalises with P23H but not WT rod opsin. SK-N-SH cells were transfected with WT rod opsin-GFP (WT-GFP) (A-D, green) or P23H rod opsin-GFP (P23H GFP) (E-H, green). 24 hours after transfection, the cells were fixed and stained with antibodies against EDEM1 (red). Cells were analysed by confocal microscopy. Areas enclosed by white boxes in C and G are enlarged in D and H to show detail of rod opsin-GFP and EDEM1 staining. Scale bars: 10 μm.
EDEM1 enhances P23H rod opsin degradation
The role of EDEM1 in enhancing the degradation of P23H rod opsin was investigated (Fig. 2A). Untagged P23H rod opsin was expressed in SK-N-SH cells with increasing EDEM1-HA plasmid concentrations and GFP was used as a transfection control. DM soluble fractions were analysed 24 hours after transfection by western blotting with antibodies against rod opsin, HA and GFP. Western blotting of rod opsin expressed in cell culture with anti-rod opsin mAb 1D4 revealed a complex pattern of specific bands. WT rod opsin was detected as a major smear of bands between 36 and 55 kDa corresponding to different glycoforms. By contrast, P23H rod opsin was expressed at lower levels and had three major bands of approximately 27-30, 36-38 and 55-60 kDa that correspond to an N-terminal truncated protein, an EndoH-sensitive glycoform and a dimer, respectively; other minor bands were also observed that corresponded to different glycoforms, such as deglycosylated rod opsin, or high molecular weight aggregates (Noorwez et al., 2004; Saliba et al., 2002). EDEM1-HA protein levels increased with increasing amount of plasmid and led to a dose-dependent reduction in all species of P23H rod opsin, whereas the levels of GFP were unchanged (Fig. 2A). This reduction correlated with a decrease in the incidence of P23H-GFP inclusions (Fig. 2B) from 29% in P23H-GFP and empty vector-transfected to 12% with a fourfold molar excess of EDEM1-HA (Fig. 2B). To confirm the effect of EDEM1 on P23H degradation, SK-N-SH cells expressing P23H rod opsin were radiolabelled with [35S]methionine/cysteine, chased for 1 hour, and rod opsin was purified by immunoprecipitation with 1D4, followed by digestion with PNGase F to resolve all the glycoforms to a single deglycosylated band that facilitated quantification (supplementary material Fig. S1). Pulse chase revealed that although 87% of the initial P23H rod opsin remained after 1 hour, cotransfection with EDEM1-HA resulted in only 56% of the mutant protein remaining after the same time interval. Hence, EDEM1-HA enhanced the degradation of P23H rod opsin (Fig. 2C).
Fig. 2.
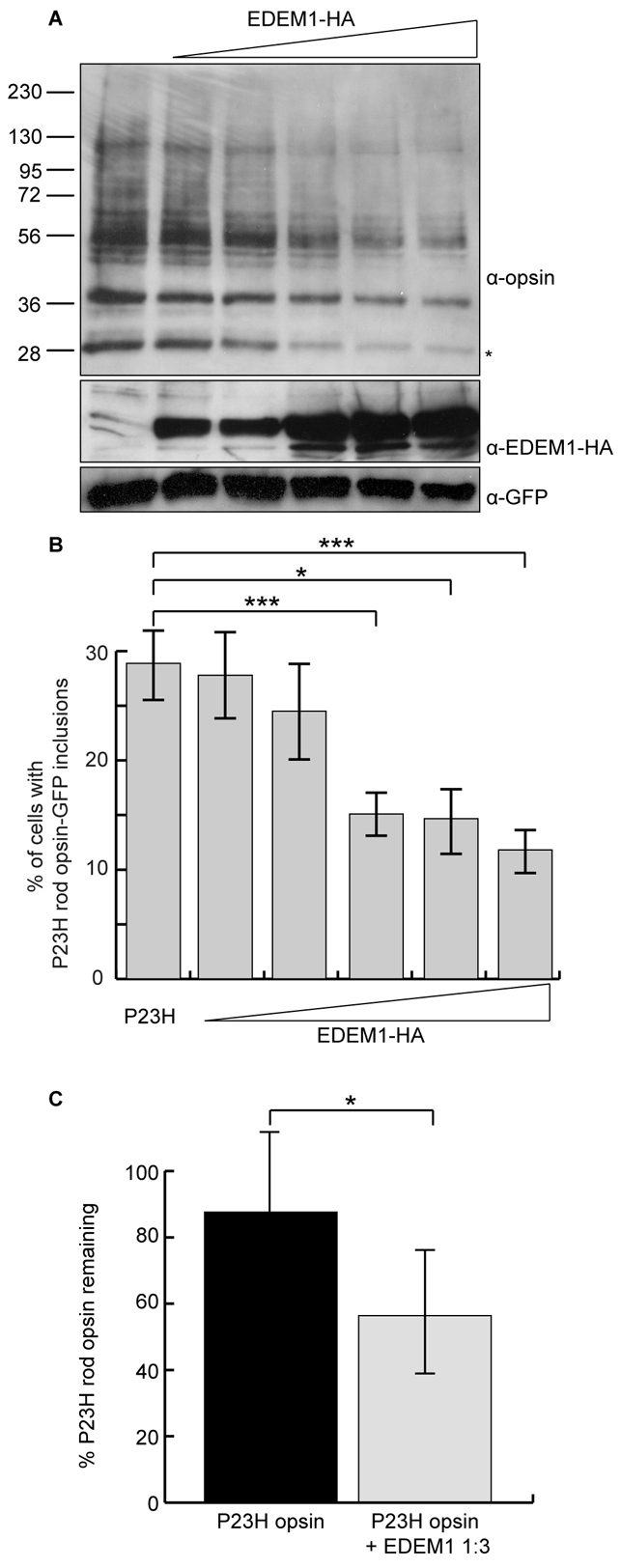
EDEM1 accelerates the degradation of mutant rod opsin. (A) Constant amounts of P23H rod opsin-pMT3 and increasing (1:0.5, 1:1, 1:2, 1:3, 1:4) ratios of EDEM1-HA plasmid were transfected in SK-N-SH cells. DM-soluble fractions were obtained 24 hours after transfection and 10 μg protein resolved by SDS-PAGE. Proteins were immunoblotted with antibodies against opsin, HA and GFP, as indicated. GFP was used as a transfection and loading control. The position of molecular mass markers in kDa are indicated on the left. Asterisk indicates the species most sensitive to EDEM1-enhanced degradation. (B) The incidence of inclusion formation for P23H rod opsin-GFP was assessed in SK-N-SH cells with increasing (1:0.5, 1:1, 1:2, 1:3, 1:4) ratios of EDEM1-HA plasmid. Error bars represent ± 2 s.e.m. ***P<0.001, *P<0.05. P23H 1:4 EDEM1, ***P=0.0009. P23H 1:3 EDEM1, *P=0.012. P23H 1:2 EDEM1, ***P=0.0005. (C) SK-N-SH cells were pulsed for 20 minutes with [35S]methionine/cysteine and were chased in unlabelled methionine for 60 minutes. P23H rod opsin 1D4 immunoprecipitates were digested with 1500 units PNGaseF overnight and resolved by SDS-PAGE. Quantification of deglycoslyated P23H rod opsin was performed using ImageJ from four replicates. Error bars represent ± 2 s.e.m. *P<0.05.
