

Introduction
Epidermolysis bullosa (EB) represents a spectrum of conditions that are characterized by blistering and mechanical fragility of the skin. There is tremendous genetic heterogeneity and marked variation in clinical phenotypes in the multiple EB disorders. The most recent classification recognizes four major EB groupings and over 30 EB subtypes 1. The four major EB groups include intraepidermal EB (Simplex), junctional EB, dermolytic EB (Dystrophic), and mixed EB (Kindler syndrome). The molecular basis is now known for 13 of EB subtypes. 1 Depending on the specific EB type there can be significant morbidity involving the soft and hard tissues of the craniofacial complex.
Individuals with EB display tremendous diversity in the various tissues and body systems involved and phenotypic severity. 2-4 Similarly, the craniofacial and oral manifestations of the different EB types vary markedly in both character and severity depending largely on the EB type.5, 6 The tissues affected and the phenotypes displayed in affected individuals are closely related to the specific abnormal or absent proteins resulting from the causative genetic mutations for these disorders. For example, Type VII collagen is critical for maintaining the integrity of the oral mucosa in the same manner it is in skin. It is not essential for normal development in the forming tooth bud. Consequently individuals with Type VII collagen mutations typically have a developmentally normal dentition but can have severely affected oral soft tissues. In contrast, laminin 332 is highly expressed during tooth development so individuals with mutations that affect laminin 332 function have defects in the enamel of their teeth.7-9 In the following chapter the major oral manifestations will be reviewed for different EB subtypes and related to the causative genetic mutations and gene expression.
Intraepidermal Epidermolysis Bullosa
The EB Simplex subtypes are caused by mutations in the PKP1,DSP, KRT5, KRT14, PLEC1, ITGA6 and genes.1 These genes all cause intra-epidermal cleavage in the skin and are all expressed by the oral mucosa which, like skin, also is comprised of a stratified epithelium.10-12 Not surprisingly individuals with EB Simplex also exhibit an increased fragility of the oral mucosa with a high percentage of individuals experiencing blistering and ulceration of the oral mucosa.5 In most cases these will be localized and occur most often secondary to trauma or tissue manipulation, however, some individuals can experience significant oral blistering and severe mucosal involvement. Typically oral soft tissue lesions heal without scarring although some severely affected EB Simplex subtypes can display some oral scarring (e.g. Dowling Meara) (Figure 1).
1.
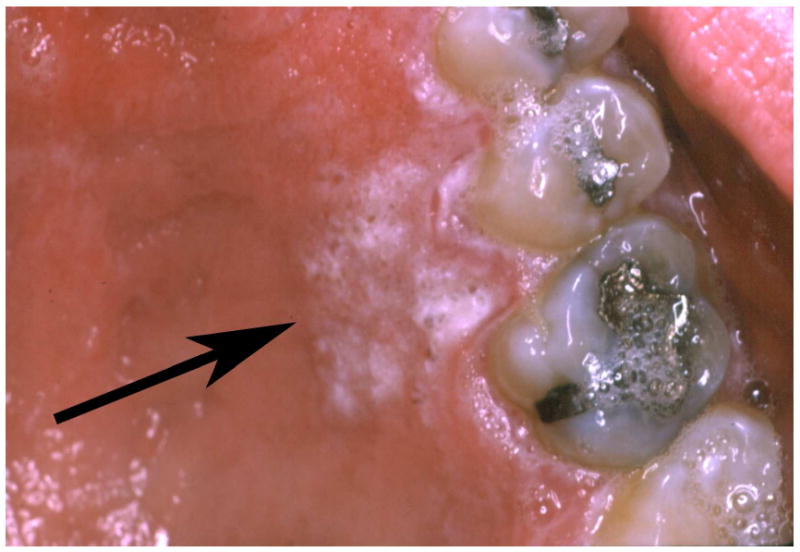
This adult with EBS Dowling Meara shows a normal soft tissue palatal architecture and a localized area of gingival hyperkeratosis (arrow).
