

Abstract
Elastin breakdown in vascular aneurysms is mediated by cytokines such as tumor necrosis factor-α (TNF-α), which induces vascular smooth muscle cell (SMC) activation and regulates their deposition of matrix. We previously demonstrated exogenous supplement of TGF-β1 (1 ng/mL) and hyaluronan oligomers (0.786 kDa; 0.2 µg/mL) cues to upregulate elastin matrix synthesis by healthy cultured SMCs. Here, we determine if these cues likewise enhance elastin matrix synthesis and assembly by TNF-α-stimulated SMCs, while restoring their healthy phenotype. Adult rat aortic SMCs were treated with TNF-α alone or together with TGF-β1/ hyaluronan oligomeric cues, and the release of inflammatory markers were monitored during over a 21 day culture. Biochemical analysis was used to quantify cell proliferation, matrix protein synthesis and crosslinking efficiency, while immunofluorescence and electron microscopy techniques were used to analyze the elastin matrix quality. It was observed that SMC activation with TNF-α (10 ng/mL) induced matrix calcification and promoted production of elastolytic MMPs-2, 9. However, these effects were attenuated by the addition of TGF-β1 and HA oligomers cues to TNF-α-stimulated cultures, which also enhanced tropoelastin and collagen production, improved elastin matrix yield and crosslinking, promoted elastin fiber formation, and suppressed elastase activity, though release of the MMPs-2, 9 was not impacted. Overall, the results suggest that TGF-β1 and HA oligomers are potentially useful to suppress SMC activation and induce regenerative elastin repair within aneurysms.
Keywords: Hyaluronan oligomers, Tumor necrosis factor-α, Transforming growth factor-β1, Elastin matrix regeneration, Aortic aneurysms
1. Introduction
Progression of aortic aneurysms (AA) is typically associated with matrix calcification, decrease in vascular smooth muscle cell (SMC) content and chronic aortal matrix degradation, leading to its gradual dilatation and rupture1. The pathogenesis of AAs is characterized by injury, infiltration of extracellular inflammatory cells (e.g., monocytes, lymphocytes, plasma cells), and their secretion of matrix metalloproteinases (MMPs) and inflammatory cytokines (e.g., TNF-α, IL-1β), which in turn can induce changes in SMC phenotype and vascular matrix remodeling2. Since elastin is a major component of the ECM in vascular connective tissues, the released MMPs degrade crosslinked fibers of elastin (and collagen) to generate soluble peptides3. These peptide fragments can activate elastin-laminin receptors present on the surface of vascular cells4 to trigger further production of elastases, increase Ca2+ influx into cells, switch of SMCs from contractile to synthetic phenotype, and enhance their proliferation and disorganized deposition of matrix5. This cascade of events is typically associated with elastin breakdown6, concomitant loss of vessel elasticity7, and ultimately leads to vascular calcification and aneurysm progression.
Among the various cytokines secreted by macrophages within calcified aneurysmal lesions, tumor necrosis factor (TNF-α) has been found to be particularly dominant8. TNF-α critically mediates inflammation and can incite SMC proliferation and MMP release9. Under inflammatory conditions, TNF-α appears to down-regulate elastin gene expression by SMCs10, while MMPs and macrophage-derived elastases have been shown to degrade existing elastin11. These events can together contribute to interruption of intact elastin-SMC signaling pathways. TNF-α has been shown to affect the synthesis and sulfation of glycosaminoglycans (GAGs) and collagen12 in SMC cultures. Despite this clinical relevance and significance, the chronic-stimulatory effects of TNF-α on SMCs and their resultant effects on the quality and quantity of basal elastin matrix repair and regeneration by SMCs are unclear.