Knockdown of EDEM1 increases P23H aggregation
RNA interference was used to reduce endogenous EDEM1 and to determine the effect on the processing of P23H rod opsin. SK-N-SH cells were transfected with shRNA to EDEM1 (Molinari et al., 2003). The ability of this shEDEM1 to interfere with the expression of EDEM1 was confirmed on endogenous EDEM1 and the dose-response of increasing amounts of shRNA was tested in cells transfected with the EDEM1-HA construct (supplementary material Fig. S2). Subsequently, shEDEM1 was used to reduce the expression of endogenous EDEM1 in SK-N-SH cells transfected with P23H-GFP (Fig. 3A,B). The cells were fixed 24 hours after transfection and analysed by confocal microscopy with the same scanning parameters for image acquisition for P23H-GFP and empty vector or shEDEM1 transfections. Silencing of EDEM1 led to an increase in ER staining, and increased inclusion formation in P23H-GFP-expressing cells (Fig. 3B) compared with the control (Fig. 3A). Intracellular inclusions were found to increase in a dose-dependent manner. Inclusion incidence increased to 47% with a fourfold molar excess of shEDEM1 from 32% in the control shRNA-treated cells (Fig. 3C). Western blotting showed that reducing the expression of endogenous EDEM1 resulted in a global increase of P23H rod opsin species, with the glycoform at 38 kDa preferentially increased (Fig. 3D).
Fig. 3.

EDEM1 knockdown results in the accumulation of P23H rod opsin. P23H rod opsin-GFP (P23H-GFP) and empty vector (A) or P23H-GFP and shEDEM1 (B) were transfected in SK-N-SH cells. 24 hours after transfection, the cells were fixed and analysed by confocal microscopy. Scanning parameters during image acquisition were kept constant. Arrows indicate increased ER staining and inclusion formation. Scale bar: 10 μm. (C) The incidence of inclusion formation of P23H-GFP was assessed with increasing shEDEM1 plasmid (1:1, 1:2, 1:3, 1:4). Error bars represent ± 2 s.e.m. *P=0.0134. (D) Constant pMT3-P23H rod opsin and increasing (1:2, 1:3, 1:4) molar plasmid equivalents of shEDEM1 were transfected in SK-N-SH cells. DM-soluble fractions were obtained 24 hours after transfection and 10 μg protein resolved by SDS-PAGE. Asterisk indicates species showing greatest accumulation. Rod opsin was detected using 1D4 mAb. The position of molecular size markers in kDa are indicated on the left.
EDEM1 prefentially binds mutant rod opsin independently of mannose trimming
An interaction between mutant rod opsin and EDEM1-HA was investigated using coimmunoprecipitation. SK-N-SH cells were transfected with P23H rod opsin and EDEM1-HA. Cells were treated 16 hours after transfection with the proteasome inhibitor MG132 for 8 hours (Fig. 4A,B). Cell lysates were immunoprecipitated with antibodies against rod opsin (1D4) and EDEM1-HA (HA). EDEM1 is thought to promote the degradation of misfolded glycoproteins via ERAD, where the terminal acceptor is the proteasome; hence proteasome inhibition was expected to enhance the interaction between the two proteins. As before, EDEM1 enhanced the degradation of P23H rod opsin (Fig. 4A). As previously described (Saliba et al., 2002), treatment of P23H rod opsin with MG132 led to increased levels of the mutant protein. MG132 reduced the EDEM1-mediated degradation of mutant rod opsin (Fig. 4A, input). Immunoprecipitation revealed that P23H rod opsin and EDEM1-HA coprecipitated, and this interaction was enhanced in the presence of MG132 (Fig. 4A,B). Importantly, EDEM1 was observed to interact only with a 38 kDa rod opsin glycoform, which was sensitive to digestion by EndoH glycosidase, and therefore corresponds to immature ER-retained P23H rod opsin (Fig. 4A,F).
Fig. 4.
