

Abstract
In Saccharomyces cerevisiae, destabilizing telomeres, via inactivation of telomeric repeat binding factor Cdc13, induces a cell cycle checkpoint that arrests cells at the metaphase to anaphase transition—much like the response to an unrepaired DNA double-strand break (DSB). Throughout the cell cycle, the multi-domain adaptor protein Rad9 is required for activation of checkpoint effector kinase Rad53 in response to DSBs and is similarly necessary for checkpoint signaling in response to telomere uncapping. Rad53 activation in G1 and S phase depends on Rad9 association with modified chromatin adjacent to DSBs, which is mediated by Tudor domains binding histone H3 di-methylated at K79 and BRCT domains to histone H2A phosphorylated at S129. Nonetheless, Rad9 Tudor or BRCT mutants can initiate a checkpoint response to DNA damage in nocodazole-treated cells. Mutations affecting di-methylation of H3 K79, or its recognition by Rad9 enhance 5' strand resection upon telomere uncapping, and potentially implicate Rad9 chromatin binding in the checkpoint response to telomere uncapping. Indeed, we report that Rad9 binds to sub-telomeric chromatin, upon telomere uncapping, up to 10 kb from the telomere. Rad9 binding occurred within 30 min after inactivating Cdc13, preceding Rad53 phosphorylation. In turn, Rad9 Tudor and BRCT domain mutations blocked chromatin binding and led to attenuated checkpoint signaling as evidenced by decreased Rad53 phosphorylation and impaired cell cycle arrest. Our work identifies a role for Rad9 chromatin association, during mitosis, in the DNA damage checkpoint response to telomere uncapping, suggesting that chromatin binding may be an initiating event for checkpoints throughout the cell cycle.
Keywords: cdc13-1, cell cycle checkpoint, H2A phosphorylation, BRCT, telomere
Introduction
In metazoans, unrepaired DNA double strand breaks (DSBs) are a deleterious form of DNA damage that can lead to inactivation of tumor suppressors, activation of oncogenes and the promotion of carcinogenesis. In response to DSBs, cells initiate a checkpoint response that senses DNA lesions and signals to downstream effectors to induce cell cycle arrest and DNA repair. Signal transduction in response to DNA damage has been reviewed extensively and many of the checkpoint signaling proteins are conserved from yeast to humans [1–3]. In Saccharomyces cerevisiae, DSB induction recruits the Mre11-Rad50-Xrs2 complex (MRE11-RAD50-NBS1 in humans) and Tel1 (ATM) to breaks [1,4] where Tel1 may phosphorylate histone H2A (H2AX) at S129 [5,6]. Next, the multi-domain adaptor protein Rad9 (53BP1, MDC1, BRCA1) localizes to chromatin adjacent to DSBs [7–10] and DNA ends undergo 5' to 3' resection in the late S and G2/M phases. The Rfa1-Rfa2-Rfa3 heterotrimer (RPA) binds to ssDNA and recruits the Rad24 (RFC) clamp loader to assemble the Rad17-Mec3-Ddc1 clamp (Rad9-Rad1-Hus1) at the junction of ssDNA and dsDNA [11]. The RFA, Rad24 and Rad17 complexes recruit Ddc2 (ATRIP) and the Mec1 (ATR) kinase to breaks [4,12–15]. Mec1 phosphorylates Rad9, which recruits Rad53 (CHK2) to DSBs and leads to Mec1-dependent phosphorylation and auto-phosphorylation of Rad53 [16–18]. Finally, Rad53 kinase activity propagates a DNA damage signal that leads to cell cycle arrest and DNA repair.
The nucleosome, the basic unit of chromatin, is an octameric DNA-protein complex consisting of histones H3, H4, H2A, H2B and 146 bps of DNA [19,20]. Histones can be modified by ubiquitination, phosphorylation, methylation and acetylation [21]. These chromatin modifications alter the interaction among histones, DNA and other proteins, and thereby play significant roles in the DNA damage response [1,22–27]. For example, the mammalian phosphoinositide-3-kinase-related protein kinases (PIKKs) ATM, ATR and DNA-PK, phosphorylate H2A variant H2AX in response to DNA damage [28–30]. In budding yeast, histone H2A is phosphorylated by Mec1 and Tel1 kinases to form a chromatin domain that extends up to 50 kb from DNA lesions and recruits chromatin modifiers including NuA4, Ino80 and SWR [23,31–33]. H2A phosphorylation may lead to changes in chromatin structure [34,35] but also mediates recruitment and retention of checkpoint proteins, including Rad9 homologs 53BP1, MDC1 and BRCA1, at DNA lesions [36]. Recruitment of MDC1, like Schizosaccharomyces pombe homolog Crb2, is mediated by direct binding to phosphorylated H2A via tandem BRCA1 carboxyl-terminal (BRCT) domains [37,38]. Moreover, hta1, 2-S129A point mutants are sensitive to DSB-inducing agents, deficient in non-homologous end joining and have G1 and S phase checkpoint defects [9,10,35].
The biology of telomeres, specialized chromatin domains that protect chromosome ends, has been reviewed extensively [39–43]. When the telomere is “uncapped” by loss of one or more protein components that bind to G1–3T::C1–3A repeats, the resulting structure resembles one end of a natural DSB and activates a checkpoint response during mitosis. Cdc13 [44], a telomere-specific protein in yeast that binds to single stranded G1–3T::C1–3A overhangs, plays a key role in the recruitment of telomerase [45] and telomere capping [46]. The temperature sensitive cdc13-1 P371S allele binds single stranded telomeric repeats at permissive temperatures (< 25 °C) but dissociates at restrictive temperatures (>26 °C). When held in G1 with α-factor and shifted to non-permissive temperature, cdc13-1 mutants do not activate Rad53 [47]. However, with the accumulation of Cdc28 activity in late S or G2/M phases, uncapped telomeres undergo 5' to 3'resection to generate long regions of ssDNA [46], activating Rad53 and a sustained mitotic checkpoint arrest at the metaphase to anaphase transition. Thus, the characteristic changes in gene expression and cell cycle progression in cdc13-1 mutants at non-permissive temperature resemble the response to ionizing radiation [46,48–50]. As such, the cdc13-1 mutation has served as a powerful tool to elucidate molecular mechanisms mediating the G2/M checkpoint response to DNA damage [39,44,51–53].
