

Summary
All aspects of transcription are controlled by complexes that modify or remodel chromatin at the level of individual genes, gene clusters, or whole chromosomes. The MSL complex that is responsible for dosage compensation in Drosophila is an example of complexes that operate at the whole-chromosome level on the transcription of individual genes. Recent experiments using traditional genetic analysis, molecular cytology, chromatin immunoprecipitation, or microarray technology have characterized the function of the two known enzymatic components of the MSL core complex and have identified the sequence characteristics that allow spreading of the complex along the X chromosome and a specific histone modification of active X-linked genes to which it is attracted. Further progress in understanding the function of this complex will benefit from biophysical approaches.
Introduction
Dosage compensation refers to the equalization of most X-linked gene products between males and females. In Drosophila, it is mediated by the MSL complex consisting of a core of five proteins, as well as one of two non-coding RNAs. The complex preferentially associates with numerous sites on the X chromosome in somatic cells of males but not of females. It is responsible for an enhancement of the transcriptional rate of a substantial number of X-linked genes, thereby mediating a compensatory effect for the difference in dosage of these genes between males and females [1]. Although all of the genes that encode the MSL complex protein subunits are transcribed in females, the complex is absent because the SXL protein that is responsible for female differentiation prevents the translation of the msl2 gene transcript [2, 3].
The presence of the MSL complex on the male X chromosome is correlated with a significant increase of histone H4 acetylated at lysine 16 (H4K16ac; [4]). This acetylation is the result of the activity of MOF - a histone acetyltransferase of the MYST family [5]. In order to display maximal activity and strict specificity, MOF must be included in the MSL complex [6] and, in particular must associate with the MSL1 and MSL3 subunits [7]. In addition to this enzyme, the MSL complex of Drosophila includes an ATP-dependent DEXH-box RNA/DNA helicase (MLE) that prefers double-stranded RNA or RNA/DNA hybrid substrates with a short 3′ overhang [8].
In Drosophila males, the complex is believed to assemble at the locus of the two roX genes and then spread to numerous additional sites along the X chromosome for which it has a complete range of affinity levels [9, 10]. Approximately 40 of these sites were defined as “high-affinity” because a partial complex can bind to them [11]. The nature of these sites was poorly understood until Gilfillan et al. [12•] identified a core sequence and proposed that dispersed along the X chromosome are clusters of several distinct but degenerated sequence motifs for which the MSL complex exhibits a complete range of affinity. Recently, using immunoprecipitation and microarray hybridization or sequencing, from 130 to 150 X-chromosome sites predominantly enriched for a GA repeated sequence for which the MSL complex has particular affinity have been identified [13••, 14••].
To define the role of a chromatin remodeling complex on gene function it is necessary to develop a detailed understanding of its association with target genes and of the mechanism that it uses to modulate the transcription of these genes not only at the molecular but also at the biophysical level. The purpose of this review is to set the stage for the study of the function of the MSL complex at the level of chromatin architecture.
Spreading of the MSL complex along the X chromosome
The generally accepted model has the MSL complex initially associating with chromatin entry sites and subsequently accessing active genes in order to enhance their transcription (Figure 1). Spreading beyond the 50 or so high-affinity sites to the numerous, newly characterized entry sites requires the complex to be fully assembled [15] and for its two enzymatic components - MOF and MLE - to be functional [16]. The mutation in MOF that prevents spreading is in the acetyl-CoA binding site and it is not clear whether the effect on spreading is caused by the loss of acetyltransferase function or by a conformational change in the protein, due to its failure to bind the co-enzyme, that in turn affects the general conformation of the complex.
Figure 1. Spreading of the MSL complex along the X chromosome.