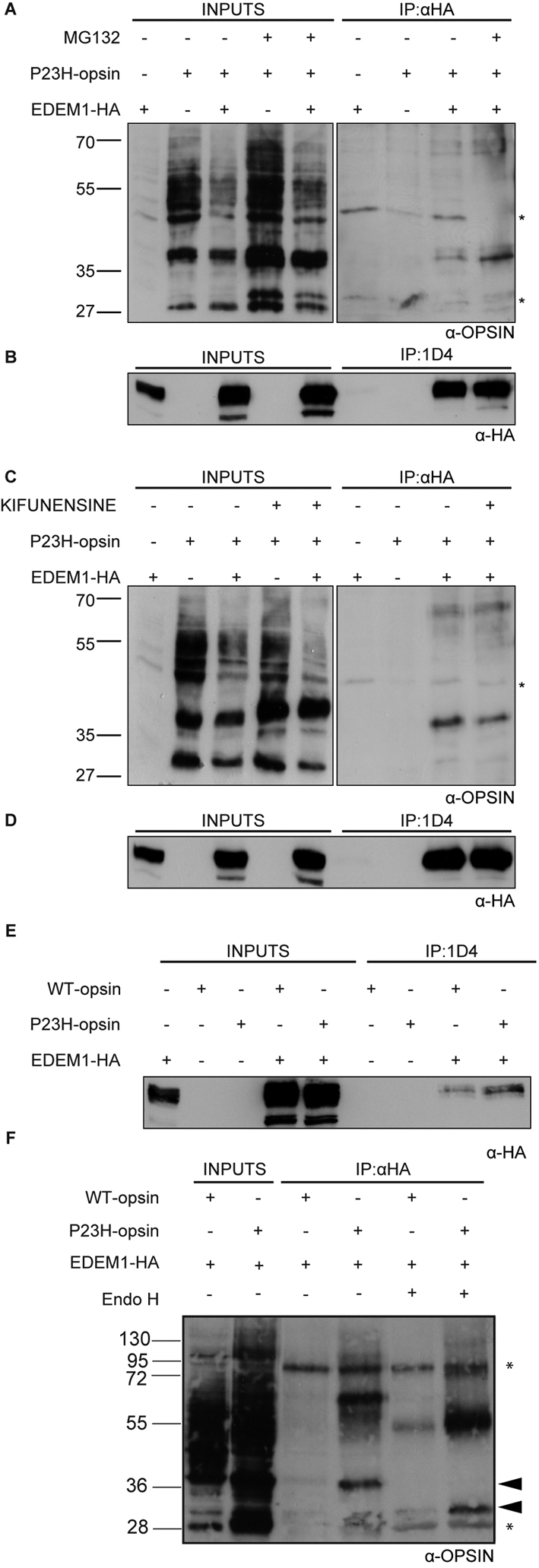
EDEM1-HA interacts preferentially with P23H rod opsin independently of mannosidase cleavage. SK-N-SH cells were transfected with EDEM1-HA and WT (E,F) or P23H rod opsin (A-F) expression constructs as indicated. Cells were treated with MG132 (50 μM) for 8 hours (A,B) or kifunensine (50 μg/ml) for 4 hours (C,D), as indicated, before cell lysis. Soluble postnuclear fractions were obtained and incubated overnight with either anti-HA (A,C,F) or 1D4 (B,D,E), and digested with EndoH (F) as indicated. 10 μg (A-D) or 7 μg (E,F) of total protein was loaded as input fraction. Immunoprecipitation lane loading was equivalent to ∼15 μg (A-D) or ∼10 μg (E,F) of input material. Blots were probed using reciprocal antibodies to detect coimmunoprecipitated species. Asterisks represents H+L IgG bands. The position of molecular size markers in kDa are indicated on the left.
EDEM1 is proposed to interact with substrate glycoproteins depending on the extent of mannose trimming (Hosokawa et al., 2001; Nakatsukasa et al., 2001; Olivari et al., 2006). To test the role of mannose trimming in the opsin interaction, kifunensine was used to prevent hydrolysis of the Mannose-9 (Man9) structure, which specifically inhibits ER mannosidase I (ERManI) (Hosokawa et al., 2003). If the interaction between P23H rod opsin and EDEM1 was mediated via mannose-trimmed glycan chains, then kifunensine should abolish the interaction. P23H rod opsin was treated with kifunensine for 4 hours before cell lysis, which resulted in an accumulation of P23H rod opsin species (Fig. 4C, input). The interaction between P23H rod opsin and EDEM1-HA in the presence of kifunensine, however, was only slightly diminished. The interaction was confirmed by reciprocal immunoprecipitation with 1D4 and western blotting with anti-HA antibodies (Fig. 4B,D). Hence, P23H rod opsin and EDEM1-HA are found in a complex that does not require mannose trimming.
The ability of EDEM1 to bind control WT rod opsin was compared with mutant P23H rod opsin by reciprocal co-immunoprecipitation (Fig. 4E,F). Similarly to P23H rod opsin, only the 38 kDa EndoH-sensitive form of WT rod opsin was bound to EDEM1-HA. The yield of P23H rod opsin recovered with EDEM1-HA was much higher than with WT rod opsin, despite the lower levels of P23H rod opsin input. Therefore, EDEM1 appears to bind the mutant misfolded form of rod opsin preferentially to normal WT protein.
EDEM1 and rod opsin in the retina
Immunohistochemistry of mouse retina was performed with antibodies against rhodopsin, calnexin and EDEM1 to investigate the localisation of EDEM1 in the retina. Rhodopsin was observed mainly in the rod OS, with a small amount visible in the IS, the major biosynthetic organelle of the rod photoreceptor cell (Fig. 5A,B). Calnexin staining was confined to the ER within the IS (Fig. 5A), whereas EDEM1 occupied a subset of the IS as shown by the punctate staining obtained (Fig. 5B). These data are consistent with the localisation of EDEM1 in cells and might represent vesicles budding from the ER (Zuber et al., 2007). As EDEM1-HA can bind WT rod opsin in cell culture, we used coimmunoprecipitation to investigate whether rhodopsin and EDEM1 form a physiological complex in porcine retina. Rhodopsin was detected as a major band of approximate molecular weight 38 kDa (Fig. 5C). Immunoprecipitation with antibodies to EDEM1 revealed a 1D4 immunoreactive band at 38 kDa, which was not detected with control antibody precipitation. The immunoprecipitated material was treated with EndoH glycosidase. The rhodopsin band was found to be sensitive to EndoH, suggesting that rhodopsin and EDEM1 interact in the ER of rod photoreceptor cells (Fig. 5C). Antibodies to rhodopsin (1D4) were used for the reciprocal co-immunoprecipitation of EDEM1 (Fig. 5D).
Fig. 5.