Seminal work characterizing the DNA damage checkpoint in budding yeast identified Rad9 as a key mediator in signaling [54,55]. When challenged with genotoxic agents or ionizing radiation, mutants deleted for the RAD9 gene display DNA damage sensitivity, fail to activate Rad53, and display checkpoint defects at each phase of the cell cycle. Similarly, rad9Δ cdc13-1 cells fail to arrest, instead dividing to form microcolonies of inviable cells after temperature shift [44,51,56–60]. Rad9, 53BP1 and Crb2 share a domain structure characterized by tandem BRCT and Tudor domains at the carboxyl terminus. Although the Rad9 BRCT domains have been implicated in oligomerization [61,62], the Rad9 BRCT domains also interact with phosphorylated H2A peptides in vitro and disrupting this interaction leads to G1 and intra-S phase checkpoint defects [9,10,61]. Consistent with dual recognition of chromatin by Rad9, chromatin immunoprecipitation studies demonstrate that H3 K79 di-methylation and H2A S129 phosphorylation are both necessary for Rad9 chromatin association [9,10,61]. The Rad9, 53BP1 and Crb2 Tudor domains have been shown to bind di-methylated histone H3 at K79 and/or H4 K20 in vitro [7,63]. Yeast strains deficient in the conserved H3 K79 methyl-transferase Dot1 or expressing the Tudor domain mutant rad9 Y798Q display G1 and intra-S phase checkpoint defects after ionizing radiation [8]. In addition, rad9Δ, rad9-Y798Q Tudor domain mutants and dot1Δ strains harboring the cdc13-1 mutation also have an increased rate of ssDNA accumulation at uncapped telomeres [51,56,57,59].
While Rad9 is necessary for checkpoint signaling in response to telomere uncapping, a role for Rad9 chromatin association in signaling damage to telomeres remains unclear. In this study, we targeted the putative Rad9 phospho-H2A binding site to analyze the role of H2A phosphorylation in checkpoint signaling at uncapped telomeres. Comparative protein modeling was used to identify residues in the Rad9 BRCT domains that define a phospho-H2A binding pocket. As expected, mutation of these residues resulted in checkpoint defects in G1 and S phase after ionizing radiation. However, contrary to nocodazole-based assays, rad9 BRCT mutants displayed an attenuated or impaired checkpoint response to telomere uncapping. In addition, chromatin immunoprecipitation analysis shows that Rad9 binds to sub-telomeric chromatin and this interaction is dependent on both chromatin-binding domains. Taken together, these results demonstrate that Rad9 chromatin association at sub-telomeric chromatin is important for DNA damage checkpoint activation in the response to telomere uncapping during mitosis.
Materials and Methods
Western Blot Analysis
For Western blot analysis, OD600 of yeast cultures were measured and volumes corresponding to 2–2.5 OD600 units were collected. Cell pellets were treated with 0.2 N NaOH for at least 5 min and resuspended in 100 μl of 1X SDS sample buffer (125 mM Tris-HCl pH 6.8, 20% glycerol, 4% SDS, 1.43 M β-Mercaptoethanol). Samples were incubated at 95 °C for 5 min and centrifuged at 16,100 × g for 1 min to clarify the lysate. 15 μl of cell lysate was fractionated with NUPAGE 3–8% TA gels (Invitrogen) and transferred to nitrocellulose. Rad53-FLAG blots were probed using M2 mouse anti-FLAG (Sigma, 1:1000), and Rad9-13Myc blots using 9E10 mouse anti-MYC (1:200, Santa Cruz), and detected with goat anti-mouse IgG HRP conjugate secondary antibody. Phospho-H2A Western blotting was performed with rabbit anti-yeast histone H2A phospho-Ser129 (Millipore 1:100 in 1X PBS) and tubulin was detected using 1:500 YL1/2 rat anti-tubulin (Millipore) and HRP conjugated goat anti-rat IgG secondary antibody. Figures are representative of 2 or more replicate experiments.
cdc13-1 Checkpoint Assays
Rad53 activation and flow cytometry analysis
Strains carrying the cdc13-1 allele were incubated at 25 °C in α-factor (WHWLQLKPGQPNleY) [64] for 1 h, transferred to 37 °C for 1 h and then released into 37 °C media for an additional 3 h. Samples were collected hourly and analyzed for Rad53 activation and DNA content.
Microcolony analysis
Sonicated and serially diluted cdc13-1 strains were plated and incubated at 25, 30 or 37 °C for 6.5–7 h. Next, plates were transferred to 4 °C until scored, as previously described [55]. Briefly, microcolonies with ≤ four cell bodies were scored as arrested and microcolonies with > four cell bodies were scored as cycling. The data shown are the average of 3 replicates, mean ± SD.
Chromatin Immunoprecipitation Assays
Chromatin immunoprecipitation (ChIP) assays were performed as described previously [8] with minor modifications. Briefly, overnight cultures grown in minimal media were pelleted, washed with sterile water and grown in YPD to OD600 0.3–0.4 at 20 °C. α-factor was added to the final concentration of 15 μg/ml and the culture was grown for one hour at room temperature and then shifted to 37 °C for 30 min. Cells were pelleted and washed once with sterile water and twice with pre-warmed media. Cells were resuspended in 200 ml YPD and grown at 37 °C. Samples were collected at 30 min, 1 h and 2 h time points and cross-linked with 1% formaldehyde for 20 min at 20 °C. Cross-linked chromatin was sonicated (Diagenode Bioruptor) to yield DNA fragments of an average size of 500 bp, 100 μg of sonicated sample was used for immunoprecipitations with 9E10 antibody (Babco). Primers used in the PCR reactions were analyzed for linearity range and efficiency with a LightCycler (Roche) in order to evaluate protein occupancy (% of IP/input). All PCR reactions were performed at least as duplicates, and variation was less than 15%. The numbers presented with standard errors are based on two independent experiments.