Although many of the genes that are known to be causative of EB Simplex are known to be expressed in the odontogenic epithelium of developing teeth, the dentition in EB Simplex tends to form normally.9 These genes are also known to be expressed by the epithelial glands of the salivary tissues. Salivary function in appears to be normal in people with the EB Simplex subtypes that have been tested.13 The limited intra oral soft tissue morbidity in EB Simplex and the normal tooth formation are likely the primary reasons that individuals with these EB subtypes have a prevalence of dental caries that is similar to unaffected populations.14
Junctional Epidermolysis Bullosa
The Junctional forms of EB are caused by mutations in LAMA3, LAMB3, LAMC3, COL17A1, ITG6A, and ITGB4 that are important in basement membrane mediated cell adhesion.1, 15, 16 The proteins transcribed from these genes are important in epithelia cell adhesion in both the oral mucosa and the developing tooth bud.8, 17 The tissue fragility resulting from these mutations is variable but almost all individuals with the Junctional forms of EB have an increased fragility of the oral mucosa that is accompanied by blister formation and ulceration.5, 18 In some individuals this can be relatively severe. Despite the high prevalence of oral soft tissue lesions in the different Junctional EB subtypes, most affected individuals do not have significant oral scarring. The soft tissue mobility and architecture remains relatively normal. One noted exception to this is the Herlitz EB subtype that is characterized by exuberant peri-oral granulation tissue (Figure 2). This frequently results in a reduction in the oral opening (microstomia) and some loss of tissue mobility in the lips and peri-oral tissues.5
2.
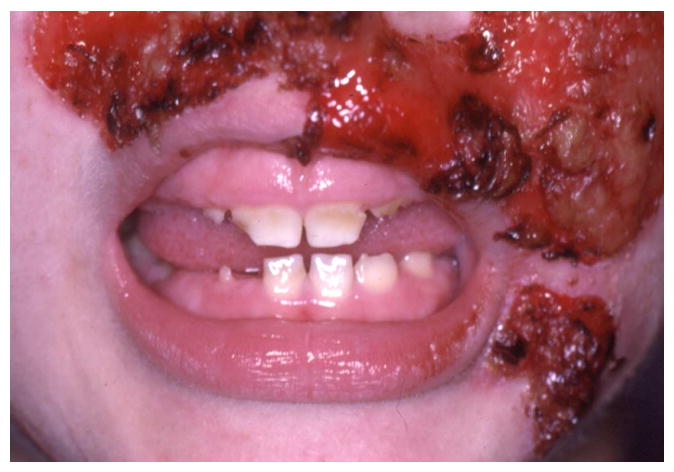
The Herlitz Junctional EB subtype is characterized by generalized enamel hypoplasia that results in yellow brown coloration of the teeth that are frequently spaced as seen in this child (A). Scanning electron microscopy of the tooth surfaces demonstrates the severe pitting that is often seen with JEB (B). Individuals that are heterozygous for JEB associated genes can have enamel defects as seen in this male heterozygous for a COL17A1 mutation (C compliments of Dr. Dedee Murrell and Dr Richard Widmer).