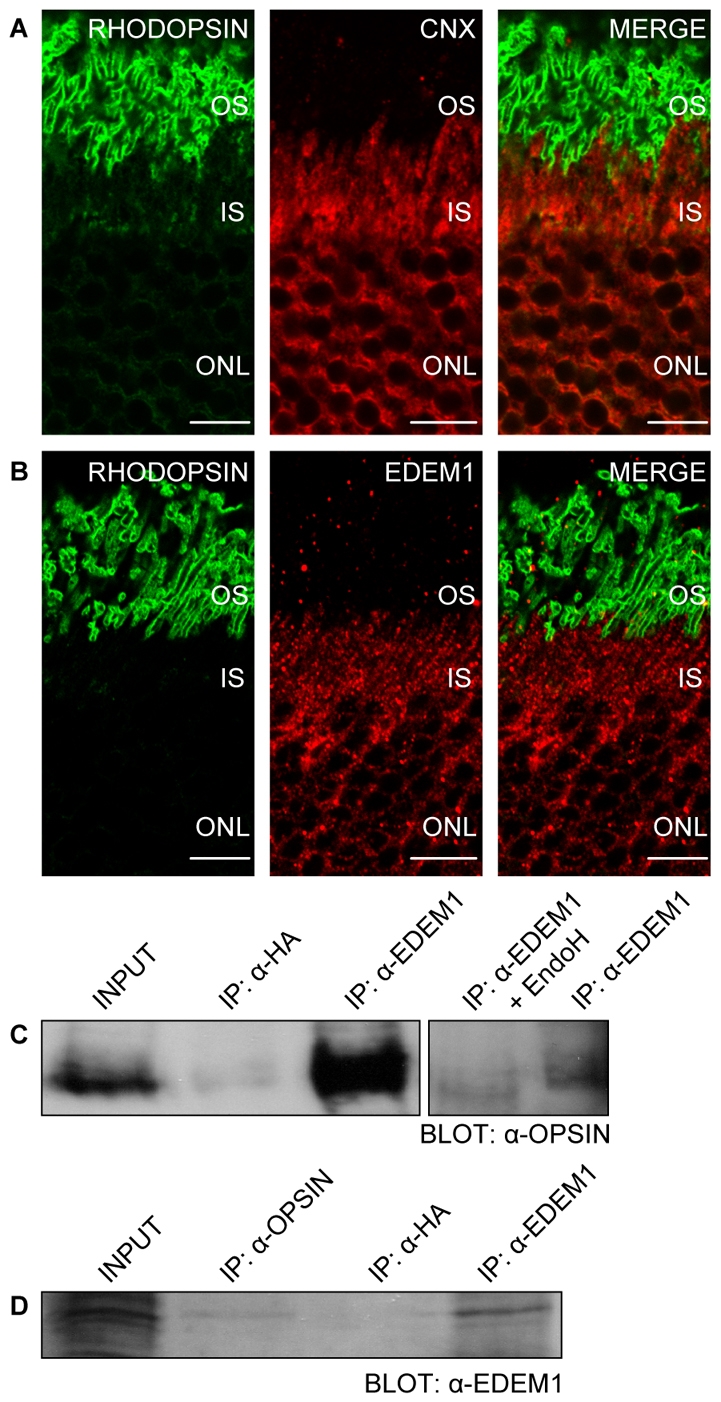
EDEM1 interacts with rhodopsin in photoreceptors. Immunofluorescent confocal microscopy of peripheral mouse retina using 4D2 antibody against rod opsin (green) and calnexin (red) (A) or EDEM1 (red) (B). Scale bars: 10 μm. Immunoprecipitation of porcine retinal homogenates (200 μg protein) using EDEM1 (C) and 1D4 antibodies (D). Rod opsin was digested with EndoH for 1 hour at 37°C, resolved by SDS-PAGE and was immunoblotted using indicated antibodies. 0.075 μg total protein was loaded as the rhodopsin `input', whereas 1.5 μg was loaded as the EDEM1 `input' fraction. Immunoprecipitated material loading was equivalent to ∼55 μg of input material.
EDEM1 promotes the traffic of mutant rod opsin
Confocal microscopic analyses at constant intensity levels of P23H-GFP and P23H-GFP in the presence of EDEM1 confirmed that EDEM1-HA led to the reduced expression of P23H-GFP (Fig. 6A,C). Close inspection revealed that in a significant portion of cells, the remaining P23H-GFP fluorescence was present on the plasma membrane (Fig. 6B,D). An antibody targeted to the intradiscal (or extracellular) N-terminus of rod opsin (4D2) was used to confirm the observation that mutant rod opsin decorated the plasma membrane. SK-N-SH cells were fixed, but not permeabilised, and stained with 4D2 then analysed by confocal microscopy; scanning parameters optimised for WT rod opsin image acquisition were used for all remaining conditions. Cells expressing WT rod opsin showed intense plasma membrane staining (Fig. 7A). P23H rod opsin displayed no detectable immunoreactivity at the cell surface, which is consistent with the known ER retention of the mutant protein (Fig. 7B) (Sung et al., 1991; Illing et al., 2002; Saliba et al., 2002). P23H rod opsin expressed with EDEM1-HA confirmed the previous observation, because a portion of the mutant protein was found to decorate the plasma membrane of cells (Fig. 7C).
Fig. 6.

EDEM1 promotes the translocation of mutant rod opsin to the plasma membrane. SK-N-SH cells were transfected with P23H rod opsin-GFP (P23H-GFP) and empty vector (A-D) or P23H rod opsin-GFP with two molar equivalents of EDEM1-HA (P23H-GFP+EDEM1-HA) (E-H). Cells were imaged by confocal microscopy at the same scanning parameters and staining detail is shown in enlarged areas (D,H). Arrow shows potential plasma membrane staining of P23H rod opsin (green) in the presence of EDEM1-HA (red) (H). Note that in contrast to endogenous EDEM1 staining, EDEM1-HA (F) has an ER localisation (Zuber et al., 2007). Scale bars: 10 μm.
Fig. 7.