Presently, pharmacological agents are sought to stabilize surviving elastin within aneurysmal vessels by either inhibiting elastin-degrading MMPs-2 and 913, or chemically stabilizing elastin structures14. However, since mature elastic fibers rarely undergo active remodeling in adults, loss of elastin is not compensated by synthesis of new elastin. Thus, though AAs may be potentially stabilized against further growth, they cannot be regressed to restore native ultrastructure. To enable such regeneration of elastin structures in diseased adult vessels in vivo and within tissue-engineered constructs, we have identified and optimized a combination of elastogenic biomolecular cues based on hyaluronan (HA) fragments and growth factors (TGF-β1, IGF-1)15,16. While HA is hypothesized to mediate elastogenesis via intimate binding with elastin-associated proteins to form macromolecules vital to elastic fiber assembly17, TGF-β1 appears to mildly upregulate elastin synthesis by adult SMCs18. We recently showed that HA oligomers (MW∼756–1220 Da; 0.2 µg/mL) and TGF-β1 (1 ng/mL), henceforth referred to as elastogenic cues, attenuated proliferation of adult vascular SMCs and enhanced elastin production, matrix yield, maturation, and stability15. We hypothesize that delivery of TGF-β1 and HA oligomers will likewise coax elastin matrix regeneration and minimize inflammation within aneurysms.
Despite their benefits to elastin production by adult vascular SMCs, it is unknown if the cues will suppress pro-calcific and elastolytic activities of chronically-stimulated cells within aneurysms, such as those incited by TNF-α, and simultaneously enhance their synthesis and assembly of elastin matrix. To test this, we investigated efficacy of elastogenic cues, in TNF-α-stimulated rat aortic SMC cultures. Since parameters influencing AAs in vivo (e.g., source of injury stimulus, heterogeneity in ECM composition, proximity to site of aneurysmal rupture) can render establishing the efficacy of these cues difficult, a simple, yet established12, 19, culture model of TNF-α stimulated cells may be more useful. Our study outcomes can potentially lead to new aneurysm treatments based on both regeneration and stabilization of elastin matrices, and which may be employed as a stand-alone option or in consort with existing surgical/ pharmacological approaches.
2. Materials and Methods
2.1 Cell Isolation and Culture
Aortae from adult healthy Sprague-Dawley rats (n = 3) were excised from the arch to the celial axis, the intimal layers were scraped off, and the medial layers were dissected from the underlying adventitial layers and chopped into small slices. The tissue slices were then degraded in DMEM-F12 containing 125 U/mg collagenase and 3 U/mg elastase (30 min, 37 °C), centrifuged (400 g, 5 min), washed and cultured in DMEM-F12 containing 10% v/v FBS for 7 days. Primary rat aortic SMCs (RASMCs) outgrowing from these tissue explants were further propagated. The animal surgeries were performed in accordance with the Guide for the care and use of laboratory animals published by the US National Institutes of Health (NIH Publication No. 85-23, 1996). For matrix synthesis experiments, SMCs from passages 3–5 were seeded onto 6-well tissue culture plates (A = 10 cm2 ) at a seeding density of 4 × 104 cells/ well and cultured in 5 mL of DMEM-F12 with 10% FBS and 1% Penstrep. HA oligomers used in this study contained 75 ± 15% w/w of HA 4-mers, with 6-mers and 8-mers forming the balance, and were prepared using protocols we have previously described20.
Experimental cultures either received: controls with no-additives, TNF-α alone, or TGF-β and HA oligomeric cues with or without TNF-α. TNF-α (Sigma Aldrich, MO) was supplemented at 10 ng/mL, HA oligomers at 0.2 µg/mL, and TGF-β1 (Peprotech Inc., NJ) at 1 ng/mL. TNF-α concentration was chosen based on previous reports which suggest its pro-inflammatory effects at doses as low as 10ng/mL21, 22, while the concentrations of TGF-β1 and HA oligomers were based on doses that we previously demonstrated to elastogenically stimulate healthy RASMCs15. Fresh medium was added to cultures twice weekly, and the spent medium aliquots collected from each well at different time points, pooled and stored at −20 °C. These aliquots and the corresponding harvested cell layers were biochemically analyzed at 21 days of culture.