Results
Modeling and mutagenesis of the Rad9 BRCT domain
A rad9 BRCT domain mutant, K1088M, disrupted H2A phosphopeptide binding and altered G1 and S phase checkpoint responses [9]. These data are consistent with prior observations in H2A phosphorylation mutants [10]. We sought to further characterize the interaction between the Rad9 BRCT domains and phosphorylated H2A by examining the role of H2A binding in G2/M checkpoint activation. We therefore performed a sequence alignment of BRCT domains from Rad9, MDC1, BRCA1 and Crb2 using the T-Coffee algorithm [65] and comparative protein modeling to identify a region in Rad9 that corresponds to the phospho-H2A binding pocket of the MDC1 BRCT domains. Next, we generated a Rad9 BRCT model using the crystal structure of MDC1 [66]. Then the Rad9 BRCT domain was modeled with I-Tasser software [67–69] and visualized with Swiss-PDBviewer (http://www.expasy.org/spdbv/) [67–70]. In the sequence alignment, Rad9 residues R1085 and K1088 were highly conserved (Fig. 1A). Furthermore, the orientation of residues analogous to R1085 and K1088 in Rad9 are conserved in the crystal structures of MDC1 (R1933/K1936) (Fig. 1B) and Crb2 (R616/K619) [71] (Fig. 1C). In Rad9, R1085 was of particular interest because of its potential to form contacts with the carboxyl terminus of H2A. In turn, K1088 was positioned to contact the phosphorylated H2A S129 residue. Thus, we mutated residues R1085, K1088, and the other residues identified in the sequence alignment in order to better characterize the potential phospho-H2A binding pocket. The mutations R1085E, S1086A, L1087A, K1088E, Y1089A, L1090A and E1091K, which lead to charge reversal, the exchange of large hydrophobic residues for alanine, or the removal of potential phosphorylation targets were constructed to disrupt an interaction with phosphorylated H2A. We focused on R1085E and K1088E, where charge reversal may eliminate favorable interactions with the H2A carboxyl terminus and phosphorylated S129 respectively. The R1085E and K1088E mutants as well as most of the other BRCT point mutants phenocopied the rad9Δ mutant by failing to perform a G1 checkpoint delay after ionizing radiation (Supplemental Figs. S1, S2). However, when cells were irradiated in early S phase, the R1085E allele progressed through replication more slowly than rad9Δ strains but faster than the RAD9 wildtype strain. K1088E phenocopied the S phase defect of the hta1, 2-S129A mutant and completed replication faster than rad9Δ cells (Supplemental Fig. S3). When cells were arrested in nocodazole, irradiated and then released from nocodazole-induced arrest, rad9Δ reentered G1 immediately but the R1085E and K1088E mutants activated a DNA damage checkpoint and displayed a large budded arrest (data not shown). Like hta1, 2-S129A mutant, the rad9-R1085E and rad9-K1088E mutations remain checkpoint competent when tested in nocodazole. Although it is possible that the R1085E and K1088E mutations might disrupt Rad9 domain structure or oligomerization, analytical gel filtration chromatography of the analogous S. pombe Crb2 R616E and K619E charge reversal mutants revealed no effects on dimer formation [71]. Thus, like the K1088M allele [9], the R1085E and K1088E mutant phenotypes may result from altered binding to the phosphorylated H2A tail.
Figure 1.

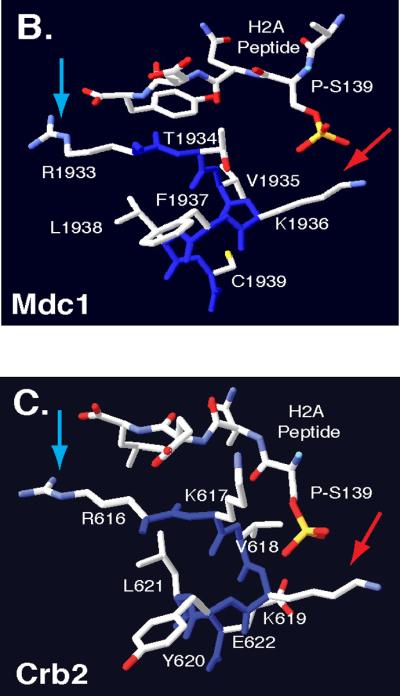
Comparative Protein Modeling for the Rad9 BRCT domain. (A)T-Coffee alignment of BRCT sequences from multiple checkpoint adaptor proteins identified a functionally conserved region, highlighted by a yellow box. Adaptor protein BRCT sequences include: S. pombe Crb2 (537–778), S. cerevisiae Rad9 (994–1122), Homo sapiens 53BP1 (1719–1977), H. sapiens BRCA1 (1649–1859) and H. sapiens MDC1 (1719–1977). Conserved arginine and lysine residues are highlighted in red. Crystal structures of MDC1 (B) and Crb2 (C) BRCT domains, in complex with a phospho-H2A peptide [66, 71]. Blue arrows denote conserved arginine residues that may contact the H2A carboxyl-terminal tail. Red arrows denote conserved lysine residues that may contact phosphoserine residues within the H2A tail. Backbone molecules are highlighted in dark blue, Nitrogen molecules are featured in light blue, oxygen molecules are featured in red and sulfur molecules are featured in yellow.
rad9 BRCT mutants have a G2/M checkpoint defect in response to uncapped telomeres
While H2A is rapidly phosphorylated after DNA damage at each stage of the cell cycle, H2A phosphorylation has not been ascribed a role in mediating G2/M checkpoint arrest [9,10]. Thus, we sought to examine the integrity of the G2/M checkpoint response in the hta1, 2-S129A and rad9 BRCT mutants. G2/M checkpoint assays in yeast commonly use nocodazole, a benzimidazole drug that destabilizes tubulin dimers [72]. Nocodazole treatment disrupts mitotic spindle formation and arrests cells prior to anaphase by activating the mitotic spindle checkpoint [73]. Previous analysis of the response to ionizing radiation in nocodazole-treated hta1, 2-S129A, dot1Δ, and rad9-Y798Q strains showed that each strain fully activates Rad53 and exhibits a characteristic large budded arrest [8,10,74,75]. As predicted, irradiation of nocodazole-arrested R1085E and K1088E mutants led to full Rad53 activation (Fig. 2A), suggesting that the G2/M checkpoint remains intact in these mutants. Similarly, Hammet et al. (2007) found that the K1088M mutant exhibited a competent G2/M response when subjected to bleomycin-induced damage in nocodazole-treated cells. However, nocodazole may introduce confounding factors in these experiments. There is crosstalk between the mitotic spindle and DNA damage checkpoints [76]. In addition, nocodazole induces phosphorylation of Rad53 and Rad9 independently of Mec1 and Tel1 in the absence of DNA damage [77,78]. Studying rad9 mutants in response to telomere uncapping provides an alternative method to examine the G2/M checkpoint arrest in response to DNA damage . Given that sub-telomeric H2A is constitutively phosphorylated on chromosome III in a cdc13-1 background [79,80], this modification may play a significant role in the checkpoint response to telomere uncapping. First, we determined whether H2A phosphorylation was necessary for Rad9-dependent checkpoint signaling after telomere uncapping. The hta1, 2-S129A mutant activated a G2/M checkpoint that was more robust than wildtype controls in a cdc13-1 background (Figs. 2B,C and E). On the other hand, rad9 BRCT and Tudor domain mutants displayed divergent checkpoint phenotypes (Figs. 2B–E, Supplemental Figs. S4, S5). While the rad9 K1088E mutant displayed an attenuated checkpoint response, the R1085E mutant displayed a G2/M checkpoint defect that resembled the rad9Δ mutant. In response to ionizing radiation and HO-induced DSBs the hta1, 2-S129A mutant displayed a competent checkpoint response, while the rad9-K1088E and rad9R1085E mutants exhibited partial checkpoint responses (Supplemental Fig. S6). These data suggest that the requirements for the Rad9 BRCT domain may differ in the G1 and S phases. Beyond facilitating interactions with the phosphorylated H2A carboxyl terminus, the R1085E mutation may disrupt binding to other unknown protein partners or chromatin modifications in response to telomere uncapping.