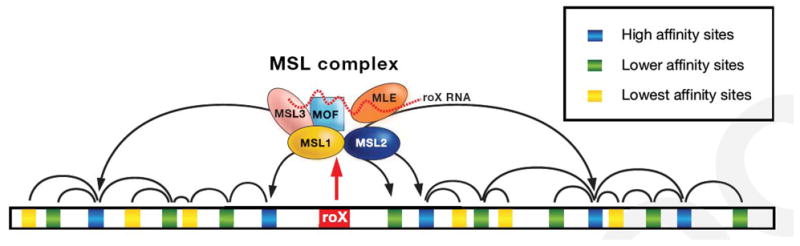
The MSL complex assembles at the loci of the two roX genes that are located on the X chromosome. This is necessary because the roX RNAs are unstable unless they associate with some of the protein subunits of the complex. Assembled complexes then access the many sites along the X chromosome for which they have different levels of affinity. Ultimately the MSL complexes associate with those X-linked genes whose transcription is thereby enhanced.
MLE is related to the ATPases present in complexes that remodel chromatin by altering the positioning or the architectural relationship between histone octamers and DNA [17]. In contrast to MLE none of these enzymatic subunits have been shown to possess double-stranded nucleic acid unwinding activity. To determine whether MLE has been subsumed by the MSL complex just for its ATPase function or for its ATP-dependent helicase activity, we analyzed mutations that allow MLE proteins to retain the ATPase but not the helicase activity. MSL complexes containing these mutant proteins could not spread along polytene chromosomes beyond the high-affinity sites but enhanced the transcription of genes immediately adjacent to these sites [18••]. These observations clearly indicate that the ATPase activity is required for MLE’s role in the transcriptional enhancement of a targeted gene while the helicase activity is necessary for the spreading of the complex along the X chromosome.
To date nothing is known of the in vivo substrate of MLE or of the mechanistic basis of its function in dosage compensation. A biophysical approach using single-molecule microscopy assays based on tethered particle motion and magnetic tweezers techniques [19] could determine whether MLE is capable of altering the torsional stress of DNA molecules or of translocating along a nucleic acid substrate.
Targeting genes for dosage compensation
Several years ago, we showed that the MSL complex is attracted to activated genes [20]. The questions that remained were: (1) What attracts the MSL complex to a transcribing gene? (2) How does the complex manage to acetylate H4K16 throughout the transcriptional unit?
Furuhashi et al. [21•] used RNAi in flies to knock down supercoiling factor (SCF) - a protein that generates negative supercoils in DNA in conjunction with topoisomerase II. They noted a lethal effect in males more pronounced than in females. They showed that depletion of SCF inhibits dosage compensation of X-linked genes but, surprisingly, does not prevent the complex from an apparently normal distribution along the X chromosome in salivary gland nuclei. Interestingly, in Drosophila SCF is present predominantly at promoter regions (Susumu Hirose, personal communication) where it may facilitate the loading of the MSL complex onto active genes. From the promoter region, the MSL complex would proceed towards the 3′ end, perhaps in association with the transcribing RNA polymerases II.
An alternative, possibility is that the complex associates with the nucleosomes of a transcribed region in an opportunistic manner, for example between successive elongating polymerases and from whatever neighboring locus where it may be concentrated (upstream or downstream of the transcribing gene). The rationale for this possibility is as follows. The presence of the MSL complex is increased at the 3′ end of transcriptional units [22, 23] as we had predicted by mapping the level of H4K16ac [24]. In contrast, following activation, the density of RNAPII on most actively transcribing genes remains skewed towards the 5′ end; this may interfere with the association of the MSL complex in this region of X-linked genes. In the body and towards the 3′ end of genes, elongating polymerases are relatively more sparsely spaced and there may be a greater opportunity for complex association. In addition, we had shown that when the MSL complex is recruited to a GAL4-induced promoter construct inserted in a region of the X chromosome normally deprived of the complex, the immunofluorescence signals generated by the GAL4 and MSL antisera do not overlap. In all cases where the site of insertion had been sequenced, the MSL protein was in a downstream position, relative to the GAL4 protein. These observations had led us to conclude that the MSL complex is localized on the transcribed portion of the region activated by the inserted promoter [20]. Evidence for this contention was provided by Larschan et al. [25•] who reported that the absence of H3K36me3, a mark of active transcriptional units, while lethal to both females and males leads to a reproducible reduction in the level of MSL binding in males and to a concomitant change in transcript levels of MSL target genes. They suggest that recognition of H3K36me3 is one contributing factor to MSL complex targeting that involves additional features of transcribed genes.
