

Abstract
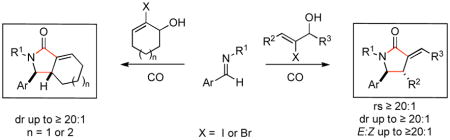
A convergent process for the assembly of stereodefined mono- and bicyclic exo-alkylidene γ-lactams is described that proceeds through the union of β-halo allylic alcohols, aromatic imines and CO. Overall, regio- and stereoselective Ti-mediated reductive cross-coupling, followed by Pd-catalyzed carbonylation can be performed in a one or two-pot procedure, defining a highly selective three-component coupling process for heterocycle synthesis.
Nitrogen-containing heterocycles are ubiquitous structural motifs in natural products and small molecules of biomedical relevance.1 Among this class, stereodefined pyrrolidines and γ-lactams are abundant (Figure 1). A wealth of chemical pathways are indeed available for the synthesis of these functionalized heterocycles.2 However, strategic considerations for the preparation of highly substituted and stereodefined systems often limit the utility of many available methods. In a program aimed at defining convergent coupling reactions for complex molecule synthesis, we have been investigating the potential of reductive cross-coupling processes between imines and alkynes, alkenes or allenes to serve as a general foundation for heterocycle synthesis.3 Recently, we set our sights on the development of a multicomponent coupling reaction suitable for the synthesis of exo-alkylidene γ-lactams (Figure 2A). These architectures, while representing interesting heterocycles in their own right, possess a rich reactivity profile suitable for diverse elaboration (Figure 2B). Herein, we report the realization of a stereoselective synthesis of exo-alkylidene γ-lactams from the convergent and stereoselective union of homoallylic alcohols, imines and carbon monoxide.
Figure 1.
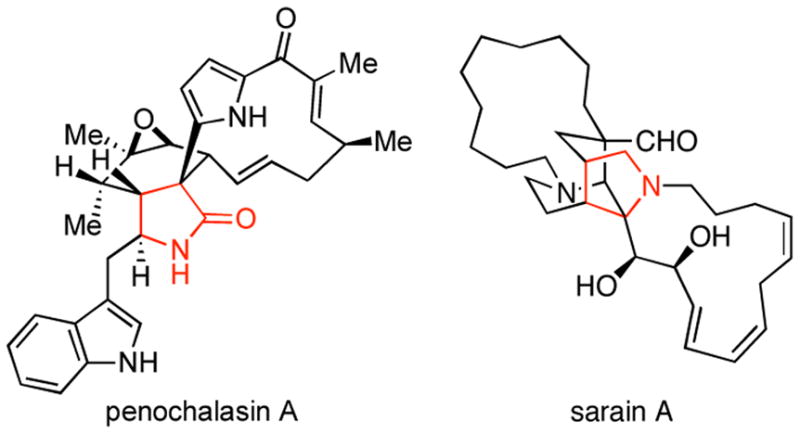
Examples of natural products bearing a substituted γ-lactam or pyrrolidine.
Figure 2.

Exo-alkylidene γ-lactams.
Recently, we reported a stereoselective synthesis of homoallylic amines that proceeds by regioselective reductive cross-coupling of allylic alcohols with aromatic imines.3d Of particular interest to our goals here, coupling of 2-halo allylic alcohols to aromatic imines was found to provide stereoselective access to anti-homoallylic amines that contain a stereodefined vinyl halide (dr ≥ 20:1; E:Z ≥ 20:1; Figure 3). While the mechanistic details that result in these high levels of stereoselection remain undefined, an emperical model has emerged to explain the patterns of reactivity and selectivity observed. The proposed model, based on a sequence of directed carbometalation and syn-elimination (A → B; Figure 3), embraces a boat-like geometry in the transition state for C–C bond formation to reflect the presumed mechanistic requirement of preassociating the allylic alkoxide to the Ti-center of the azametallacyclopropane and the orbital requirements for carbometalation (coplanarity of the σTi–C and the πC=C). A key factor for stereocontrol then derives from the minimization of A-1,2 strain in the boat like orientation A (minimize steric interaction between R3 and X). As a consequence, high selectivity is observed for the formation of products containing a pendant E-alkene. Overall, the general reactivity pattern is consistent with formal metallo-[3,3] rearrangement by way of C.4
Figure 3.