Figure 2.

Rad9 BRCT mutants initiate variable responses to uncapped telomeres. (A) rad9Δ strains were transformed with constructs harboring wild type RAD9-MYC or MYC-tagged rad9 mutant alleles, and analyzed for Rad53 activation in response to 300 Gy of ionizing irradiation. Samples were treated with nocodazole, irradiated and were harvested at 0, 15 and 30 min after irradiation. rad9Δ cdc13-1 strains were transformed with constructs harboring wild type RAD9-MYC or MYC-tagged rad9 mutant alleles, and analyzed for (B) Rad53 activation, H2A phosphorylation and (C) DNA content. (D and E) Microcolony analysis of the R1085E and K1088E mutants with the cdc13-1 allele. The data shown are the average of 3 replicates, mean ± SD. Bar = 10 μm
Rad9 binds to sub-telomeric chromatin after telomere uncapping
Previous studies identified increased rates of ssDNA accumulation at telomeres in rad9Δ cdc13-1 cells after uncapping [51,56,57,59]. This result and other studies led authors to propose that Rad9 checkpoint signaling may inhibit resection at telomeres. However, impaired DNA end resection may instead reflect direct Rad9 binding to subtelomeric chromatin. Thus, we performed chromatin immunoprecipitation (ChIP) analysis to determine whether Rad9 associates with sub-telomeric chromatin after telomere uncapping. ChIP analysis of α-factor arrested RAD9-13MYC cdc13-1 cells, incubated at 37 °C, demonstrated Rad9 binding to sub-telomeric chromatin which was apparent by 30 min and binding increased after 1 h (data not shown). Similarly, Rad9 bound to sub-telomeric chromatin 30 min after being released from α-factor into media warmed to 37 °C (Fig. 3B). At sub-telomeric regions, ~270 bps distal to the X elements of telomere VI, Rad9 chromatin association decreased ~50% by 1 h (Fig. 3B, top, 0.7 kb), likely reflecting resection through this region. Conversely, Rad9 chromatin association at regions greater than 4.4 kb from the telomere remains consistent over the duration of 2 h (Fig. 3B, see 4.4 kb, 10 kb). Given that Rad9 chromatin association is detected up to 10 kb from uncapped telomeres, it may continue to promote checkpoint signaling. Since the Rad9 BRCT mutants display checkpoint defects, we hypothesized that this might be a result of altered chromatin association at uncapped telomeres. Indeed, mutations within either of the Rad9 chromatin-binding domains (Y798Q for Tudor domains, R1085E and K1088E for BRCT domains) resulted in significantly decreased binding to chromatin at sub-telomeric regions and at distal regions of the telomere (Fig. 3B). These data demonstrate that Rad9 chromatin association is an early event and is dependent on both chromatin-binding domains.
Figure 3.
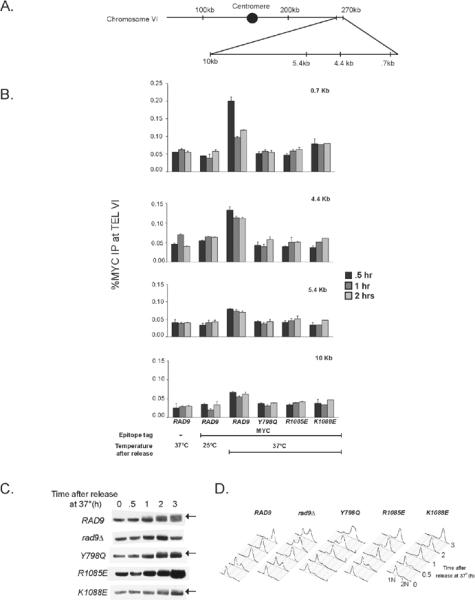
Rad9 binds to sub-telomeric chromatin after telomere uncapping. (A) A schematic representation of the sites probed within Chromosome VI for chromatin immunoprecipitation assays. (B) rad9Δ cdd13-1 strains expressing Myc-tagged RAD9 or rad9 mutant constructs were treated with α-factor at room temperature for one hour and then shifted to 37°C for 30 minutes. Next cells were released into pre-warmed YPD and incubated at 37°C. Samples were collected at 30 min, 1 h and 2 h time points and then used for ChIP analysis. Error bars represent the standard deviation between at least two replicate experiments. (C) With the same time course used in ChIP assays, Rad53 activation was detected after incubating cells at restrictive temperature. Arrows indicate high molecular weight species that correspond to phosphorylated Rad53. (D) DNA content analysis of cells from figure 3C after incubation at restrictive temperature.
Since Rad53 activation is Rad9-dependent [16,17,81], we wanted to determine whether Rad53 activation correlates with Rad9 chromatin association. Using the same time course as in the ChIP assays, we assessed Rad53 activation after incubation at restrictive temperatures. Although Rad9 binds chromatin 30 min after incubation at restrictive temperature, the characteristic Rad53 mobility shift was not observed until 1 h after incubation at 37 °C (Fig. 3C), and full Rad53 activation required 3 h. Like rad9Δ, the rad9-R1085E mutant failed to activate Rad53 while the Y798Q and K1088E mutants displayed attenuated Rad53 activation. Nonetheless, cell cycle delay was observed during mitosis in RAD9 and the rad9-Y798Q, R1085E and K1088E mutants by 2 h (Fig. 3D).