2.2 DNA Assay for Cell Proliferation
The DNA content of cell layers was quantified at 1 and 21 days of culture to assess proliferation of SMCs and to normalize matrix amounts to cell count. Briefly, cells were detached with 0.25% v/v trypsin-EDTA (Invitrogen), homogenized, pelleted by centrifugation, re-suspended in NaCl/Pi buffer, sonicated, and assayed using a fluorometric assay23. The cell density was calculated on the basis of an estimated 6 pg DNA/cell23.
2.3 Hydroxy-proline Assay for Collagen
A hydroxy-proline assay was used to estimate the collagen content within test and control cell layers, and in the medium fraction. As described earlier24, the homogenized cell layers were pelleted by centrifugation (10000 g, 10 min), and digested with 1 mL of 0.1 N NaOH (1 h, 98 °C). The digestate was centrifuged to isolate insoluble, crosslinked elastin. The supernatant containing solubilized collagen and uncrosslinked matrix elastin was neutralized with an equal volume of 12 N HCl. One half-volume was hydrolyzed at 110 °C for 16 h, dried overnight and 20 µL aliquots of the reconstituted residue assayed for hydroxy-proline content. The supernatant and matrix collagen amounts were then calculated on the basis of the 13.2% hydroxy-proline content of collagen, and normalized to DNA content of corresponding cell layers.
2.4 Fastin Assay for Elastin)
Matrix elastin (alkali-soluble and insoluble fractions) and soluble tropoelastin (in pooled spent medium) amounts were quantified using a Fastin assay (Accurate Scientific Corp, NY)24. Since the Fastin assay quantifies only soluble α-elastin, the insoluble elastin was first reduced to a soluble form by digesting with 0.25 N oxalic acid (1 h, 95 °C), and the pooled digestate was filtered in microcentrifuge tubes fitted with low molecular weight cut-off membranes (10 kDa). The insufficiently crosslinked, soluble elastin fraction retained in the oxalic acid-free fraction and in the water-reconstituted hydrolysate (from collagen assay) were also quantified using the Fastin assay. The pooled spent medium aliquots were lyophilized and subjected to the Fastin assay. Elastin amounts were normalized to corresponding DNA amounts.
2.5 Assay for Desmosine Crosslinks
Desmosine crosslink densities within elastin matrices were quantified using an ELISA kit24. At 21-days, cell layers were digested with collagenase (12 h, 37 °C) and elastase (12 h, 37 °C), the digestates were acid-hydrolyzed (6 N HCl, 110 °C, 16 h), the desmosine contents in the reconstituted residues were determined by ELISA and compared to corresponding trends in insoluble matrix elastin.
2.6 Lysyl Oxidase (LOX) Enzyme Activity
At 21 day of culture, the spent culture medium was assayed for LOX activity. H2O2 released during LOX-induced deamination was detected using an Amplex® red fluorometric assay (Molecular Probes, USA). The fluorescence intensities were recorded with excitation and emission wavelengths at 560 and 590 nm, respectively.
2.7 Western Blot Analysis for Tropoelastin and LOX Protein Synthesis
Western blot analysis was performed to semi-quantitatively confirm observed biochemical trends in tropoelastin synthesis and to compare LOX protein synthesis between cases. As previously detailed24, pooled spent medium from cultures were lyophilized, assayed for protein content using a DC protein assay kit (Bio-Rad, CA), to optimize sample volumes for SDS PAGE/ western blot. Protein bands were detected with primary rabbit anti-rat polyclonal antibodies to elastin (Elastin Products Company, MO) and the 31 kDa active LOX protein (Santa Cruz Biotechnology, CA), and visualized in a Chemi-Imager IS 4400 system (Alpha Innotech, CA).