Characterisation of EDEM1-enhanced traffic of mutant rod opsin. SK-N-SH cells were transfected with WT (A), P23H rod opsin and empty vector (B) and P23H rod opsin and 0.5 molar equivalents of EDEM1-HA (C). 24 hours after transfection, cells were fixed, not permeabilised and immunostained with 4D2 mAb to the N-terminus of rod opsin. Scale bars: 10 μm. (D) Quantification of in-cell western analysis of WT, P23H or P23H and 0.5 molar equivalents of EDEM1-HA-cotransfected cells from two independent experiments performed in triplicate. Error bars represent ± 2 s.e.m. **P<0.01 and *P<0.05. For WT and P23H, P=0.0025. P23H and P23H + EDEM1-HA, P=0.0252. (E) Cell-surface biotinylation of SK-N-SH cells transfected with WT or P23H rod opsin with a 1:1 ratio of EDEM1-HA, as indicated. 5 μg total protein was loaded as input fraction. Streptavidin precipitation lane loading was equivalent to ∼70 μg of input material. The blot was probed with 4D2 antibody. The position of molecular mass markers in kDa are indicated on the left. (F) Normalised absorption spectra of immunoaffinity chromatography purified P23H rod opsin (dotted line) or P23H rod opsin with EDEM1-HA at a ration of 1:3 (solid line), inset shows magnification of 400-650 nm region of the spectra.
A series of `in-cell western' (ICW) experiments with 4D2 were performed to confirm and quantify the plasma membrane targeting (Fig. 7D). EDEM1-HA resulted in a greater than twofold increase in fluorescence of the mutant receptor at the cell surface. The improved traffic of P23H rod opsin was characterised further using a cell surface biotinylation assay. Sulfo-NHS-SS-Biotin was used to label all cell surface proteins of transfected SK-N-SH cells, followed by precipitation with streptavidin agarose. Immunoblotting with 4D2 revealed that the precipitated WT rod opsin migrated as a group of bands between 35 and 50 kDa corresponding to mature opsin, whereas precipitated P23H rod opsin migrated as a minor component around 35-50 kDa, a discrete band of approximately 100 kDa and higher molecular weight (HMW) bands at the top of the gel (Fig. 7E). The HMW species were enriched in the precipitated mutant opsin and might correspond to rod opsin that has self-associated and/or aggregated, either at the cell surface or during the purification procedure, suggesting that the trafficked mutant rod opsin is less stable than the WT protein. The immunoblot confirmed that expression of EDEM1-HA led to a reduction in the steady state levels of P23H in the input and yet still increased the amount of precipitated P23H, particularly the HMW species (Fig. 7E). Thus, in the presence of EDEM1-HA, a greater proportion of P23H rod opsin is trafficked to the cell surface.
To investigate whether this trafficked mutant rhodopsin was correctly folded, P23H rod opsin transfected in the presence and absence of EDEM1-HA was purified using immunoaffinity chromatography with 1D4 antibody, and regenerated with 9-cis-retinal and the absorption spectrum was measured to reveal the presence of intact photopigment. As previously reported, P23H rod opsin formed little functional pigment (Noorwez et al., 2004). By contrast, EDEM1-HA expression led to a 25-30% increase in the normalised yield of pigment with a λmax of 485 nm. Therefore, expression of EDEM1 can lead to increased yields of folded mutant rod opsin, although proportionally, the yield appears to be reduced relative to the amount trafficked to the cell surface, suggesting that this mutant rod opsin is unstable or that some non-functional rod opsin is also trafficked to the cell surface.
Discussion
Protein degradation in the ER must be finely tuned because underactivity could lead to the accumulation of aberrant polypeptides (Eriksson et al., 2004) and the formation of protein aggregates. However, hyperactivity of ERAD could cause the extraction of intermediates from productive folding, resulting in their premature disposal (Sato et al., 1998). EDEM1 is involved in the regulation of protein degradation in the ER. It has been reported to accelerate the degradation of model glycoprotein substrates via ERAD (Hosokawa et al., 2001; Molinari et al., 2003; Wu et al., 2003). The data presented here suggest that there is a synergy between folding and degradation machineries, such that a physiological client rod opsin is subject to both processes mediated by EDEM1 because it can not only accelerate the degradation, but also promotes the folding and traffic of P23H rod opsin.
It was initially thought that mannose trimming was a prerequisite for glycoprotein binding with EDEM1 (Hosokawa et al., 2003). Evidence suggested that upon release from the calnexin cycle by glucosidase II, misfolded glycoproteins could either be transferred to EDEM1 for degradation or UGGT for re-entry into the calnexin cycle. Hence the trimming of the α1,2 mannose residue by ERManI was thought to establish the fate of the glycoprotein toward degradation (Oda et al., 2003). However, there is now evidence that EDEM1, and the yeast homologue Htm1p, might not require mannose trimming for the recognition of misfolded protein substrates and the interaction might also be mediated by exposed hydrophobic domains (Cormier et al., 2009; Clerc et al., 2009; Molinari et al., 2003). This is consistent with our data, because treatment with the ERManI inhibitor kifunensine did not disrupt the interaction with P23H rod opsin. However, mannosidase activity might still be required for full ERAD activity because kifunensine partially inhibited the EDEM1 enhanced degradation of P23H opsin.
EDEM1 enhances degradation by inhibiting covalent dimer formation and possibly also inhibits the aggregation of misfolded glycoproteins (Hosokawa et al., 2006; Olivari et al., 2006). It is thought that EDEM1 accomplishes this by maintaining the retrotranslocation capacity of terminally misfolded glycoproteins after reduction of sulphhydryl groups, which occurs before retrotranslocation. There are 10 cysteine residues in rod opsin, but there is only one correct disulphide bond – between C110 and C187 – and upon protein misfolding, C187 forms an incorrect disulphide linkage with C185 (Karnik et al., 1988). Therefore, rod opsin might be modified by oxidoreductase enzymes before retrotranslocation and chaperones, such as EDEM1, might have an important role in facilitating this step.
EDEM1 turnover inhibition by chloroquine resulted in enhanced degradation of glycoproteins by premature interruption of protein-folding attempts and reduced secretion of native proteins (Cali et al., 2008). By contrast, EDEM1 overexpression promoted the folding and traffic of mutant P23H rod opsin. It is possible that EDEM1 clears terminally misfolded P23H rod opsin, thereby allowing other chaperones to interact with the folding-competent mutant rod opsin and assist in their productive folding. Alternatively, the enhanced degradation of mutant rod opsin might reduce aggregation, and thereby allow productive folding to occur in the absence of aggregation seeds and proteasome inhibition.