Additional rad9 mutants demonstrate that chromatin association and SCD phosphorylation are necessary but not sufficient for checkpoint activation
Although Rad9 chromatin binding is necessary for full activation of Rad53, it is not sufficient. It is possible that both Cdc28 and Mec1/Tel1-dependent phosphorylation are necessary for activation of chromatin-bound Rad9. Thus, we assayed a series of mutants to determine whether Rad9 phosphorylation is upstream of chromatin association (Fig. 4). Previous studies have shown that Mec1/Tel1-dependent phosphorylation [16,17,81] and Cdc28 activity are each necessary for checkpoint activation. As expected, rad9 mutant alleles, with mutations in either 18 putative CDK consensus sites (rad9-18A) [82], 7 S/TQ Mec1/Tel1 consensus sites (rad9-AQ) [81] and a mutant with both Y798Q and K1088E mutations (rad9-Y798Q/K1088E) exhibited checkpoint defects. These mutants failed to activate Rad53 after telomere uncapping (Fig. 4A). Furthermore, DNA content and microcolony analysis demonstrated that these mutants also fail to arrest the cell cycle during mitosis in response to telomere uncapping (Fig. 4C–E). In addition, all three mutants failed to activate Rad53 after 300 Gy of ionizing radiation in α-factor arrested cells, where Cdc28 is inactive (Fig. 4B). Thus, it is likely that the G1 checkpoint defect of the rad9-18A mutant is independent of Cdc28 activity. Furthermore, data regarding rad9-AQ and rad9-Y798Q/K1088E mutants suggest that both Mec1/Tel1 phosphorylation and chromatin binding via the Tudor and BRCT domains are required for G2/M and G1 checkpoint signaling. Further analysis of Rad9 protein levels demonstrated that the rad9-AQ, and the rad9-Y798Q and rad9-K1088E point mutants are expressed at levels comparable to wildtype strains (Supplemental Fig. S7A and B). However, the rad9-18A and rad9-Y798Q/K1088E mutants exhibit significantly reduced protein levels (Supplemental Fig. S7B). One interpretation of these results may be that the molecular consequences of the latter mutations compromise protein stability. Alternatively, lack of CDK-mediated phosphorylation and/or chromatin association may drive physiologic processes to promote Rad9 degradation. Given that a single point mutation in either chromatin binding domain does not compromise Rad9 protein levels, it is possible that chromatin association via both domains may play a role in stabilizing Rad9 protein levels.
Figure 4.

Additional rad9 mutants demonstrate that chromatin association and SCD phosphorylation are necessary but not sufficient for checkpoint activation. A series of rad9 alleles were assayed to determine if Rad9 phosphorylation is upstream of chromatin association. rad9Δ cdc13-1 strains were transformed with additional MYC-tagged rad9 alleles and assayed for (A) Rad53 activation in response to telomere uncapping. Microcolonies of cdc13-1 strains, also harboring several rad9 alleles, were also visualized (D) and quantified (E) after telomere uncapping. The data shown are the average of 3 replicates, mean ± SD. (B) G1-arrested rad9 mutants were also assayed for Rad53 activation in response to 300 Gy of ionizing radiation. Arrows in Figs. 4A and B indicate high molecular weight species that correspond to phosphorylated Rad53. Bar = 10 μm
Discussion
In this study, we dissected the role of the interaction between H2A phosphorylation and the Rad9 BRCT domains in response to telomere uncapping. Charge reversal mutations of Rad9 BRCT residues, predicted to disrupt recognition of either phosphorylated H2A S129 or the carboxyl terminus, resulted in defective G1 checkpoint initiation, S phase delay and defective checkpoint signaling during mitosis in response to telomere uncapping.
The implications of Rad9 chromatin association at telomeres
ChIP analysis revealed that Rad9 binds to sub-telomeric chromatin up to 10 kb uncapped telomeres, and remains bound over 2 h. ChIP analysis also demonstrated that both rad9 BRCT charge-reversal mutations, in a putative H2A phosphopeptide binding cleft, and a Tudor domain di-methyl lysine binding site mutation similarly prevented binding to telomeres upon inactivation of Cdc13. However, only the R1085E mutation eliminated checkpoint signaling. Although it is possible that these mutations disrupt protein folding, analytical gel filtration chromatography of analogous Crb2 charge reversal mutants R616E and K619E indicate that BRCT dimerization remains intact [71].
Rad9 chromatin association drops quickly in the most telomere-proximal chromatin whereas regions that are more distal maintained steady levels of Rad9 2 h after telomere uncapping. It is possible that the decrease in Rad9 association, observed to be 0.7 kb 1 h after release into media at restrictive temperature, occurs as DNA end resection extends into sub-telomeric chromatin. Chromatin-bound Rad9 may impede resection, as evidenced by several studies demonstrating that the rate of telomere end resection is increased in rad9Δ cells or mutants affecting Rad9 chromatin association [51,56,57,59]. Analogous to Rad9 function in G1 and S phase, full activation of Rad9-mediated checkpoint signaling requires an association with subtelomeric chromatin that occurs before significant telomere end resection can be observed.
The implications of the interaction between Rad9 and phosphorylated H2A
Although we intended to further characterize the role of H2A phosphorylation in Rad9-dependent checkpoint signaling, our analysis of multiple Rad9 BRCT mutants has revealed a role for this domain in checkpoint signaling that is beyond that observed in mutants that merely lack H2A phosphorylation. Indeed, unlike the attenuated G1 checkpoint delay observed in the hta1, 2-S129A mutant [10], the R1085E and K1088E mutants are completely defective in Rad53 activation and G1 arrest in response to irradiation. These data suggest that the Rad9 BRCT domains facilitate interactions that are independent of H2A phosphorylation and are generally consistent with the previous analysis of the rad9-K1088M BRCT mutant [9].
In turn, the range of phenotypes observed in the rad9 BRCT mutants described here suggests a greater complexity for S phase checkpoint signaling than anticipated. The R1085E mutant displays an attenuated S phase checkpoint response to irradiation. In contrast, the K1088E mutant phenocopies the complete S phase checkpoint defect of the hta1, 2-S129A mutant observed here. Whereas the R1085E mutation may disrupt interactions required only in G1, it appears that the K1088E mutation may disrupt interactions with phosphorylated H2A, and potentially other phosphorylated proteins that are necessary in both the G1 and S phases.
It is also intriguing that rad9 BRCT mutants are checkpoint competent after nocodazole treatment, yet have variable checkpoint defects in response to telomere uncapping. Previous reports have shown that in the absence of DNA damage, nocodazole induces phosphorylation of Rad53 and Rad9, independently of Mec1 and Tel1 [77,78]. This suggests a role for Rad9 in the mitotic spindle assembly checkpoint that may not require chromatin association. Alternatively, other factors in the mitotic spindle assembly checkpoint may activate Rad53 independently of Rad9. These studies demonstrate that the mitotic spindle assembly checkpoint and the checkpoint response to telomere uncapping utilize common checkpoint signaling proteins yet diverge at the level of Rad9 chromatin association.