2.8 Immunoflourescence Detection of Elastin
Immunofluorescence techniques were used to confirm the presence of elastin within the cultured cell layers. RASMCs were cultured in 4-well sterile chamber slides under identical conditions at an initial seeding density of 5 × 103 cells/well. At 21 days of culture, the cell layers were fixed with 4% v/v paraformaldehyde for 10 min, and labeled with Alexa-488 Phalloidin (Molecular Probes; 1:20 dilution; 20 min, 25 °C), a marker for SMC F-actin. Elastin was detected with polyclonal antibody (Elastin Products Company, MO) and visualized with a Rhodamine-conjugated donkey anti-rabbit IgG secondary antibody (Chemicon). Cell nuclei were visualized with the nuclear stain 4′, 6-diamino-2--phenylindole dihydrochloride (DAPI) contained in the mounting medium (Vectashield, Vector Labs, CA).
2.9 Cytokine Array
The type and amount of cytokines produced by RASMCs in response to exogenous TNF-α alone and in presence of elastogenic cues were compared using an ELISA-based cytokine array. RASMCs were cultured to semi-confluence and then subjected to exogenous TNF-α (10 ng/mL) for 48 h, in the presence or absence of HA oligomers (0.2 µg/mL) and TGF-β (1 ng/mL). The release of different cytokines and chemokines from cultured SMCs into the media were detected by a ChemiArray™ rat cytokine array I (Millipore), consisting of the corresponding antibodies spotted in duplicate onto a membrane. The membranes were processed in accordance with the manufacturer’s protocol and imaged under chemiluminescence to quantify relative spot intensities. Intensities due to each cytokine were normalized according to the positive signal of each membrane, and the results were averaged from the outcomes of three replicate runs.
2.10 Gel Zymography
MMPs-2, 9 were detected in the culture medium by gelatin zymography methods25. Briefly, aliquots of culture medium pooled from each of the replicate test and control cell layers, over 21 days of culture, were assayed for protein content using a bicinchoninic acid (BCA)-containing protein assay (Sigma-Aldrich, St. Louis, MO), and all lanes were loaded in triplicate with 15 µg of protein from each extract alongside with pre-stained molecular weight standards (Bio-Rad, CA). After development and staining, densities of MMP-2 and 9 bands were measured using Gel Pro software (Media Cybernetics, MD), and reported as relative density units (RDU).
2.11 Elastase Assay
Elastase activity in the cell cultures was assayed using an EnzChek® Elastase Assay kit (Molecular Probes). Briefly, 50 µL of spent culture medium at 21 days of culture was mixed with 50 µL of diluted bovine neck ligament elastin, incubated for 30 min at 37 °C, and the fluorescence intensity measured at 485 nm excitation and 510 nm emission wavelengths. One unit of elastase was defined as the amount of porcine pancreatic elastase required to solubilize 1 mg of elastin at pH 8.8 and 37 °C.
2.12 Von Kossa Staining for Calcific Deposits
After 21 days of culture, cell cultures were incubated with 1% w/v AgNO3 and irradiated with UV (20 min). After several changes of distilled water, unreacted silver was removed with 5% w/v sodium thiosulfate for 5 min, and the cell layers were rinsed and counterstained with hematoxylin. The presence of black stain confirmed the presence of calcium phosphate deposits.
2.13 Matrix Ultrastructure
Transmission electron microscopy (TEM) was used to observe the ultrastructure of matrix elastin. Cell layers were fixed with 2.5% w/v glutaraldehyde, post-fixed in 1% w/v osmium tetroxide (1 h), dehydrated in a graded ethanol series, embedded in Epon 812 resin, sectioned, mounted on copper grids, stained with uranyl acetate and lead citrate, and then visualized on a Hitachi H7600T TEM.
2.14 Statistical Analysis
Data were measured in triplicate from experiments that were also performed with (n=3 samples/case), and statistically analyzed using Student’s t-test, assuming unequal variance. Significant differences between test and non-additive control cultures was deemed for p < 0.05.