EDEM1 was initially identified in a screen of genes that are upregulated during acute ER stress, but not cytoplasmic stress (Hosokawa et al., 2001). P23H rod opsin induces the UPR and persistent stress might lead to attenuation of the IRE1-XBP1 pathway and induction of the CHOP-mediated proapoptotic pathway to cause photoreceptor death (Lin et al., 2007). EDEM1 functions as a component of the IRE1-XBP1 pathway, hence EDEM1 could be cytoprotective in alleviating stress as a result of misfolded rod opsin, which could be manipulated in the development of therapies for RP.
A rod cell synthesises over 10 million molecules of rhodopsin every day, so photoreceptors are not unlike hepatocytes or antibody-secreting plasma cells. ER-stress transducers are thought to be activated in dedicated secretory cells and this might reflect the EDEM1 expression in photoreceptors. Cells involved in these processes have also displayed enhanced survival and/or differentiation, with activation of the IRE1-XBP1 pathway (Iwakoshi et al., 2003). The presence of an EDEM1-rhodopsin complex in the photoreceptor ER suggests that EDEM1 is part of the normal folding surveillance for rod opsin.
This is the first time an improvement in mutant rod opsin processing has been mediated by a molecular chaperone. Pharmacological chaperones have been known to enhance rod opsin folding (Mendes and Cheetham, 2008; Noorwez et al., 2004; Saliba et al., 2002). However, EDEM1 represents a novel mechanism for the enhanced processing of mutant P23H rod opsin. The newly elucidated dual function of enhanced degradation and improved folding is perhaps crucial for rod opsin RP and other protein misfolding diseases.
Materials and Methods
Materials
Primary antibodies 1D4 and 4D2 against rod opsin were a gift from Robert Molday (Department of Biochemistry and Molecular Biology, University of British Columbia, Vancouver, Canada). Antibodies to calnexin (C4731) and EDEM1 (E8406), anti-HA antibody (H3663) were from Sigma and AV peptide was from Clontech (St-Germain-en-Laye, France). Secondary HRP-conjugated goat anti-mouse and anti-rabbit antibodies from Pierce (Cramlington, UK). Alexa Fluor 594 anti-rabbit and Alexa Fluor 488 anti-mouse IgG (H+L) were from Invitrogen (Paisley, UK). Anti-mouse Cy3-conjugated was obtained from Jackson ImmunoResearch (Stratech Scientific, Suffolk, UK). Goat anti-mouse IgG antibody for ICW analysis (IRDye 800CW) from LI-COR Biosciences (Cambridge, UK). Lipofectamine and Plus reagent were purchased from Invitrogen. 4′,6-diamidino-2-phenylindole (DAPI) for nuclear staining, and Protease Inhibitor Cocktail in DMSO for mammalian cell extracts were purchased from Sigma. The BCA protein assay kit was from Pierce. Rod opsin constructs, untagged rod opsin in pMT3 and rod opsin-GFP, were as described previously (Saliba et al., 2002). MG-132 from BIOMOL (Exeter, UK). Kifunensine was from Calbiochem (Nottingham, UK).
Immunocytochemistry, immunohistochemistry and ICW
SK-N-SH cells were maintained and transfected as described (Mendes and Cheetham, 2008). 24 hours after transfection, cells were washed twice with HBSS and fixed with 4% paraformaldehyde (PFA) for 15 minutes. Cells were permeabilised in 0.5% Triton X-100 for 5 minutes. Nonspecific binding was blocked using 10% FBS, 10% serum of the secondary antibodies species, in phosphate-buffered saline (PBS) (Oxoid, Basingstoke, UK) for 1 hour. Anti-HA (1:1000), anti-EDEM1 (1:250), anti-calnexin (1:600) and 4D2 (1:100) antibodies were incubated in blocking buffer, before washing in PBS. Secondary antibodies conjugated to Cy3 (1:100), Alexa Fluor 488 (1:2000) and Alexa Fluor 594 (1:1000) were used. The staining was analysed using a Nikon Eclipse 80i microscope and images were taken using a Carl Zeiss LSM 510 laser-scanning confocal microscope. Excitation and emission conditions were: GFP and Alexa Fluor 488, 488/505-530 nm; Cy3, 550/570; Alexa Fluor 594, 594/620. The images were exported from LSM Browser and prepared using Adobe Photoshop and Illustrator CS2. Cell morphology studies scored the predominant localisation of rod opsin on the plasma membrane, ER or inclusions as a percentage of total transfected cells. Four fields of ∼100 cells were counted for each condition. Statistical analysis used SPSS software (Chicago, IL). For ICW analysis, SK-N-SH cells were transfected with a 1:0.5 rod opsin:EDEM1-HA ration and fixed in 3% PFA prepared in HBSS for 15 minutes. Nonspecific binding was blocked using Odyssey Blocking Buffer (LI-COR, Cambridge, UK). Rod opsin immunoreactivity was detected with 4D2 (1:100) and secondary antibody (LI-COR, Cambridge, UK; 1:10,000). The cells were scanned using an Odyssey Imager (LI-COR). Eyes from C57BI6 mice were fixed, cryopreserved and cryosections cut at 14 μm. Sections were blocked for 30 minutes in 10% normal goat serum, 3% BSA in PBS. 4D2 was used at 1:250, EDEM1 (1:250) and calnexin (1:600) and Alexa-Fluor-conjugated secondary antibody concentrations were as above. Sections were washed in PBS with 5 μg/ml DAPI in the final wash. The sections were mounted in Vectashield mounting medium (Vector Laboratories, Peterborough, UK). Imaging was carried out as above.