While hta1, 2-S129A mutants activate a cdc13-1 checkpoint that is more robust than even the RAD9 strains, rad9 Tudor and BRCT domain mutants display a range of distinct checkpoint responses. Specifically, the Y798Q and K1088E mutants display a partial G2/M checkpoint response, while the R1085E mutant exhibits a completely defective checkpoint response. One possibility is that these mutants form an allelic series, but the complex patterns suggest that their distinct phenotypes are attributable to separate factors. Conversely, the hyper-checkpoint phenotype of the hta1, 2-S129A mutant may be due to disruption of constitutive phosphorylation of H2A in sub-telomeric chromatin [79]. In this case, phosphorylated H2A may serve a DNA-damage independent function that, along with telomere end binding proteins, limits Rad9 binding or activation while telomeres are capped.
In support of an alternative role for H2A phosphorylation adjacent to telomeres, our ChIP analysis demonstrated that Rad9 does not bind to sub-telomeric chromatin in cdc13-1 mutants at 24 °C but is instead recruited to telomeres within 30 min of incubation at non-permissive temperatures. It is certainly possible that H2A phosphorylation may be necessary for full Rad9 recruitment, but it is not sufficient. Constitutive H2A phosphorylation may compensate for reduced Rad9 chromatin binding sites that might result from H3 K79 hypo-methylation at sub-telomeric chromatin [80]. On the other hand, data from studies of 53BP1 suggest that foci formation adjacent to DSBs, mediated by constitutive binding of Tudor domains to di-methylated H3 K79 or dimethylated H4 K20, requires a change in chromatin structure [63,83]. Different studies have placed H2A phosphorylation upstream, downstream or parallel to chromatin remodeling at sites of bona fide DNA double strand breaks [10,32,84,85]. Telomeres display a position effect, such that otherwise active promoters located in sub-telomeric chromatin are commonly silenced. This is apparently associated with a dense, compact form of chromatin that may limit access of Rad9 to its binding site(s), irrespective of H2A S129 and H3 K79 modification states. Perhaps upon telomere uncapping, this repressive activity may be relaxed, chromatin remodeling may ensue and Rad9 may then be able to access the di-methylated H3 K79 and/or phosphorylated H2A S129, promoting stable association. Taken together, our analysis of functional roles for rad9 BRCT phospho-H2A binding residues reveals unanticipated roles in the DNA damage checkpoint response to telomere uncapping during mitosis, suggesting that Rad9 chromatin association can mediate checkpoint signaling throughout the cell cycle.
Supplementary Material
Table 1.
Yeast strains used in this study
| Strain | Description | Source |
|---|---|---|
| W303-1A | MATa ade2-1 can1-100 ura3-1 leu2-3, 112, his3-11, 15, trp1-1 69 | Thomas et al. 1989 |
| SKY2998 | W303-1A, rad9Δ::kanMX6 RAD53-3xFLAG | This study |
| SKY2999 | W303-1A, rad9Δ::kanMX6 RAD53-3xFLAG hta1S129A::his3MX6 hta2S129A::TRP1 | This study |
| SKY3000 | W303-1A, rad9Δ::kanMX6 cdc13-1 RAD53-3xFLAG | This study |
| SKY3001 | W303-1A, rad9Δ::kanMX6 cdc13-1RAD53-3xFLAG hta1S129A::his3MX6 hta2S129A::TRP1 | This study |
| SKY3002 | W303-1A, rad9Δ::kanMX6 cdc13-1RAD53-3xFLAG dot1Δ::TRP1 | This study |
| SKY3041 | MATa rad9Δ::kanMX6 hoΔ hml Δ::ADE1 hmrΔ::ADE1 ade1-110 leu2, 3-112 lys5 trp1::hisG ura3-52 ade3::GAL1,10:HO | This study |
| QY375 | MATa hoΔ hmlΔ::ADE1 hmrΔ::ADE1 ade1-110 leu2,3-112 lys5 trp1::hisG ura3-52 ade3::GAL1, 10:HO RAD9-HA::kanMX6 hta1::S129* hta2::S129* | Javaheri et. al 2006 |
Acknowledgements
We would like to thank Fiyinfolu Balogun and Drs. Satoe Takahashi and Latishya Steele for their thoughtful comments. We also thank Drs. David Toczyski and Katsunori Sugimoto for generously providing reagents. This study was supported by RO1 GM60443. C. C. N. was supported by F31 CA110277. S. J. K. was a Leukemia and Lymphoma Society Scholar.
Abbreviations
- DSBs
DNA double strand breaks
- BRCT
BRCA1 C-terminal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement The authors declare that there are no conflicts of interest.
References
- [1].Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- [2].Melo J, Toczyski D. A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol. 2002;14:237–245. doi: 10.1016/s0955-0674(02)00312-5. [DOI] [PubMed] [Google Scholar]
- [3].McGowan CH, Russell P. The DNA damage response: sensing and signaling. Curr Opin Cell Biol. 2004;16:629–633. doi: 10.1016/j.ceb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- [4].Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- [5].Nakada D, Matsumoto K, Sugimoto K. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003;17:1957–1962. doi: 10.1101/gad.1099003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Redon C, Pilch DR, Rogakou EP, Orr AH, Lowndes NF, Bonner WM. Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep. 2003;4:678–684. doi: 10.1038/sj.embor.embor871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Huyen Y, Zgheib O, Ditullio RA, Jr., Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- [8].Wysocki R, Javaheri A, Allard S, Sha F, Cote J, Kron SJ. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol. 2005;25:8430–8443. doi: 10.1128/MCB.25.19.8430-8443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hammet A, Magill C, Heierhorst J, Jackson SP. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 2007;8:851–857. doi: 10.1038/sj.embor.7401036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Javaheri A, Wysocki R, Jobin-Robitaille O, Altaf M, Cote J, Kron SJ. Yeast G1 DNA damage checkpoint regulation by H2A phosphorylation is independent of chromatin remodeling. Proc Natl Acad Sci U S A. 2006;103:13771–13776. doi: 10.1073/pnas.0511192103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A. 2003;100:13827–13832. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Paciotti V, Clerici M, Lucchini G, Longhese MP. The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev. 2000;14:2046–2059. [PMC free article] [PubMed] [Google Scholar]