3 Results
3.1 TNF-α Stimulation of SMCs
Figure 1A shows how 10 ng/mL TNF-α prompted release of inflammatory cytokines (TNF-α), interleukins (IL-1, 4, 6, 10) and chemokines (FNK, LIX, MCP-1, MIP-3α), and of tissue inhibitor of MMPs (TIMP-1) by RASMCs, relative to non-stimulated control RASMCs (p < 0.05 in all the cases). Von kossa staining also indicated a significantly greater number of calcific deposits within TNF-α treated cell layers (Figure 1B) compared to controls. Gel zymography showed that production of elastolytic MMPs-9, 2 were enhanced by 43 ± 14% and 51 ± 17% respectively in TNF-α-supplemented cultures and control cultures (Figure 1C; p < 0.001). These results suggest that TNF-α (10 ng/mL) chronically-activates cultured SMCs. Also, as shown in Figure 1D, TNF-α supplements enhanced elastase production within non-additive control cultures by 39 ± 4 % (p < 0.01).
Figure 1.
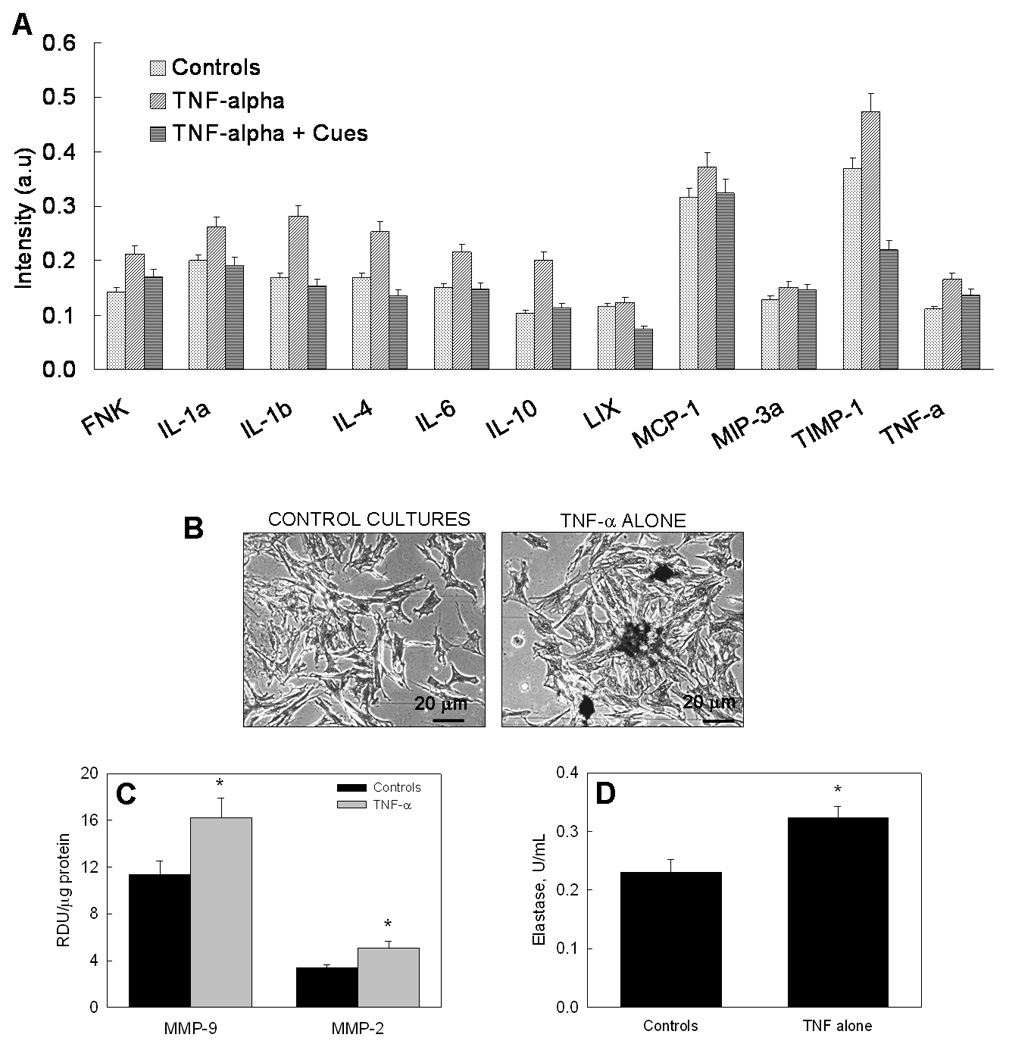
(A) Cytokine array analysis of inflammatory factors released by supplement-free RASMC cultures, and cultures treated with TNF-α in the absence or presence of elastogenic cues. (B) Von kossa staining of RASMC cultures stimulated with TNF-α shows several calcific deposits, while control cultures show none. (C) Production of elastolytic MMPs-2 and 9 by RASMC cultures is enhanced by exposure to TNF-α. (D) Elastase enzyme activity is enhanced in TNF-a-stimulated cultures relative to non-additive control cultures. In panels 1C and 1 D, (*) represents significant differences from controls, deemed for p<0.05.