What is the function of the H4K16 acetylation mediated by MOF?
The undisputed correlation between enrichment in H4K16ac in the X chromosome and the enhanced transcription of X-linked genes in males has been known for a number of years. As is true in regard to most epigenetic events described to date, the mechanistic link between this particular histone modification and its effect on transcription – the link between cause and effect – was a proverbial black box. The basis for beginning to understand its content is Karoline Luger’s seminal description of the structure of the nucleosome (Figure 2) and of a probable inter-nucleosomal interaction involving the acidic patch formed by an H2A/H2B dimer in one nucleosome and the basic tail of histone H4 from a neighboring nucleosome [26, 27]. These observations led us to the hypothesis that the acetylation of lysine 16 on the tail of histone H4 lessens inter-nucleosomal interactions thereby facilitating nucleosomal eviction by the RNAPII complex, thereby enhancing the rate of elongation [24]. In support of this hypothesis, Dorigo et al. [28] first reported the effect of histone tails on electrostatic interactions between neighboring nucleosomes. More recently, the acetylation of H4 at lysine 16 was shown to prevent the conversion of reconstituted nucleosomal arrays into 30 nanometer fibers that are thought to represent a level of compaction of native chromatin unfavorable to transcription [29••].
Figure 2. Structure of a chromatin fiber.
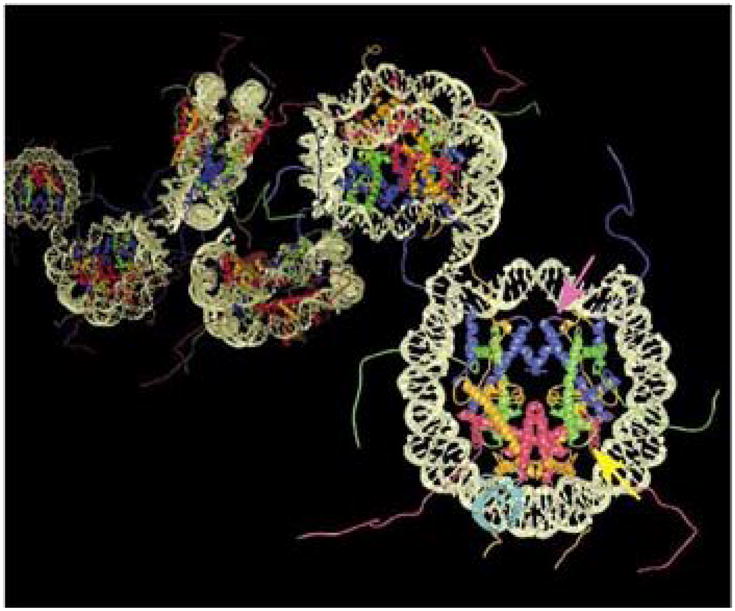
The interaction of neighboring nucleosomes via the N-termilal tails of their core histones can be modified by covalent modifications such as the acetylation of histone H4 at lysine 16 (Reprinted with permission [51]).
The very act of transcription is associated with alterations in the chromatin organization of transcriptional units. Are there features of chromatin reorganization that are uniquely associated with the enhanced level of gene expression mediated by the MSL complex? This question could be addressed by determining differences in restriction enzyme sites accessibility and nucleosome positioning on compensated and control genes. Unfortunately, given the average transcription enhancement of two-fold, these approaches may not yield meaningful signal to noise ratios. In contrast, the faithful reproduction of dosage compensation of a reporter gene carried on a plasmid introduced into S2 cultured cells by transfection [30••] provides the opportunity to investigate the effect of H4K16 acetylation on chromatin structure by probing for topological changes (Box 1). The type of results that can be obtained are illustrated in Figure 3.
Box 1. Topoisomers distribution.