- [13].Rouse J, Jackson SP. LCD1: an essential gene involved in checkpoint control and regulation of the MEC1 signalling pathway in Saccharomyces cerevisiae. Embo J. 2000;19:5801–5812. doi: 10.1093/emboj/19.21.5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wakayama T, Kondo T, Ando S, Matsumoto K, Sugimoto K. Pie1, a protein interacting with Mec1, controls cell growth and checkpoint responses in Saccharomyces cerevisiae. Mol Cell Biol. 2001;21:755–764. doi: 10.1128/MCB.21.3.755-764.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Majka J, Niedziela-Majka A, Burgers PM. The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol Cell. 2006;24:891–901. doi: 10.1016/j.molcel.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF. Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell. 2002;9:1055–1065. doi: 10.1016/s1097-2765(02)00532-4. [DOI] [PubMed] [Google Scholar]
- [17].Sun Z, Hsiao J, Fay DS, Stern DF. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science. 1998;281:272–274. doi: 10.1126/science.281.5374.272. [DOI] [PubMed] [Google Scholar]
- [18].Gilbert CS, Green CM, Lowndes NF. Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol Cell. 2001;8:129–136. doi: 10.1016/s1097-2765(01)00267-2. [DOI] [PubMed] [Google Scholar]
- [19].Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- [20].White CL, Suto RK, Luger K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. Embo J. 2001;20:5207–5218. doi: 10.1093/emboj/20.18.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Millar CB, Grunstein M. Genome-wide patterns of histone modifications in yeast. Nat Rev Mol Cell Biol. 2006;7:657–666. doi: 10.1038/nrm1986. [DOI] [PubMed] [Google Scholar]
- [22].Vidanes GM, Bonilla CY, Toczyski DP. Complicated tails: histone modifications and the DNA damage response. Cell. 2005;121:973–976. doi: 10.1016/j.cell.2005.06.013. [DOI] [PubMed] [Google Scholar]
- [23].Peterson CL, Cote J. Cellular machineries for chromosomal DNA repair. Genes Dev. 2004;18:602–616. doi: 10.1101/gad.1182704. [DOI] [PubMed] [Google Scholar]
- [24].Foster ER, Downs JA. Histone H2A phosphorylation in DNA double-strand break repair. FEBS J. 2005;272:3231–3240. doi: 10.1111/j.1742-4658.2005.04741.x. [DOI] [PubMed] [Google Scholar]
- [25].Chambers AL, Downs JA. The contribution of the budding yeast histone H2A C-terminal tail to DNA-damage responses. Biochem Soc Trans. 2007;35:1519–1524. doi: 10.1042/BST0351519. [DOI] [PubMed] [Google Scholar]
- [26].van Attikum H, Gasser SM. The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol. 2005;6:757–765. doi: 10.1038/nrm1737. [DOI] [PubMed] [Google Scholar]
- [27].Bilsland E, Downs JA. Tails of histones in DNA double-strand break repair. Mutagenesis. 2005;20:153–163. doi: 10.1093/mutage/gei031. [DOI] [PubMed] [Google Scholar]
- [28].Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Histone H2A variants H2AX and H2AZ. Curr Opin Genet Dev. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- [29].Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- [30].Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- [31].Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, Kron SJ, Jackson SP, Cote J. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell. 2004;16:979–990. doi: 10.1016/j.molcel.2004.12.003. [DOI] [PubMed] [Google Scholar]
- [33].Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol. 2004;14:1703–1711. doi: 10.1016/j.cub.2004.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fink M, Imholz D, Thoma F. Contribution of the serine 129 of histone H2A to chromatin structure. Mol Cell Biol. 2007;27:3589–3600. doi: 10.1128/MCB.02077-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000;408:1001–1004. doi: 10.1038/35050000. [DOI] [PubMed] [Google Scholar]
- [36].Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- [38].Nakamura TM, Du LL, Redon C, Russell P. Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol Cell Biol. 2004;24:6215–6230. doi: 10.1128/MCB.24.14.6215-6230.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lydall D, Weinert T. Use of cdc13-1-induced DNA damage to study effects of checkpoint genes on DNA damage processing. Methods Enzymol. 1997;283:410–424. doi: 10.1016/s0076-6879(97)83034-0. [DOI] [PubMed] [Google Scholar]
- [40].Lydall D. Hiding at the ends of yeast chromosomes: telomeres, nucleases and checkpoint pathways. J Cell Sci. 2003;116:4057–4065. doi: 10.1242/jcs.00765. [DOI] [PubMed] [Google Scholar]
- [41].Kanoh J, Ishikawa F. Composition and conservation of the telomeric complex. Cell Mol Life Sci. 2003;60:2295–2302. doi: 10.1007/s00018-003-3245-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004;73:177–208. doi: 10.1146/annurev.biochem.73.071403.160049. [DOI] [PubMed] [Google Scholar]
- [43].Blasco MA. The epigenetic regulation of mammalian telomeres. Nat Rev Genet. 2007;8:299–309. doi: 10.1038/nrg2047. [DOI] [PubMed] [Google Scholar]
- [44].Lydall D, Weinert T. G2/M checkpoint genes of Saccharomyces cerevisiae: further evidence for roles in DNA replication and/or repair. Mol Gen Genet. 1997;256:638–651. doi: 10.1007/s004380050612. [DOI] [PubMed] [Google Scholar]
- [45].Nugent CI, Hughes TR, Lue NF, Lundblad V. Cdc13p: a single-strand telomeric DNA-binding protein with a dual role in yeast telomere maintenance. Science. 1996;274:249–252. doi: 10.1126/science.274.5285.249. [DOI] [PubMed] [Google Scholar]
- [46].Garvik B, Carson M, Hartwell L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol. 1995;15:6128–6138. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vodenicharov MD, Wellinger RJ. DNA degradation at unprotected telomeres in yeast is regulated by the CDK1 (Cdc28/Clb) cell-cycle kinase. Mol Cell. 2006;24:127–137. doi: 10.1016/j.molcel.2006.07.035. [DOI] [PubMed] [Google Scholar]
- [48].Grandin N, Damon C, Charbonneau M. Cdc13 prevents telomere uncapping and Rad50-dependent homologous recombination. EMBO J. 2001;20:6127–6139. doi: 10.1093/emboj/20.21.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jia X, Weinert T, Lydall D. Mec1 and Rad53 inhibit formation of single-stranded DNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics. 2004;166:753–764. doi: 10.1534/genetics.166.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- [51].Lydall D, Weinert T. Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- [52].Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science. 1999;286:1166–1171. doi: 10.1126/science.286.5442.1166. [DOI] [PubMed] [Google Scholar]