The genes that are causative of the Junctional EB subtypes are all critical for normal tooth formation.8, 17 Specifically these genes produce proteins that are involved in cell adhesion of the odontogenic epithelium that gives rise to the cells that produce the dental enamel, ameloblasts. Ameloblasts secrete an extracellular matrix and maintain contact and adhesion to the adjacent ameloblasts and thereby control the microenvironment that is critical for allowing normal mineralization of the enamel. When cell adhesion between ameloblasts is lost then enamel defects are created. Dysfunctional ameloblast adhesion can result in leaking of serum fluids into the developing enamel resulting in a retention of albumin and decreased mineralization.19 In the case of individuals with Junctional EB the enamel lesions can vary from generalized pitting to a generalized hypoplasia leaving only a very thin layer of enamel on the tooth surface (Figure 2). Some cases of Herlitz EB subtype also exhibit abnormal tooth eruption.20 This is often most notable in the molar regions but can affect anterior teeth as well. Abnormal tooth eruption could occur due dysfunction of the odontogenic epithelium that is known to play an important role in tooth eruption. Heterozygous carriers of COL17A1 mutations have been shown to have enamel defects that range from horizontal hypoplastic bands to white mottled enamel.21
Individuals with Junctional EB are at increased risk for developing dental caries.14 This is thought to be primarily a function of their having marked enamel defects. The presence of extensive pitting over the tooth surface creates non-cleansable areas that are ideal for microbial growth and substrate retention that are known to cause dental caries. Generalized thin enamel that is frequently rough in nature also reduces the tooth's primary resistance to the development and progression of caries. Salivary function appears to be normal in most individuals with Junctional EB subtypes.13 Occasional affected individuals can develop lesions resulting in transient occlusion of a salivary duct. These episodes typically self correct with no specific treatment.
Dermolytic Epidermolysis Bullosa
The Dystrophic EB types are caused by mutations in COL7A1 gene that codes for anchoring fibril protein that are located below the basal lamina at the dermal-epidermal basement membrane zone.22 The dominant subtype tends to be less severe than the generalized recessive subtype with severity being related to the amount and functionality of the anchoring fibril protein that is present. Type VII collagen is present in the basement membrane zone of the oral mucosa and is present during the early stages of tooth formation.23 The COL7A1 gene is not expressed by the ameloblasts.
The soft tissue manifestations of Dystrophic EB range from relatively mild to extremely severe.5, 24-27 For example, in Dominant Dystrophic EB the oral manifestations can involve an increased tissue fragility but infrequent blistering. Blistering often can be induced relatively easily and dental treatments in even mildly affected Dominant Dystrophic EB should be approached with extra diligence to reduce soft tissue trauma. Individuals with severe generalized recessive dystrophic EB typically have extreme fragility of their oral and perioral mucosa. This is usually evident shortly after birth and can interfere with the neonate's ability to suckle. The oral ulcerations can affect all areas of the oral mucosa including the tongue. The lesions heal with scarring. The continual process of blister formation and healing with scarring results in marked changes in the oral architecture. The tongue looses the lingual papillae and becomes bound down to the floor of the mouth (ankyloglossia) (Figure 3). Anatomical structures such as the palatal rugae are ablated (Figure 4). The oral vestibules that normally form corridors for food clearance between the teeth and lips and cheeks become obliterated with the soft tissue attachment advancing till it is just below the crowns of the teeth. The soft tissues defining the oral opening fail to grow normally due to scarring resulting in a typically markedly restricted oral aperture. The presence of severe microstomia can impede the degree to which affected individuals can open their mouth dramatically limiting the distance between the teeth even when they are fully open (Figure 5).
3.
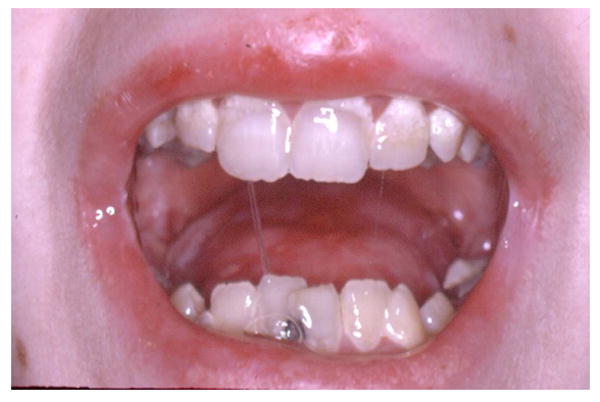
Severe generalized RDEB is associated with ankyloglossia and a loss or the normal lingual papillae that cover the dorsal surface of the tongue leaving a smoothened appearance that is frequently decorated with ulcerations.
4.