The wrapping of DNA around a histone octamer, induces one negative supercoil in the DNA molecule (a linking number change of −1) that is protected from relaxation by topoisomerase I. As the plasmid is extracted, endogenous topoisomerase I activity relaxes the linker DNA between nucleosome cores and the number of negative supercoils that remain is an indication of the level of protection conferred to the DNA by its association with nucleosomes. The difference in linking number of two plasmids of equal size can be resolved by either one-dimensional or two-dimensional electrophoresis in the presence of chloroquine. Binding of chloroquine unwinds the DNA helix and results in the loss of negative supercoils. At different chloroquine concentrations, each plasmid can be resolved as a normal distribution of topoisomers whose center reflects the plasmid’s linking number. A chloroquine concentration that gives the best distribution of topoisomers for both plasmids is chosen to carry out the linking number comparison.
Figure 3. Example of a topological analysis of a plasmid subjected to dosage compensation.

(a) Topoisomers are resolved in a chloroquine-containing gel. (b) Histogram representation of the topoisomers. Upper panel corresponds to the compensated plasmid; lower panel is a non-compensated control plasmid. The height of a bar represents the intensity of the corresponding bands in the gel. NC is the position of the nicked plasmid in the gel. The difference in the relative position of the two topoisomer distributions indicates a difference in the nucleosomal organization in the two plasmids.
Does the MSL complex act by reducing the level of pausing of RNAPII on X-linked genes?
Since the seminal observation by John Lis that uninduced heat shock genes carry a transcriptionally engaged polymerase that has stalled after synthesizing a very short RNA transcript [31] evidence has accumulated that numerous genes in the genome display paused polymerases [32–34]. Surprisingly, a number of active genes have a greater concentration of RNAPII in their promoter region than within their body indicating that pausing is not just used to shut off uninduced genes but is also used to regulate the transcriptional output of active genes. These observations lead to the hypothesis that the chromatin modifications mediated by the MSL complex may exert their effect on transcription by reducing the pause of X-linked genes thereby increasing the number of polymerases that are engaged in elongation.
A characteristic of gene promoters with stalled or pausing polymerases is a significant enrichment in GAGA factor binding sites [35]. The GAGA factor exerts both positive and negative effects on gene transcription by facilitating chromatin remodeling and maintaining promoters in a conformation accessible to other regulatory factors. A reduction in the level of this factor affects the viability of males to a greater extent than females [36]. In addition, the small number of autosomal sites that are normally targeted by the MSL complex (5 to 7) is approximately doubled. These effects could be simply a reflection of the general role played by the GAGA factor on transcription: the necessity for males to hyper-transcribe their X-linked genes would render them more sensitive to general transcription disturbances than females. Yet, the similarity of the GAGA factor binding sites with the GA-rich X-chromosome binding sites of the MSL complex reopens the question of a functional role for this factor in dosage compensation [13••, 14••].
Interactions of the MSL complex with general chromatin assembly and nucleosome positioning complexes
The X chromosome in males responds dramatically to the loss-of-function of the general chromatin assembly complexes ACF and CHRAC and the nucleosome repositioning complex NURF. In vitro, ACF and CHRAC establish regularly ordered arrays of nucleosomes [37, 38] while NURF disrupts nucleosome periodicity [39]. Loss-of-function mutations in ISWI, the ATPase common to all three complexes, or in a subunit unique to the NURF complex [40] transform the male X chromosome in salivary gland polytene chromosome preparations into a chromatin mass that has lost all morphological features. X-chromosome morphology can be rescued in males by preventing the occurrence of H4K16ac. In vitro, the interaction of purified ISWI with nucleosomes is abrogated if H4 is acetylated at lysine16 [41]. Recently, Bai et al. [42] reported that in a mutant nurf background, loss-of-function mutations in either roX1 or roX2 lead to a more normal appearance of the polytenic X in the general region of the mutation; conversely, a wild type roX transgene relocated to an ectopic autosomal location nucleated a region of disorganization at its site of insertion. These authors also provided evidence that the NURF complex inhibits the synthesis of the roX RNAs in wild type females and prevents the over-transcription of roX2 in males. Grau et al. [43] have reported that mutations in the dAda3 gene cause a defect in the banding organization of polytene chromosomes in both males and females; once again, the X chromosome in males is more severely affected. Finally, Carre et al. [44] have shown that mutations in the histone acetyl transferase Gcn5 and the ATAC complex component Ada2a induce a specific decondensation of the X chromosome in mutant males. In contrast, the Drosophila RSF (remodeling and spacing factor) complex consisting of dRsf1 and ISWI does not affect the appearance of polytene chromosomes in either sex [45].