Imine–allylic alcohol coupling reaction.
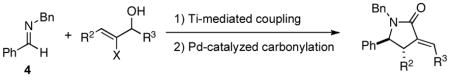
While having a stereoselective coupling reaction in place for the synthesis of highly functionalized homoallylic amines, we were aware of the potential of halogenated homoallylic amines to participate in Pd-catalyzed carbonylation chemistry.5, 6 As depicted in Figure 4, this was indeed the case. Carbonylation of vinylbromide 1 and vinyliodide 2 resulted in the production of the exo-methylene γ-lactam 3 in ≥ 93% yield.
Figure 4. Pd-catalyzed carbonylative cyclization.

a CO was introduced by a balloon.
With the knowledge gleaned from these initial studies, we moved on to explore the compatibility of more complex substrates in this two-step reductive cross-coupling/carbonylation process as a means to access a variety of stereodefined γ-lactams (Table 1).7 As depicted in entries 1–3, the size of the alkyl group at the allylic position plays an important role in stereoselection. While reductive cross coupling of allylic alcohol 5 with imine 4 proceeds in a fairly unselective manner (E:Z = 1.5:1), union of imine 4 with allylic alcohol 7 occurs with increased levels of stereoselection and produces the homoallylic amine 8 in 76% yield (E:Z ≥ 4:1). Subsequent carbonylation then delivers the stereodefined unsaturated γ-lactam 9 in 99% yield. As depicted in entry 3, branched alkyl substitution on the allylic alcohol leads to the highest levels of E-selectivity in this coupling reaction. Here, the homoallylic amine 11 is forged in 58% yield with greater than 20:1 selectivity for the formation of the stereodefined E-alkene. Palladium catalyzed carbonylation then furnishes γ-lactam 12 in 94% yield. Moving on to a more highly substituted allylic alcohol, the conversion of 13 to homoallylic amine 14 proceeds in 53% yield and delivers the stereodefined anti-product as essentially a single isomer. Similarly, carbonylation then provides the highly substituted exo-alkylidene γ-lactam 15 in 66% yield.
Table 1.
Exo-alkylidene γ-lactams via imine–allylic alcohol reductive cross-coupling
 | |||||
|---|---|---|---|---|---|
| entry | allylic alcohol | % yielda | homoallylic amine | % yielde | γ-lactam |
| 1 |
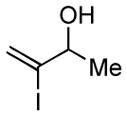 5 |
81b |
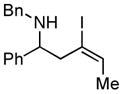 6 E:Z = 1.5:1 |
– | – |
| 2 |
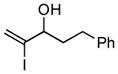 7 |
76b |
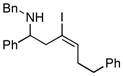 8 E:Z = 4:1 |
99c |
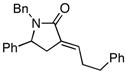 9 |
| 3 |
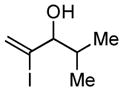 10 |
58d |
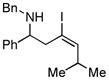 11 E:Z ≥ 20:1 |
94 |
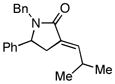 12 |
| 4 |
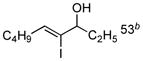 13 |
53b |
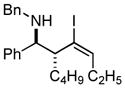 14 dr ≥ 20:1 |
66 |
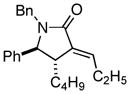 15 |
| 5 |
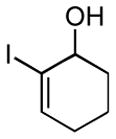 16 |
67d |
 17 dr ≥ 20:1 |
87 |
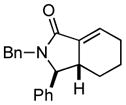 18 |
| 6 |
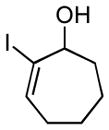 19 |
53d |
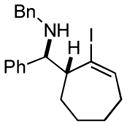 20 dr ≥ 13:1 |
92 |
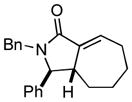 21 |
General reaction conditions for the Ti-mediated coupling: imine, ClTi(Oi-Pr)3, c-C5H9MgCl, Et2O or PhMe (−70 to −40 °C), then the sodium alkoxide of the allylic alcohol was added as a solution in THF.7
4 equiv of imine were employed.