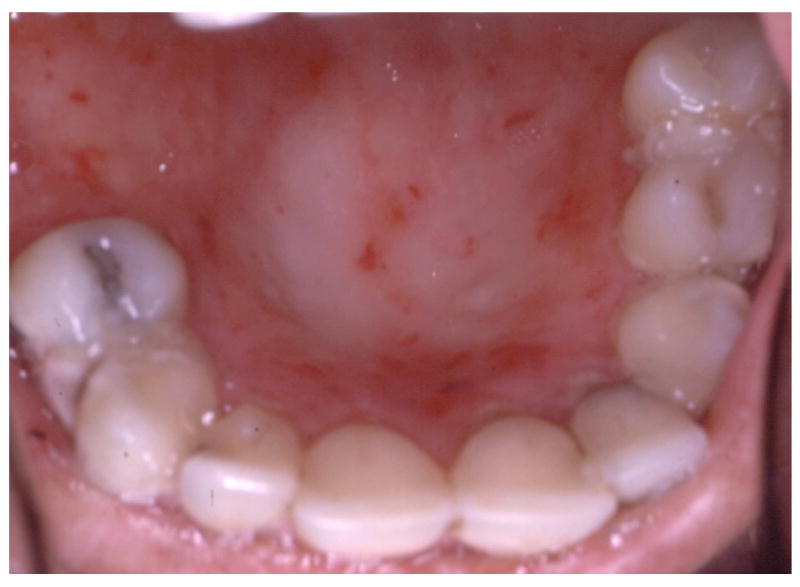
Continual blistering and healing associated with severe generalized RDEB results in loss of normal anatomical oral features such as the palatal rugae leaving a smooth and ulcerated roof of the mouth.
5.
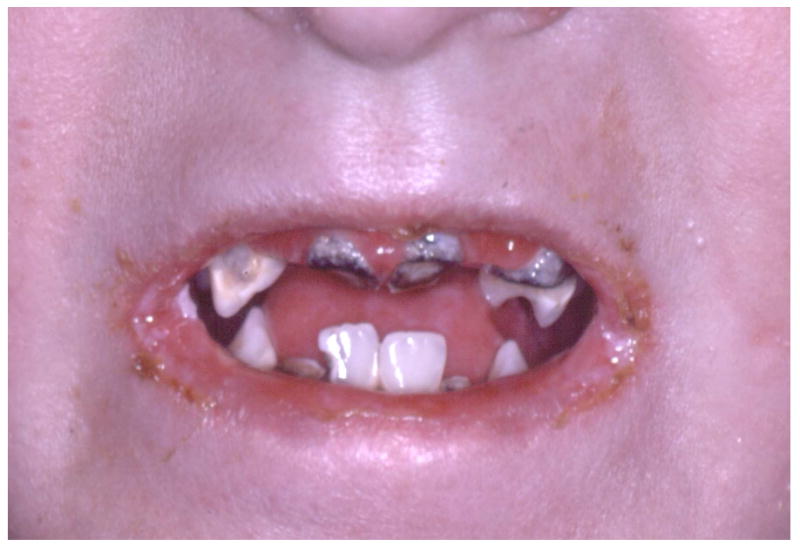
Microstomia and reduced mobility of the oral tissues are typical features of severe generalized RDEB that limits oral access and can contribute to the formation of rampant dental caries as seen in this child.
Milia are frequently observed on the skin of individuals with EB and these can occur intra-orally as well. These intra-oral keratocysts occur most commonly in the dermolytic forms of EB with about 50% of individuals with dominant and recessive forms having milia compared with about 10 to 20% of individuals with EB Simplex.5
It is well-known that individuals with certain forms of EB are at increased risk for developing squamous cell carcinomas and this is clearly the case in severe generalized recessive dystrophic EB.28, 29 While carcinomas occur with far greater frequency on the skin in individuals with EB, they can occur intra-orally.5, 18, 29, 30 Individuals with severe generalized recessive dystrophic EB are at increased risk of oral squamous cell carcinoma formation and should therefore be extra vigilant in monitoring changes in oral ulcerations such as the development of raised, indurated borders.