The MSL complex also interacts specifically with structural heterochromatin components Su(var)3–7 and Su(var)2–5/HP1, with the histone H3K9-specific methyltransferase Su(var)3–9 and with the tandem kinase JIL-1. Over-expression of Su(var)3–7 results in morphological effects in the larval salivary gland polytene chromosomes of both males and females, but the male X is most affected as it assumes a very small and highly compacted shape [46]. Loss of this heterochromatin protein or loss of HP1 results in a polytene X chromosome phenotype in males that is similar to the one induced by ISWI knockdown [47]. In their most recent paper, Spierer et al. [48••] report that the distribution of the MSL complex is abnormal on male polytene X chromosomes when the latter are enriched in heterochromatin by over-expression of Su(var)3–7. JIL-1 localizes to all polytene chromosomes but is substantially more abundant on the X chromosome in males [49]. Loss-of-function alleles result in global changes in the morphology of polytene chromosomes: the X chromosomes of females and the autosomes of both sexes exhibit some abnormal coiling, while the male X is once again shorter, fatter and without any evidence of banding [50].
The general similarity of the effects of very different complexes, remodeling activities or structural proteins strongly suggests that the common denominator may be the unique characteristic of the chromatin of the male X chromosome which renders it more sensitive to disturbances than autosomes or X chromosomes in females. These effects have been described by means of genetic and cytological experiments; their molecular basis should be sought at the biophysical level, using reconstituted chromatin fibers and approaches pioneered by Jeffrey Hansen and used effectively, for example, by Shogren-Knaak et al. [29].
Conclusions
In this review, I have highlighted the progress that has been made recently in understanding the function of the two known enzymatic components of the MSL complex, as well as in characterizing some of the parameters that allow the complex to spread along the X chromosome. A complete definition of these features and of the mechanism used by the complex to achieve transcriptional enhancement will rely increasingly on biophysical approaches.
Acknowledgments
Supported by grant GM15691 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
- 1.Gelbart ME, Kuroda MI. Drosophila dosage compensation: a complex voyage to the X chromosome. Development. 2009;136:1399–410. doi: 10.1242/dev.029645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bashaw GJ, Baker BS. The msl-2 dosage compensation gene of Drosophila encodes a putative DNA-binding protein whose expression is sex specifically regulated by Sex-lethal. Development. 1995;121:3245–3258. doi: 10.1242/dev.121.10.3245. [DOI] [PubMed] [Google Scholar]
- 3.Kelley RL, Solovyeva I, Lyman LM, Richman R, Solovyev V, Kuroda MI. Expression of msl-2 causes assembly of dosage compensation regulators on the X chromosomes and female lethality in Drosophila. Cell. 1995;81:867–877. doi: 10.1016/0092-8674(95)90007-1. [DOI] [PubMed] [Google Scholar]
- 4.Bone JR, Lavender J, Richman R, Palmer MJ, Turner BM, Kuroda MI. Acetylated histone H4 on the male X chromosome is associated with dosage compensation in Drosophila. Genes Dev. 1994;8:96–104. doi: 10.1101/gad.8.1.96. [DOI] [PubMed] [Google Scholar]
- 5.Hilfiker A, Hilfiker-Kleiner D, Pannuti A, Lucchesi JC. mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J. 1997;16:2054–2060. doi: 10.1093/emboj/16.8.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, Allis CD, Lucchesi JC. The drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol Cell Biol. 2000;20:312–318. doi: 10.1128/mcb.20.1.312-318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morales V, Straub T, Neumann MF, Mengus G, Akhtar A, Becker PB. Functional integration of the histone acetyltransferase MOF into the dosage compensation complex. EMBO J. 2004;23:2258–2268. doi: 10.1038/sj.emboj.7600235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee CG, Chang KA, Kuroda MI, Hurwitz J. The NTPase/helicase activities of Drosophila maleless, an essential factor in dosage compensation. Embo J. 1997;16:2671–2681. doi: 10.1093/emboj/16.10.