The major isomer of 8 was employed in the carbonylation reaction.
1.5 equiv of allylic alcohol were used.
Reaction conditions: Cl2Pd(PPh3)2 (3–5 mol %), Et3N, PhMe or MeOH (entry 4), CO (balloon), 70 °C.
Finally, this two-step three-component heterocycle synthesis is useful for the synthesis of stereodefined bicyclic lactams. As illustrated in entries 5 and 6, reductive cross-coupling of cyclic allylic alcohols 16 and 17 with imine 4 can be accomplished in a highly stereoselective manner to deliver vinyliodides 17 and 20 (dr up to ≥ 20:1) These substrates are equally effective in the Pd-catalyzed carbonylative cyclization and deliver the bicyclo[4.3.0] and [5.3.0] systems 18 and 21 in 87% and 92% yield.
While this two-step procedure is effective for the stereoselective convergent synthesis of mono- and bicyclic γ-lactams, this multi-component coupling sequence can be streamlined. Specifically, we have defined a one-pot three-component coupling reaction that converts 2-halo allylic alcohols, imines and carbon monoxide directly to stereodefined exo-alkylidene γ-lactams. Aware of the the compatibility of Pd-catalyzed coupling processes with water and base, we speculated that aqueous quenching of the titanium-mediated reductive cross-coupling reaction may directly furnish a suitable environment for Pd-catalyzed carbonylation. This expectation was indeed the case.
As depicted in Figure 5, this sequential multicomponent coupling process for the synthesis of substituted γ-lactams can be conducted in a single reaction vessel. Here, union of imine 4 with allylic alcohol 2 furnishes lactam 3 in 69% yield. Similarly, union of imine 4 with allylic alcohol 22 provides the stereodefined bicyclic lactam 18 in 73% yield.8 Notably, avoiding the requirement for purification of the intermediate homoallylic amines substantially improves the overall yield for this γ-lactam forming annulation process.
Figure 5.

One-pot three-component coupling for heterocycle synthesis.
Overall, we have described studies that have culminated in the elucidation of a multi-component coupling process for the synthesis of stereodefine exo alkylidene γ-lactams. In short, titanium-mediated regio- and stereoselective coupling of aromatic imines with 2-halo allylic alcohols furnishes intermediate homoallylic amines that are well-suited for palladium-catalyzed carbonylation. This two-step process has been demonstrated with a variety of allylic alcohols and has defined a convergent and stereoselective pathway to mono- and bicyclic γ-lactams. Finally, a one-pot procedure has been developed that enables direct preparation of stereodefined lactams from allylic alcohols and imines. Given the flexibility of the titanium-mediated coupling with respect to imine structure,3d and the ability to translate stereochemical information from the allylic alcohol to the homoallylic amine intermediates,3d we anticipate that this heterocycle-forming annulation will be of utility in medicinal chemistry and natural product synthesis.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support of this work by the Arnold and Mabel Beckman Foundation, JSPS Research Fellowship for Young Scientists (to SU), the G-COE in Chemistry from Nagoya University, and the National Institutes of Health -–NIGMS (GM80266 and GM80266-04S1).