Type VII collagen is not expressed by ameloblasts and the enamel appears to generally form normally in individuals with the dermolytic forms of EB.9, 31 Despite having relatively normal tooth formation the prevalence of dental caries and resulting dental morbidity in severe generalized recessive dystrophic EB can be severe.14, 32, 33 The tremendous oral soft tissue involvement results in the need to consume relatively soft diets that are frequently high in calories to meet the nutritional needs of the individual. In the presence of marked oral blistering affected individuals frequently eat slowly and with increased frequency. The loss of normal tongue mobility and obliteration of the oral vestibule decrease the normal food clearance causing additional prolongation of the dental surfaces to potentially cariogenic substrates. Despite normal salivary secretion in most people with dermolytic EB subtypes, the oral cavity tends to be inoculated with high numbers of bacteria and there tends to be excessive tooth plaque formation that further promote the formation of dental caries.33 Taken together these factors produce an extremely high risk for dental caries in individuals with severe generalized recessive dystrophic EB that can be challenging to prevent and difficult to treat. Many severely affected individuals have tremendous difficulty performing normal oral hygiene due to their extreme soft tissue fragility and even the use of anticariogenic mouth rinses can be unpleasant due to the presence of alcohol or strong flavoring agents.34 Mixed Epidermolysis Bullosa (Kindler Syndrome)
Kindler syndrome is an autosomal recessive genodermatosis caused by mutations in the KIND1 gene that encodes for Kindlin-1 that is a component of focal contacts in basal keratinocytes.35 Kindlin-1 is known to be expressed by the oral epithelium including the surface of the tongue.4, 6 It is not known whether it is expressed by the odontogenic epithelium. Oral blistering in the neonate and infant can be severe in Kindler syndrome.1 The severity of the oral involvement regarding tissue fragility diminishes with age. Individuals affected with Kindler syndrome are at risk for developing marked periodontal disease that can have its onset during the teenage years. The Kindlin-1 protein is expressed by the epithelium that attaches the oral mucosa to the tooth.36 The abnormal functioning of kindlin-1 in forming normal focal adhesion in the basal keratinocytes appears to cause abnormal attachment and a predisposition to early onset periodontal disease. Inflammation and gingival hyperplasia have been noted even in young children with Kindler syndrome suggesting that periodontal health may be compromised well before clinical signs of periodontal disease and alveolar bone loss.37
It is not known if kindling-1 is expressed by the odontogenic epithelium, however the dentition of affected individuals appears to be normal. Similarly, the salivary function and risk of dental caries is unknown in this rare syndrome.
Summary
The diverse conditions classified as EB share skin fragility and blistering and can have marked oral involvement. The genes that cause EB are also involved in development and maintenance of a variety of oral tissues. The specific oral manifestations and their severity are defined by the expression of genes that are important in cell adhesion and integrity. Some of the EB associated proteins are critical for normal enamel formation (eg laminin 332) and when abnormal result in varying degrees of enamel hypoplasia. Other proteins such as Kindlin-1 are important in maintaining mucosal integrity but appear not to affect tooth formation. Some of the oral morbidity associated with the EB conditions results from secondary effects of the condition such as dental caries in the severe generalized recessive dystrophic EB subtype. Phenotype-genotype studies evaluating specific EB subtypes and their allelic mutations have are complicated by the multitudes of mutations in the different genes known to cause EB and the many variable resulting phenotyps.38, 39 While much has been learned over the past 30 years regarding the molecular basis of these conditions and their association with specific phenotypes, further phenotype- genotype studies regarding the oral manifestations of these conditions would not only advance our understanding of EB but also the role of these and yet to be discovered EB associated genes with oral development and health.
Table 1.