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fagegaltier D, Baker BS. X chromosome sites autonomously recruit the dosage compensation complex in Drosophila males. PLos Biol. 2004;2:e341. doi: 10.1371/journal.pbio.0020341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oh H, Bone JR, Kuroda MI. Multiple classes of MSL binding sites target dosage compensation to the X chromosome of Drosophila. Curr Biol. 2004;14:481–7. doi: 10.1016/j.cub.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Lyman LM, Copps K, Rastelli L, Kelley RL, Kuroda MI. Drosophila male-specific lethal-2 protein: structure/function analysis and dependence on MSL-1 for chromosome association. Genetics. 1997;147:1743–1753. doi: 10.1093/genetics/147.4.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12•.Gilfillan GD, Konig C, Dahlsveen IK, Prakoura N, Straub T, Lamm R, Fauth T, Becker PB. Cumulative contributions of weak DNA determinants to targeting the Drosophila dosage compensation complex. Nucleic Acids Res. 2007;35:3561–3572. doi: 10.1093/nar/gkm282. The authors use a clever “one-hybrid” transfection assay to examine the recruitment of the MSL complex to candidate X-chromosome sequences contained in previously described high affinity sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13••.Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, Kuroda MI. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell. 2008;134:599–609. doi: 10.1016/j.cell.2008.06.033. The authors performed chromatin immunoprecipitation with an MSL complex subunit followed by hybridization to a genomic microarray or by sequencing. They identified 150 potential entry sites containing various repeats of a GA-rich recognition element. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14••.Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB. The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet. 2008:e1000302. doi: 10.1371/journal.pgen.1000302. This paper reached the same conclusions as reference 13. Here the authors use the long-standing observation that mutations in all but two of the MSL subunits allow an incomplete complex to bind to a small number of sites that were visible by IF on polytene chromosomes. A second strategy is based on the assumption that at reduced levels of cross-linking, only the sites where the complex is present in greater abundance, will be enriched following immunoprecipitation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley RL, Meller VH, Gordadze PR, Roman G, Davis RL, Kuroda MI. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell. 1999;98:513–522. doi: 10.1016/s0092-8674(00)81979-0. [DOI] [PubMed] [Google Scholar]
- 16.Gu W, Wei X, Pannuti A, Lucchesi JC. Targeting the chromatin-remodeling MSL complex of Drosophila to its sites of action on the X chromosome requires both acetyl transferase and ATPase activities. Embo J. 2000;19:5202–5211. doi: 10.1093/emboj/19.19.5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hogan C, Varga-Weisz P. The regulation of ATP-dependent nucleosome remodelling factors. Mutat Res. 2007;618:41–51. doi: 10.1016/j.mrfmmm.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 18••.Morra R, Smith ER, Yokoyama R, Lucchesi JC. The MLE subunit of the Drosophila MSL complex uses its ATPase activity for dosage compensation and its helicase activity for targeting. Mol Cell Biol. 2008;28:958–966. doi: 10.1128/MCB.00995-07. Mutations that inactivate the helicase activity of MLE while allowing ATPase activity are used to show the ATPase activity is required for the hypertranscription of genes that are located near a site of concentration of the MSL complex; these mutations prevent the complex from spreading beyond the high affinity sites along the X chromosome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lia G, Praly E, Ferreira H, Stockdale C, Tse-Dinh YC, Dunlap D, Croquette V, Bensimon D, Owen-Hughes T. Direct observation of DNA distortion by the RSC complex. Mol Cell. 2006;21:417–425. doi: 10.1016/j.molcel.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sass GL, Pannuti A, Lucchesi JC. Male-specific lethal complex of Drosophila targets activated regions of the X chromosome for chromatin remodeling. Proc Natl Acad Sci U S A. 2003;100:8287–8291. doi: 10.1073/pnas.1332749100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21•.Furuhashi H, Nakajima M, Hirose S. DNA supercoiling factor contributes to dosage compensation in Drosophila. Development. 2006;133:4475–4483. doi: 10.1242/dev.02620. These authors use RNA interference in vivo to show that the supercoiling factor protein (SCF) is required for the function of the MSL complex in dosage compensation. [DOI] [PubMed] [Google Scholar]