Footnotes
Supporting Information Available Experimental procedures and tabulated spectroscopic data for new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fischer J, Ganellin CR, editors. Analogue-based Drug Discovery. Wiley-VCH; Weinheim: 2002. [Google Scholar]
- 2.For reviews that cover the synthesis of nitrogen heterocycles, see: Nakamura I, Yamamoto Y. Chem Rev. 2004;104:2127–2198. doi: 10.1021/cr020095i.Deiters A, Martin SF. Chem Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872.Barluenga J, Santamaría J, Tomás M. Chem Rev. 2004;104:2259–2283. doi: 10.1021/cr0306079.Zeni G, Larock RC. Chem Rev. 2004;104:2285–2309. doi: 10.1021/cr020085h.Royer J, Bonin M, Micouin L. Chem Rev. 2004;104:2311–2352. doi: 10.1021/cr020083x.Mangelinckx S, Giubellina N, De Kimpe N. Chem Rev. 2004;104:2353–2399. doi: 10.1021/cr020084p.Bur SK, Padwa A. Chem Rev. 2004;104:2401–2432. doi: 10.1021/cr020090l.Mitchinson A, Nadin A. J Chem Soc Perkin Trans. 1999;1:2553–2581.Nadin A. J Chem Soc Perkin Trans. 1998;1:3493–3513.Harrison T. Contemp Org Synth. 1995;2:209–224.
- 3.For the coupling of homoallylic alcohols to imines, see: Takahashi M, Micalizio GC. J Am Chem Soc. 2007;129:7514–7416. doi: 10.1021/ja071974v.Lysenko IL, Lee HG, Cha JK. Org Lett. 2009;11:3132–3134. doi: 10.1021/ol901006c.For the coupling of homopropargylic alcohols to imines, see: McLaughlin M, Takahashi M, Micalizio GC. Angew Chem Int Ed. 2007;46:3912–3914. doi: 10.1002/anie.200605060.For the coupling of allenic alcohols to imines, see: McLaughlin M, Shimp HL, Navarro R, Micalizio GC. Synlett. 2008:735–738. doi: 10.1055/s-2008-1042808.For the coupling of allylic alcohols to imines, see: Takahashi M, McLaughlin M, Micalizio GC. Angew Chem Int Ed. 2009;48:3702–3706. doi: 10.1002/anie.200900236.
- 4.For related reductive cross-coupling reactions that appear to proceed by formal metallo-[3,3] rearrangement, see: Kolundzic F, Micalizio GC. J Am Chem Soc. 2007;129:15112–15113. doi: 10.1021/ja075678u.Shimp HL, Hare A, McLaughlin M, Micalizio GC. Tetrahedron. 2008;64:6831–6837. doi: 10.1016/j.tet.2008.02.015.Belardi JK, Micalizio GC. J Am Chem Soc. 2008;130:16870–16872. doi: 10.1021/ja8074242.Lysenko IL, Kim K, Lee HG, Cha JK. J Am Chem Soc. 2008;130:15997–16002. doi: 10.1021/ja806440m.
- 5.For a review of Pd-catalyzed carbonylation for the synthesis of lactams and lactones, see: Farina V, Magnus E. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E-I, editor. John Wiley & Sons, Inc; Hoboken, NJ: 2002. pp. 2351–2375.
- 6.For early examples of Pd-catalyzed carbonylation for lactam synthesis, see: Miwako M, Chiba K, Ban Y. J Org Chem. 1978;43:1684–1687.Miwako M, Washioka Y, Urayama T, Yoshiura Y, Chiba K, Ban Y. J Org Chem. 1983;48:4058–4067.Crisp GT, Meyer AG. Tetrahedron. 1995;51:5585–5596.