Oral Manifestations in Common Epidermolysis Bullosa Subtypes
| EB Type | OMIM # | Oral Blistering | Oral Scarring | Microstomia | Enamel Defects |
|---|---|---|---|---|---|
| Simplex | |||||
| Localized | #131800 | + | - | - | - |
| Generalized | #131900 | + | - | - | - |
| Dowling Meara | #131760 | + | -+ | - | - |
| Junctional | |||||
| Non-herlitz | #226650 | + | - | - | ++ |
| Herlitz | #226700 | + | -* | + | ++ |
| Dystrophic | |||||
| Dominant | #131750 | + | -+ | - | - |
| Recessive | #226600 | ++ | ++ | ++ | - |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008 Jun;58(6):931–950. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Gedde-Dahl T., Jr Sixteen types of epidermolysis. On the clinical discrimination, therapy, and prenatal diagnosis. Acta Dermatovener (Stockholm) 1981 95:74–87. [PubMed] [Google Scholar]
- 3.Fine JD, Wright JT. Epidermolysis Bullosa. In: Demis JD, editor. Clinical Dermatology. Vol. 2. Philadelphia: Lippincott-Raven; 1995. pp. 1–35. [Google Scholar]
- 4.Lai-Cheong JE, Tanaka A, Hawche G, et al. Kindler syndrome: a focal adhesion genodermatosis. Br J Dermatol. 2009 Feb;160(2):233–242. doi: 10.1111/j.1365-2133.2008.08976.x. [DOI] [PubMed] [Google Scholar]
- 5.Wright JT, Fine JD, Johnson LB. Oral soft tissues in hereditary epidermolysis bullosa. Oral Surg Oral Med Oral Pathol. 1991;71:440–446. doi: 10.1016/0030-4220(91)90426-d. [DOI] [PubMed] [Google Scholar]
- 6.Wiebe CB, Petricca G, Hakkinen L, Jiang G, Wu C, Larjava HS. Kindler syndrome and periodontal disease: review of the literature and a 12-year follow-up case. J Periodontol. 2008 May;79(5):961–966. doi: 10.1902/jop.2008.070167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meneguzzi B, Marinkovich MP, Aberdam D, Pisani A, Burgeson R, Ortonne JP. Kalinin is abnormally expressed in epithelial basement membranes of Herlitz's Junctional epidermolysis bullosa patients. Exp Dermatol. 1992;1:221–229. doi: 10.1111/j.1600-0625.1992.tb00080.x. [DOI] [PubMed] [Google Scholar]
- 8.Aberdam D, Aguzzi A, Baudoin C, Galliano MF, Ortonne JP, Meneguzzi G. Developmental expression of nicein adhesion protein (laminin-5) subunits suggests multiple morphogenic roles. Cell Adhes Communic. 1994;2:115–129. doi: 10.3109/15419069409004431. [DOI] [PubMed] [Google Scholar]
- 9.Wright JT, Fine JD, Johnson LB. Developmental defects of enamel in humans with hereditary epidermolysis bullosa. Archs Oral Biol. 1993;38:945–955. doi: 10.1016/0003-9969(93)90107-w. [DOI] [PubMed] [Google Scholar]
- 10.Pelissier A, Ouhayoun JP, Sawaf MH, Forest N. Evolution of cytokeratin expression in developing human tooth germ. J Biol Buccale. 1990;18:99–108. [PubMed] [Google Scholar]
- 11.Modolo F, Martins MT, Loducca SV, de Araujo VC. Expression of integrin subunits alpha2, alpha3, alpha5, alphav, beta1, beta3 and beta4 in different histological types of ameloblastoma compared with dental germ, dental lamina and adult lining epithelium. Oral Dis. 2004 Sep;10(5):277–282. doi: 10.1111/j.1601-0825.2004.01028.x. [DOI] [PubMed] [Google Scholar]
- 12.Kieffer-Combeau S, Meyer JM, Lesot H. Cell-matrix interactions and cell-cell junctions during epithelial histo-morphogenesis in the developing mouse incisor. Int J Dev Biol. 2001 Sep;45(56):733–742. [PubMed] [Google Scholar]