- 22.Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI. High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev. 2006;20:848–857. doi: 10.1101/gad.1400206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilfillan GD, Straub T, de Wit E, Greil F, Lamm R, van Steensel B, Becker PB. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev. 2006;20:858–870. doi: 10.1101/gad.1399406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith ER, Allis CD, Lucchesi JC. Linking global histone acetylation to the transcription enhancement of X-chromosomal genes in Drosophila males. J Biol Chem. 2001;276:31483–31486. doi: 10.1074/jbc.C100351200. [DOI] [PubMed] [Google Scholar]
- 25•.Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, Yang P, Workman JL, Park PJ, Kuroda MI. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol Cell. 2007;28:121–133. doi: 10.1016/j.molcel.2007.08.011. Taking a page from published work in yeast, these authors tested whether MSL3 – a chromodomain-containing subunit of the MSL complex – is attracted to histone H3 trimethylated at lysine 36 (H3K36me3) and whether this may play a role in targeting the complex to genes that are being transcribed. [DOI] [PubMed] [Google Scholar]
- 26.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 27.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 28.Dorigo B, Schalch T, Bystricky K, Richmond TJ. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J Mol Biol. 2003;327:85–96. doi: 10.1016/s0022-2836(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 29••.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. In this seminal paper, the authors use chemical ligation of histone tails to obtain nucleosomes that are uniformly acetylated at lysine 16 of their H4 subunits. Single chromatin fibers reconstituted with these nucleosomes resist condensation. [DOI] [PubMed] [Google Scholar]
- 30••.Yokoyama R, Pannuti A, Ling H, Smith ER, Lucchesi JC. A plasmid model system shows that Drosophila dosage compensation depends on the global acetylation of histone H4 at lysine 16 and is not affected by depletion of common transcription elongation chromatin marks. Mol Cell Biol. 2007;27:7865–70. doi: 10.1128/MCB.00397-07. This paper describes a plasmid-based model of dosage compensation in cultured S2 cells that allows new experimental approaches for the study of this regulatory mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rougvie AE, Lis JT. The RNA polymerase II molecule at the 5′ end of the uninduced hsp70 gene of D. melanogaster is transcriptionally engaged. Cell. 1998;54 :795–804. doi: 10.1016/s0092-8674(88)91087-2. [DOI] [PubMed] [Google Scholar]
- 32.Rougvie AE, Lis JT. Postinitiation transcriptional control in Drosophila melanogaster. Mol Cell Biol. 1990;10:6041–5. doi: 10.1128/mcb.10.11.6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, Adelman K. RNA polymerase is poised for activation across the genome. Nat Genet. 2007;39:1507–1511. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, Young RA. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet. 2007;39:1512–1516. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hendrix DA, Hong JW, Zeitlinger J, Rokhsar DS, Levine MS. Promoter elements associated with RNA Pol II stalling in the Drosophila embryo. Proc Natl Acad Sci U S A. 2008;105:7762–7. doi: 10.1073/pnas.0802406105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenberg AJ, Yanowitz JL, Schedl P. The Drosophila GAGA factor is required for dosage compensation in males and for the formation of the male-specific-lethal complex chromatin entry site at 12DE. Genetics. 2004;166:279–289. doi: 10.1534/genetics.166.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito T, Bulger M, Pazin MJ, Kobayashi R, Kadonaga JT. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell. 1997;90:145–155. doi: 10.1016/s0092-8674(00)80321-9. [DOI] [PubMed] [Google Scholar]