- 7.General experimental procedure for the two-step g-lactam synthesis described in Table 1: Synthesis of (S*)-N-benzyl-1-((R*)-2-iodocyclohex-2-enyl)-1-phenylmethanamine 17: To a solution of imine 4 (563 μL, 586 mg, 3.00 mmol) and ClTi(Oi-Pr)3 (1.0 M in diethyl ether, 3.75 mmol) in diethyl ether (12 mL) at −70 °C was added c-C5H9MgCl (2.00 M in diethyl ether, 7.50 mmol) in a drop-wise manner with a syringe. The brown solution was slowly warmed to −40 °C over 30 minutes and stirred at −40 °C for a further 1.5 hours. A solution of the sodium alkoxide, generated from the deprotonation of alcohol 16 (1.01 g, 4.50 mmol) with NaH (60 % suspension, 225 mg, 5.63 mmol), in THF (15 mL) at 0 °C, was added in a drop-wise manner via Teflon cannula to the brown solution of imine–Ti complex at −40 °C. The reaction was allowed to warm to ambient temperature and stirred overnight. Next, saturated aqueous NH4Cl (5 mL) was added and the resulting biphasic mixture was stirred rapidly. The resulting solution was further diluted with saturated aqueous NaHCO3 (150 mL) and extracted with ether (3 × 150 mL). The combined organic layers were washed with brine (50 mL), dried over MgSO4, and concentrated in vacuo. The crude material was purified by column chromatography on silica gel (1/40→1/30 EtOAc/Hexanes) to yield haloallylic amine 17 as a colorless oil, (811 mg, 67%, dr ≥ 20:1). Synthesis of (3S*,3S*)-2-benzyl-3-phenyl-2,3,3a,4,5,6-hexahydro-1H-isoindol-1-one 18: To a round bottom flask equipped with a reflux condenser was sequentially added amine 17 (169 mg, 0.420 mmol), toluene (4.2 mL), Cl2Pd(PPh3)2 (14 mg, 0.021 mmol) and Et3N (114 μL, 83 mg, 0.820 mmol). The reaction was placed under an atmosphere of CO by briefly exposing the reaction vessel to vacuum and backfilling with carbon monoxide from a balloon. The atmosphere of CO was maintained by a balloon for the remainder of the reaction. The reaction vessel was then submerged in a preheated oil bath (70 °C), and stirred for 12 hours. The reaction mixture was then allowed to cool to ambient temperature, diluted with EtOAc, filtered through cotton and concentrated in vacuo. The crude material was purified by column chromatography on silica gel (1/10→1/5 EtOAc/Hexanes) to yield γ-lactam 18 as a white solid, (111 mg, 87%).
- 8.General experimental procedure for the one-pot g-lactam synthesis described in Figure 5: Synthesis of (3S,3aS)-2-benzyl-3-phenyl-2,3,3a,4,5,6-hexahydro-1H-isoindol-1-one 18: To a solution of imine 4 (74 μL, 78 mg, 0.400 mmol) and ClTi(Oi-Pr)3 (1.0 M in diethyl ether, 0.500 mmol) in toluene (1.6 mL) at −70 °C was added c-C5H9MgCl (2.00 M in diethyl ether, 1.00 mmol) in a drop-wise manner with a syringe. The orange-brown solution was slowly warmed to −40 °C over 30 minutes and stirred at −40 °C for a further 1.5 hours. A solution of the sodium alkoxide, generated from the deprotonation of allylic alcohol 22 (106 mg, 0.600 mmol) with NaH (60 % suspension, 30 mg, 0.750 mmol), in THF (1.6 mL) at 0 °C, was added in a drop-wise manner via Teflon cannula to the brown solution of imine-Ti complex at −40 °C. The reaction was allowed to warm to ambient temperature and stirred overnight. The following morning H2O (36 μL, 36 mg, 2.00 mmol) was added and then rapidly stirred for 2 hours at ambient temperature. To the yellow solution of the reaction mixture was added PdCl2 (1 mg, 0.008 mmol), t-Bu3P (1.0 M toluene, 0.024 mmol) and Et3N (223 μL, 161 mg, 1.60 mmol). The reaction was placed under an atmosphere of CO by briefly exposing the reaction vessel to vacuum and backfilling with carbon monoxide from a balloon. The atmosphere of CO was maintained by a balloon for the remainder of the reaction. The reaction vessel was then submerged in a preheated oil bath (70 °C), and stirred for 12 hours. The reaction vessel was then allowed to cool to ambient temperature. The reaction mixture was diluted with EtOAc (10 mL), the solids ware removed by filtration through celite. The filtrate was further diluted with EtOAc (75 mL) and washed with 1 M HCl aq. (75 mL), saturated aqueous NaHCO3 (75 mL) and brine (100 mL). The organic layer was dried over MgSO4, and concentrated in vacuo. The crude material was purified by column chromatography on silica gel (1/10→1/5 EtOAc/Hexanes) to yield γ-lactam 18 as a white solid, (89 mg, 73%, dr ≥20:1).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.