- 13.Wright JT, Childers NK, Evans KL, Johnson LB, Fine JD. Salivary function of persons with hereditary epidermolysis bullosa. Oral Surg Oral Med Oral Pathol. 1991;71:553–559. doi: 10.1016/0030-4220(91)90361-f. [DOI] [PubMed] [Google Scholar]
- 14.Wright JT, Fine JD, Johnson L. Dental caries risk factors in hereditary epidermolysis bullosa. Pediatr Dent. 1994;16:427–432. [PubMed] [Google Scholar]
- 15.Uitto J, Pulkkinen L, Christiano AM. Molecular basis of the dystrophic and junctional forms of epidermolysis bullosa: Mutations in the type VII collagen and kalinin (Laminin 5) genes. J Invest Dermatol. 1994;103:39S–46S. doi: 10.1111/1523-1747.ep12398967. [DOI] [PubMed] [Google Scholar]
- 16.McGrath JA, Gatalica B, Christiano AM, et al. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat Genet. 1995 Sep;11(1):83–86. doi: 10.1038/ng0995-83. [DOI] [PubMed] [Google Scholar]
- 17.Asaka T, Akiyama M, Domon T, et al. Type XVII collagen is a key player in tooth enamel formation. Am J Pathol. 2009 Jan;174(1):91–100. doi: 10.2353/ajpath.2009.080573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sedano HO, Gorlin RJ. Epidermolysis bullosa. Oral Surg Oral Med Oral Pathol. 1989;67:555–563. doi: 10.1016/0030-4220(89)90272-7. [DOI] [PubMed] [Google Scholar]
- 19.Kirkham J, Robinson C, Strafford SM, et al. The chemical composition of tooth enamel in junctional epidermolysis bullosa. Arch Oral Biol. 2000 May;45(5):377–386. doi: 10.1016/s0003-9969(00)00003-0. [DOI] [PubMed] [Google Scholar]
- 20.Brooks JK, Bare LC, Davidson J, Taylor LS, Wright JT. Junctional epidermolysis bullosa associated with hypoplastic enamel and pervasive failure of tooth eruption: Oral rehabilitation with use of an overdenture. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008 Apr;105(4):e24–28. doi: 10.1016/j.tripleo.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 21.Murrell DF, Pasmooij AM, Pas HH, et al. Retrospective diagnosis of fatal BP180-deficient non-Herlitz junctional epidermolysis bullosa suggested by immunofluorescence (IF) antigen-mapping of parental carriers bearing enamel defects. J Invest Dermatol. 2007 Jul;127(7):1772–1775. doi: 10.1038/sj.jid.5700766. [DOI] [PubMed] [Google Scholar]
- 22.Christiano AM, Greenspan DS, Hoffman GG, et al. A missense mutation in type VII collagen in two affected siblings with recessive dystrophic epidermolysis bullosa. Nat Genet. 1993 May;4(1):62–66. doi: 10.1038/ng0593-62. [DOI] [PubMed] [Google Scholar]
- 23.Heikinheimo K, Morgan PR, Happonen RP, Stenman G, Virtanen I. Distribution of extracellular matrix proteins in odontogenic tumors and developing teeth. Virchows Archiv B Cell Pathol. 1991;61:101–109. doi: 10.1007/BF02890411. [DOI] [PubMed] [Google Scholar]
- 24.Album MM, Gaisin A, Lee KWT, Buck BE, Sharrar WG, Gill FM. Epidermolysis bullosa dystrophica polydysplastica. Oral Surg Oral Med Oral Pathol. 1977;43:859–872. doi: 10.1016/0030-4220(77)90078-0. [DOI] [PubMed] [Google Scholar]
- 25.Crawford E, Burkes EJ, Briggaman R. Herediatry epidermolysis bullosa: oral manifestations and dental therapy. Oral Surg. 1976;42:490–500. doi: 10.1016/0030-4220(76)90296-6. [DOI] [PubMed] [Google Scholar]