- [53].Addinall SG, Downey M, Yu M, Zubko MK, Dewar J, Leake A, Hallinan J, Shaw O, James K, Wilkinson DJ, Wipat A, Durocher D, Lydall D. A genomewide suppressor and enhancer analysis of cdc13-1 reveals varied cellular processes influencing telomere capping in Saccharomyces cerevisiae. Genetics. 2008;180:2251–2266. doi: 10.1534/genetics.108.092577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Toh GW, Lowndes NF. Role of the Saccharomyces cerevisiae Rad9 protein in sensing and responding to DNA damage. Biochem Soc Trans. 2003;31:242–246. doi: 10.1042/bst0310242. [DOI] [PubMed] [Google Scholar]
- [55].Weinert TA, Hartwell LH. Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics. 1993;134:63–80. doi: 10.1093/genetics/134.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zubko MK, Guillard S, Lydall D. Exo1 and Rad24 differentially regulate generation of ssDNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics. 2004;168:103–115. doi: 10.1534/genetics.104.027904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lazzaro F, Sapountzi V, Granata M, Pellicioli A, Vaze M, Haber JE, Plevani P, Lydall D, Muzi-Falconi M. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J. 2008;27:1502–1512. doi: 10.1038/emboj.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Foster SS, Zubko MK, Guillard S, Lydall D. MRX protects telomeric DNA at uncapped telomeres of budding yeast cdc13-1 mutants. DNA Repair (Amst) 2006;5:840–851. doi: 10.1016/j.dnarep.2006.04.005. [DOI] [PubMed] [Google Scholar]
- [59].Booth C, Griffith E, Brady G, Lydall D. Quantitative amplification of single-stranded DNA (QAOS) demonstrates that cdc13-1 mutants generate ssDNA in a telomere to centromere direction. Nucleic Acids Res. 2001;29:4414–4422. doi: 10.1093/nar/29.21.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tsolou A, Lydall D. Mrc1 protects uncapped budding yeast telomeres from exonuclease EXO1. DNA Repair (Amst) 2007;6:1607–1617. doi: 10.1016/j.dnarep.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Soulier J, Lowndes NF. The BRCT domain of the S. cerevisiae checkpoint protein Rad9 mediates a Rad9-Rad9 interaction after DNA damage. Curr Biol. 1999;9:551–554. doi: 10.1016/s0960-9822(99)80242-5. [DOI] [PubMed] [Google Scholar]
- [62].Usui T, Foster SS, Petrini JH. Maintenance of the DNA-damage checkpoint requires DNA-damage-induced mediator protein oligomerization. Mol Cell. 2009;33:147–159. doi: 10.1016/j.molcel.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Raths SK, Naider F, Becker JM. Peptide analogues compete with the binding of alpha-factor to its receptor in Saccharomyces cerevisiae. J Biol Chem. 1988;263:17333–17341. [PubMed] [Google Scholar]
- [65].Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- [66].Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- [67].Wu S, Skolnick J, Zhang Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007;5:17. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 2007;69(Suppl 8):108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- [69].Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- [71].Kilkenny ML, Dore AS, Roe SM, Nestoras K, Ho JC, Watts FZ, Pearl LH. Structural and functional analysis of the Crb2-BRCT2 domain reveals distinct roles in checkpoint signaling and DNA damage repair. Genes Dev. 2008;22:2034–2047. doi: 10.1101/gad.472808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lee JC, Field DJ, Lee LL. Effects of nocodazole on structures of calf brain tubulin. Biochemistry. 1980;19:6209–6215. doi: 10.1021/bi00567a041. [DOI] [PubMed] [Google Scholar]
- [73].Straight AF, Murray AW. The spindle assembly checkpoint in budding yeast. Methods Enzymol. 1997;283:425–440. doi: 10.1016/s0076-6879(97)83035-2. [DOI] [PubMed] [Google Scholar]
- [74].Toh GW, O'Shaughnessy AM, Jimeno S, Dobbie IM, Grenon M, Maffini S, O'Rorke A, Lowndes NF. Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair (Amst) 2006;5:693–703. doi: 10.1016/j.dnarep.2006.03.005. [DOI] [PubMed] [Google Scholar]
- [75].Grenon M, Costelloe T, Jimeno S, O'Shaughnessy A, Fitzgerald J, Zgheib O, Degerth L, Lowndes NF. Docking onto chromatin via the Saccharomyces cerevisiae Rad9 Tudor domain. Yeast. 2007;24:105–119. doi: 10.1002/yea.1441. [DOI] [PubMed] [Google Scholar]
- [76].McSherry TD, Kitazono AA, Javaheri A, Kron SJ, Mueller PR. Non-catalytic function for ATR in the checkpoint response. Cell Cycle. 2007;6:2019–2030. doi: 10.4161/cc.6.16.4553. [DOI] [PubMed] [Google Scholar]
- [77].Clemenson C, Marsolier-Kergoat MC. The spindle assembly checkpoint regulates the phosphorylation state of a subset of DNA checkpoint proteins in Saccharomyces cerevisiae. Mol Cell Biol. 2006;26:9149–9161. doi: 10.1128/MCB.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Krishnan V, Surana U. Taming the spindle for containing the chromosomes. Cell Cycle. 2005;4:376–379. doi: 10.4161/cc.4.3.1537. [DOI] [PubMed] [Google Scholar]
- [79].Kim JA, Kruhlak M, Dotiwala F, Nussenzweig A, Haber JE. Heterochromatin is refractory to gamma-H2AX modification in yeast and mammals. J Cell Biol. 2007;178:209–218. doi: 10.1083/jcb.200612031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ng HH, Ciccone DN, Morshead KB, Oettinger MA, Struhl K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: a potential mechanism for position-effect variegation. Proc Natl Acad Sci U S A. 2003;100:1820–1825. doi: 10.1073/pnas.0437846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Naiki T, Wakayama T, Nakada D, Matsumoto K, Sugimoto K. Association of Rad9 with double-strand breaks through a Mec1-dependent mechanism. Mol Cell Biol. 2004;24:3277–3285. doi: 10.1128/MCB.24.8.3277-3285.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bonilla CY, Melo JA, Toczyski DP. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol Cell. 2008;30:267–276. doi: 10.1016/j.molcel.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zgheib O, Pataky K, Brugger J, Halazonetis TD. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol Cell Biol. 2009;29:1050–1058. doi: 10.1128/MCB.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Shim EY, Hong SJ, Oum JH, Yanez Y, Zhang Y, Lee SE. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol Cell Biol. 2007;27:1602–1613. doi: 10.1128/MCB.01956-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Liang B, Qiu J, Ratnakumar K, Laurent BC. RSC functions as an early double-strand-break sensor in the cell's response to DNA damage. Curr Biol. 2007;17:1432–1437. doi: 10.1016/j.cub.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.