Western blotting, immunoprecipitation and pulse-chase analysis
SK-N-SH cells were transfected with 2.5 μg plasmid DNA/well, 0.5 μg of which was rod opsin and the remainder was made up with chaperones or stuffer plasmid, with 8 μl PLUS and 4 μl Lipofectamine to a total volume of 750 μl, following the method previously described (Mendes and Cheetham, 2008). 24 hours after the end of transient transfection, cells were lysed for 15 minutes at 4°C in 1% n-dodecyl-β-D-maltoside (DM) buffer with 2% protease inhibitor cocktail (Sigma) in PBS. Protein concentration was determined by BCA protein assay kit and modified Laemmli sample buffer added (Mendes and Cheetham, 2008). For deglycosylation reactions, 15 μg total protein in DM soluble cell lysate was digested with EndoH or PNGase F (New England Biolabs, Hitchin, UK). Digestions were carried out overnight at 37°C before resolving by SDS-PAGE. For co-immunoprecipitations, cells were transfected with a threefold molar excess of EDEM1, cell lysates were sonicated (Soniprep 150; Wolf Laboratories, York, UK) and 1D4 (1:50) or anti-EDEM1 (1:250) added overnight at 4°C. Porcine retina was lysed in DM lysis buffer with 2% mammalian protease inhibitor cocktail and 2% phosphatase inhibitors (Sigma). The lysate was precleared and incubated with antibodies as above. Immune complexes were eluted with modified Laemmli sample buffer. For pulse-chase analysis, cells were depleted of L-methionine or L-cysteine at 37°C for 20 minutes. Depletion medium containing 250 μCi/ml of [35S]methionine/cysteine TRAN35S-LABEL (MP Biomedical, Illkirch, France) was added for 20 minutes. Chase medium (50 mM methionine and 50 mM cycloheximide) was added at the end of the labelling interval and ice-cold stop buffer containing 20 mM N-ethylmaleimide at the end of the chase interval. The cells were lysed in DM lysis buffer. Precleared lysates were incubated with 1D4 (1:50) overnight at 4°C. The immune complexes were eluted with 50 μM peptide I (DEASTTVSKTETSQVAPA; Biomatik, Wilmington, DE), corresponding to the 1D4 epitope. Eluted proteins were analysed by SDS-PAGE, radioactive bands detected by Amersham Hyperfilm MP (GE Healthcare).
Cell-surface biotinylation
SK-N-SH cells were transfected with a 1:1 ratio of P23H rod opsin and EDEM1-HA as described above, 24 hours after transient transfection, cells were washed three times with PBS solution containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBS/CM). Immediately before use, a 10 mM Sulpho-NHS-SS-biotin (Perbio, Cramlington, UK) solution was prepared and added to each well and incubated for 30 minutes at room temperature. The biotinylation solution was removed and the cells were washed once with ice-cold PBS/CM and quenched with PBS containing 0.1 M glycine, followed by three washes with ice-cold PBS. Cells were harvested in 200 μl cold DM buffer containing 2% protease inhibitor cocktail (Sigma). The samples were centrifuged at 16,060 g for 15 minutes at 4°C. Resulting supernatants, less 50 μl, which was kept aside as input sample for western analysis, were incubated with 100 μl of 50% streptavidin agarose (Perbio) prepared by washing three times in DM buffer. After overnight incubation with rotation at 4°C, the samples were centrifuged at 3000 g for 1 minute at 4°C, washed three times in cold DM buffer containing protease inhibitor cocktail, and eluted in 50 μl modified reducing Laemmli sample buffer, which will release the biotin from the precipitated proteins. Eluted proteins were analysed by SDS-PAGE, probed with the 4D2 opsin antibody and detected with ECL (GE Healthcare).
Rod opsin purification
Mutant P23H rod opsin was expressed, either alone or co-transfected with EDEM1 (1:3 ratio) in transiently transfected monkey kidney COS-1 cells, essentially as described (Aguilà et al., 2009). Briefly, 50 μM 9-cis-retinal was added to a cell suspension, in the dark, 48 hours after transfection. The regenerated cells were subsequently solubilised in 1% DM, and the proteins were purified by immunoaffinity chromatography by using the 1D4 monoclonal antibody. Proteins were eluted in 2 mM Na2HPO4, pH 6, 0.05% DM and spectroscopically characterised. UV-visible absorption spectra were measured with a Unicam UV 500 spectrophotometer. All spectra were recorded with a bandwidth of 2 nm, a response time of 1 second, and a scan speed of 240 nm/minute. Baseline correction was performed with elution buffer (2 mM Na2HPO4, pH 6, 0.05% DM) in 150 μl quartz cuvettes, and all measurements were carried out in a darkroom under dim red light using Kodak No2 filters. Spectra were normalised for protein absorbance at 280 nm and corrected for light scattering with SigmaPlot version 8.0 (SPSS).
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/24/4465/DC1
We would like to thank Professor Molinari for the gift of EDEM1-HA, sh-EDEM1 plasmids and Professor Molday for rhodopsin antibodies. This work was supported by Fight for Sight (to M.E.C.), the Wellcome Trust (to M.E.C.), the National Institute of Health Research (NIHR; to M.E.C.) and Grant SAF2008-04943-C02-02 from the Spanish Ministry of Science (to P.G.). M.K. is a Fight for Sight Prize student, M.A. is supported by a FPI Predoctoral Fellowship from the Spanish Ministry of Science. Deposited in PMC for release after 6 months.