- 26.Winter G. Dental Problems in Epidermolysis Bullosa. In: Priestley GC, Tidman MJ, Weiss JB, Eady RAJ, editors. Epidermolysis Bullosa: A Comprehensive Review of Classification, Management and Laboratory Studies. Berkshire UK: D.E.B.R.A.; 1990. pp. 21–27. [Google Scholar]
- 27.Wright JT, Fine JD, Johnson LB, Steinmetz TT. Oral involvement of recessive dystrophic epidermolysis bullosa inversa. Am J Med Genet. 1993;47:1184–1188. doi: 10.1002/ajmg.1320470811. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz RA, Birnkrant AP, Rubenstein DJ, Kim U, Burgess BH, Stoll HL. Squamous cell carcinoma in dominant type epidermolysis bullosa dystrophica. Cancer. 1981;47:615–620. doi: 10.1002/1097-0142(19810201)47:3<615::aid-cncr2820470332>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 29.Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol. 2009 Feb;60(2):203–211. doi: 10.1016/j.jaad.2008.09.035. [DOI] [PubMed] [Google Scholar]
- 30.Reed WB, College J, Francis MJO, et al. Epidermolysis bullosa dystrophica with epidermal neoplasms. Arch Dermatol. 1974;110:894–902. [PubMed] [Google Scholar]
- 31.Kirkham J, Robinson C, Strafford SM, Bonass WA, Brookes SJ, Wright JT. The chemical composition of tooth enamel in recessive dystrophic epidermolysis bullosa: significance with respect to dental caries. J Dent Res. 1996;75:1672–1678. doi: 10.1177/00220345960750090901. [DOI] [PubMed] [Google Scholar]
- 32.De Benedittis M, Petruzzi M, Favia G, Serpico R. Oro-dental manifestations in Hallopeau-Siemens-type recessive dystrophic epidermolysis bullosa. Clin Exp Dermatol. 2004 Mar;29(2):128–132. doi: 10.1111/j.1365-2230.2004.01485.x. [DOI] [PubMed] [Google Scholar]
- 33.Harris JC, Bryan RA, Lucas VS, Roberts GJ. Dental disease and caries related microflora in children with dystrophic epidermolysis bullosa. Pediatr Dent. 2001 Sep-Oct;23(5):438–443. [PubMed] [Google Scholar]
- 34.Wright JT, Fine JD, Johnson LB. Hereditary epidermolysis bullosa: oral manifestations and dental management. Pediatr Dent. 1993;15:242–248. [PubMed] [Google Scholar]
- 35.Jobard F, Bouadjar B, Caux F, et al. Identification of mutations in a new gene encoding a FERM family protein with a pleckstrin homology domain in Kindler syndrome. Hum Mol Genet. 2003 Apr 15;12(8):925–935. doi: 10.1093/hmg/ddg097. [DOI] [PubMed] [Google Scholar]
- 36.Abdallah BM, Jensen CH, Gutierrez G, Leslie RG, Jensen TG, Kassem M. Regulation of human skeletal stem cells differentiation by Dlk1/Pref-1. J Bone Miner Res. 2004 May;19(5):841–852. doi: 10.1359/JBMR.040118. [DOI] [PubMed] [Google Scholar]
- 37.Wiebe CB, Penagos H, Luong N, et al. Clinical and microbiologic study of periodontitis associated with Kindler syndrome. J Periodontol. 2003 Jan;74(1):25–31. doi: 10.1902/jop.2003.74.1.25. [DOI] [PubMed] [Google Scholar]
- 38.Varki R, Sadowski S, Pfendner E, Uitto J. Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J Med Genet. 2006 Aug;43(8):641–652. doi: 10.1136/jmg.2005.039685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varki R, Sadowski S, Uitto J, Pfendner E. Epidermolysis bullosa. II. Type VII collagen mutations and phenotype-genotype correlations in the dystrophic subtypes. J Med Genet. 2007 Mar;44(3):181–192. doi: 10.1136/jmg.2006.045302. [DOI] [PMC free article] [PubMed] [Google Scholar]