- 38.Varga-Weisz PD, Wilm M, Bonte E, Dumas K, Mann M, Becker PB. Chromatin-remodelling factor CHRAC contains the ATPases ISWI and topoisomerase II. Nature. 1997;388:598–602. doi: 10.1038/41587. [DOI] [PubMed] [Google Scholar]
- 39.Tsukiyama T, Daniel C, Tamkun J, Wu C. ISWI, a member of the SWI2/SNF2 ATPase family, encodes the 140 kDa subunit of the nucleosome remodeling factor. Cell. 1995;83:1021–1026. doi: 10.1016/0092-8674(95)90217-1. [DOI] [PubMed] [Google Scholar]
- 40.Deuring R, Fanti L, Armstrong JA, Sarte M, Papoulas O, Prestel M, Daubresse G, Verardo M, Moseley SL, Berloco M, et al. The ISWI chromatin-remodeling protein is required for gene expression and the maintenance of higher order chromatin structure in vivo. Mol Cell. 2000;5:355–365. doi: 10.1016/s1097-2765(00)80430-x. [DOI] [PubMed] [Google Scholar]
- 41.Corona DF, Clapier CR, Becker PB, Tamkun JW. Modulation of ISWI function by site-specific histone acetylation. EMBO Rep. 2002;3:242–247. doi: 10.1093/embo-reports/kvf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bai X, Larschan E, Kwon SY, Badenhorst P, Kuroda MI. Regional control of chromatin organization by noncoding roX RNAs and the NURF remodeling complex in Drosophila melanogaster. Genetics. 2007;176:1491–9. doi: 10.1534/genetics.107.071571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grau B, Popescu C, Torroja L, Ortuno-Sahagun D, Boros I, Ferrus A. Transcriptional adaptor ADA3 of Drosophila melanogaster is required for histone modification, position effect variegation, and transcription. Mol Cell Biol. 2008;28:376–385. doi: 10.1128/MCB.01307-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carre CA, Ciurciu A, Komonyi O, Jacquier C, Fagegaltier D, Pidoux J, Tricoire H, Tora L, Boros IM, Antoniewski C. The Drosophila NURF remodelling and the ATAC histone acetylase complexes functionally interact and are required for global chromosome organization. EMBO Rep. 2008;9:187–192. doi: 10.1038/sj.embor.7401141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanai K, Furuhashi H, Yamamoto T, Akasaka K, Hirose S. RSF governs silent chromatin formation via histone H2Av replacement. PLoS Genet. 2008;4:e1000011. doi: 10.1371/journal.pgen.1000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delattre M, Spierer A, Jaquet Y, Spierer P. Increased expression of Drosophila Su(var)3–7 triggers Su(var)3–9-dependent heterochromatin formation. J Cell Sci. 2004;117:6239–6247. doi: 10.1242/jcs.01549. [DOI] [PubMed] [Google Scholar]
- 47.Spierer A, Seum C, Delattre M, Spierer P. Loss of the modifiers of variegation Su(var)3–7 or HP1 impacts male X polytene chromosome morphology and dosage compensation. J Cell Sci. 2005;118:5047–5057. doi: 10.1242/jcs.02623. [DOI] [PubMed] [Google Scholar]
- 48••.Spierer A, Begeot F, Spierer P, Delattre M. SU(VAR)3–7 links heterochromatin and dosage compensation in Drosophila. PLoS Genet. 2008;4:e1000066. doi: 10.1371/journal.pgen.1000066. These authors report that reduced or increased amounts of the heterochromatin protein SU(VAR)3–7 alter the distribution of the MSL complex and of the histone H4 acetylated at lysine 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin Y, Wang Y, Walker DL, Dong H, Conley C, Johansen J, Johansen KM. JIL-1: a novel chromosomal tandem kinase implicated in transcriptional regulation in Drosophila. Mol Cell. 1999;4:129–35. doi: 10.1016/s1097-2765(00)80195-1. [DOI] [PubMed] [Google Scholar]
- 50.Deng H, Zhang W, Bao X, Martin JN, Girton J, Johansen J, Johansen KM. The JIL-1 kinase regulates the structure of Drosophila polytene chromosomes. Chromosoma. 2005;114:173–82. doi: 10.1007/s00412-005-0006-8. [DOI] [PubMed] [Google Scholar]
- 51.Luger L. The tail does not always wag the dog. Nature Genetics. 2002;32:221–222. doi: 10.1038/ng1000. [DOI] [PubMed] [Google Scholar]