References
- Aguilà., M, Toledo, D., Morillo, M., Dominguez, M., Vaz, B., Alvarez, R., de Lera, A. R. and Garriga, P. (2009). Structural coupling of 11-cis-7-methyl-retinal and amino acids at the ligand binding pocket of rhodopsin. Photochem. Photobiol. 85, 485-493. [DOI] [PubMed] [Google Scholar]
- Bennett, E. J., Bence, N. F., Jayakumar, R. and Kopito, R. R. (2005). Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell 17, 351-365. [DOI] [PubMed] [Google Scholar]
- Cali, T., Galli, C., Olivari, S. and Molinari, M. (2008). Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys Res Commun. 371, 405-410. [DOI] [PubMed] [Google Scholar]
- Clerc, S., Hirsch, C., Oggier, D. M., Deprez, P., Jakob, C., Sommer, T. and Aebi, M. (2009). Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J. Cell Biol. 184, 159-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier, J. H., Tamura, T., Sunryd, J. C. and Hebert, D. N. (2009). EDEM1 recognition and delivery of misfolded proteins to the SEL1L-containing ERAD complex. Mol. Cell 34, 627-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja, T. P., McGee, T. L., Reichel, E., Hahn, L. B., Cowley, G. S., Yandell, D. W., Sandberg, M. A. and Berson, E. L. (1990). A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 343, 364-366. [DOI] [PubMed] [Google Scholar]
- Eriksson, K. K., Vago, R., Calanca, V., Galli, C., Paganetti, P. and Molinari, M. (2004). EDEM contributes to maintenance of protein folding efficiency and secretory capacity. J. Biol. Chem. 279, 44600-44605. [DOI] [PubMed] [Google Scholar]
- Hosokawa, N., Wada, I., Hasegawa, K., Yorihuzi, T., Tremblay, L. O., Herscovics, A. and Nagata, K. (2001). A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2, 415-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa, N., Tremblay, L. O., You, Z., Herscovics, A., Wada, I. and Nagata, K. (2003). Enhancement of endoplasmic reticulum (ER) degradation of misfolded Null Hong Kong alpha1-antitrypsin by human ER mannosidase I. J. Biol. Chem. 278, 26287-26294. [DOI] [PubMed] [Google Scholar]
- Hosokawa, N., Wada, I., Natsuka, Y. and Nagata, K. (2006). EDEM accelerates ERAD by preventing aberrant dimer formation of misfolded alpha1-antitrypsin. Genes Cells 11, 465-476. [DOI] [PubMed] [Google Scholar]
- Illing, M. E., Rajan, R. S., Bence, N. F. and Kopito, R. R. (2002). A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J. Biol. Chem. 277, 34150-34160. [DOI] [PubMed] [Google Scholar]
- Iwakoshi, N. N., Lee, A. H., Vallabhajosyula, P., Otipoby, K. L., Rajewsky, K. and Glimcher, L. H. (2003). Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4, 321-329. [DOI] [PubMed] [Google Scholar]
- Karnik, S. S., Sakmar, T. P., Chen, H. B. and Khorana, H. G. (1988). Cysteine residues 110 and 187 are essential for the formation of correct structure in bovine rhodopsin. Proc. Natl. Acad. Sci. USA 85, 8459-8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal, S. and Khorana, H. G. (1994). Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry 33, 6121-6128. [DOI] [PubMed] [Google Scholar]
- Kosmaoglou, M. and Cheetham, M. E. (2008). Calnexin is not essential for mammalian rod opsin biogenesis. Mol. Vis. 14, 2466-2474. [PMC free article] [PubMed] [Google Scholar]
- Lin, J. H., Li, H., Yasumura, D., Cohen, H. R., Zhang, C., Panning, B., Shokat, K. M., Lavail, M. M. and Walter, P. (2007). IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X., Garriga, P. and Khorana, H. G. (1996). Structure and function in rhodopsin: correct folding and misfolding in two point mutants in the intradiscal domain of rhodopsin identified in retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 93, 4554-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes, H. F. and Cheetham, M. E. (2008). Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 17, 3043-3054. [DOI] [PubMed] [Google Scholar]
- Mendes, H. F., van der, S. J., Chapple, J. P. and Cheetham, M. E. (2005). Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol. Med. 11, 177-185. [DOI] [PubMed] [Google Scholar]
- Molinari, M., Calanca, V., Galli, C., Lucca, P. and Paganetti, P. (2003). Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299, 1397-1400. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa, K., Nishikawa, S., Hosokawa, N., Nagata, K. and Endo, T. (2001). Mnl1p, an alpha-mannosidase-like protein in yeast Saccharomyces cerevisiae, is required for endoplasmic reticulum-associated degradation of glycoproteins. J. Biol. Chem. 276, 8635-8638. [DOI] [PubMed] [Google Scholar]
- Noorwez, S. M., Malhotra, R., McDowell, J. H., Smith, K. A., Krebs, M. P. and Kaushal, S. (2004). Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J. Biol. Chem. 279, 16278-16284. [DOI] [PubMed] [Google Scholar]
- Oda, Y., Hosokawa, N., Wada, I. and Nagata, K. (2003). EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394-1397. [DOI] [PubMed] [Google Scholar]
- Olivari, S., Cali, T., Salo, K. E., Paganetti, P., Ruddock, L. W. and Molinari, M. (2006). EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem. Biophys. Res. Commun. 349, 1278-1284. [DOI] [PubMed] [Google Scholar]
- Saliba, R. S., Munro, P. M., Luthert, P. J. and Cheetham, M. E. (2002). The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J. Cell Sci. 115, 2907-2918. [DOI] [PubMed] [Google Scholar]
- Sato, S., Ward, C. L. and Kopito, R. R. (1998). Cotranslational ubiquitination of cystic fibrosis transmembrane conductance regulator in vitro. J. Biol. Chem. 273, 7189-7192. [DOI] [PubMed] [Google Scholar]
- Sung, C. H., Schneider, B. G., Agarwal, N., Papermaster, D. S. and Nathans, J. (1991). Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 88, 8840-8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, C. H., Davenport, C. M. and Nathans, J. (1993). Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Clustering of functional classes along the polypeptide chain. J. Biol. Chem. 268, 26645-26649. [PubMed] [Google Scholar]
- Wu, Y., Swulius, M. T., Moremen, K. W. and Sifers, R. N. (2003). Elucidation of the molecular logic by which misfolded alpha 1-antitrypsin is preferentially selected for degradation. Proc. Natl. Acad. Sci. USA 100, 8229-8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, H., Matsui, T., Hosokawa, N., Kaufman, R. J., Nagata, K. and Mori, K. (2003). A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 4, 265-271. [DOI] [PubMed] [Google Scholar]
- Zuber, C., Cormier, J. H., Guhl, B., Santimaria, R., Hebert, D. N. and Roth, J. (2007). EDEM1 reveals a quality control vesicular transport pathway out of the endoplasmic reticulum not involving the COPII exit sites. Proc. Natl. Acad. Sci. USA 104, 4407-4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}