3.2. Effect of TNF-α and Elastogenic Cues on SMC Proliferation, Matrix Synthesis
Control cultures proliferated 14.9 ± 2.1 –fold over 21 days. TNF-α supplementation had no effect on basal RASMC proliferation rate (0.94 ± 0.22–fold increase vs. controls; p = 0.58). In the presence of the cues, TNF-α significantly suppressed cell proliferation relative to controls (0.74 ± 0.23–fold; p = 0.03), although this decrease was less relative to cultures that received the cues alone (0.54 ± 0.1–fold increase vs. controls, p < 0.001).
Over 21 days of culture, control cultures generated 35774 ± 4662 ng and 18931 ± 1966 ng of collagen and tropoelastin precursors, respectively, per nanogram of DNA. TNF-α had no effect on collagen (0.97 ± 0.07-fold) and tropoelastin synthesis (1.06 ± 0.08-fold), relative to control cultures. Collagen synthesis was however increased by 1.16 ± 0.1 –fold and 1.84 ± 0.1 -fold relative to controls, when cues were supplemented in the presence and absence of TNF-α, respectively (p = 0.04 and p = 0.005 respectively vs. controls). Addition of cues also enhanced tropoelastin synthesis by 2.01 ± 0.19–fold (p < 0.001 vs. controls; Figure 2A) when no TNF-α was added, and by 2.06 ± 0.12–fold, when TNF-α was also supplemented (p < 0.001 vs. controls).
Figure 2.
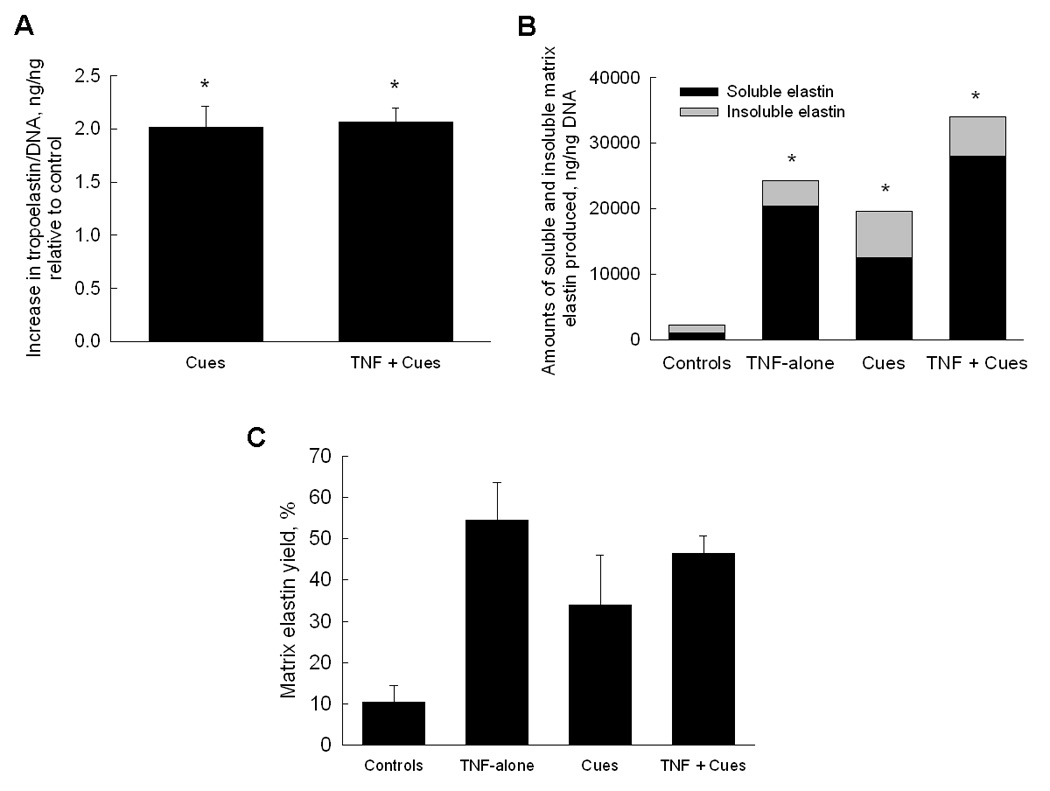
(A) Effects of cues alone or together with TNF-α on tropoelastin synthesis by adult RASMCs. Data shown (mean ± SD) are normalized to cellular DNA content at 21 days of culture and represented as fold change in protein production relative to non-additive control cultures (n = 3/case). (B) Relative amounts of alkali-soluble and crosslinked matrix elastin produced by RASMCs in all the cases. (C) Matrix elastin yields (i.e., ratio of matrix elastin to total elastin) were significantly higher in cultures that received TNF-α alone, cues alone or together with TNF-α. P < 0.05 represents significant differences from controls (*).
Elastin deposited within cell layers was measured as the sum of a highly-crosslinked alkali-insoluble fraction, and an alkali-soluble fraction. Figure 2B shows the relative proportions of these elastin fractions in each of the tested cases. Synthesis of soluble and insoluble matrix elastin increased by 20 ± 3.5-fold and 3.23 ± 0.2-fold in TNF-α–treated cultures, relative to their production levels in controls (1026 ± 269 ng/ng and 1186 ± 546 ng/ng respectively, p < 0.001 vs. controls). The elastogenic cues enhanced production of soluble and insoluble matrix elastin by 12.3 ± 4.6 and 5.9 ± 1.9–fold, respectively, vs. controls (p < 0.001). The addition of cues to the TNF-α-stimulated cultures furthered these increases to 27.3 ± 1.7 and 4.9 ± 1.1–fold vs. controls, respectively (Figure 2B; p < 0.001 in both cases). Overall, relative to controls, total matrix elastin (sum of both elastin fractions) synthesis increased by 11 ± 1.8, 8.8 ± 3.1 and 15.3 ± 1.4–fold, respectively, on addition of TNF-α alone, cues alone or the cues together with TNF-α.
Figure 2C shows the elastin matrix yields [matrix yield=matrix elastin/ (tropoelastin + matrix elastin)]. While only 10.5 ± 3.9% of total elastin produced in non-additive control cultures was deposited as a matrix, this yield was increased to 54 ± 9% by addition of TNF-α alone, 34 ± 12% with the addition of cues alone, and 46.5 ± 4.3% upon supplementation of both cues and TNF-α.
3.3 Effects of TNF-α and Cues on Elastin Matrix Crosslinking
As shown in Figure 3A, relative to control cell layers (12.8 ± 1.7 pg desmosine/ng DNA), cells cultured with TNF-α alone did not show any significant increase in desmosine synthesis (1.06 ± 0.17-fold; p = 0.31 vs. controls), while cues significantly enhanced desmosine synthesis by 1.91 ± 0.42–fold and 1.42 ± 0.3–fold respectively in the absence and presence of TNF-α (p < 0.01 and p < 0.02 vs. controls).
Figure 3.
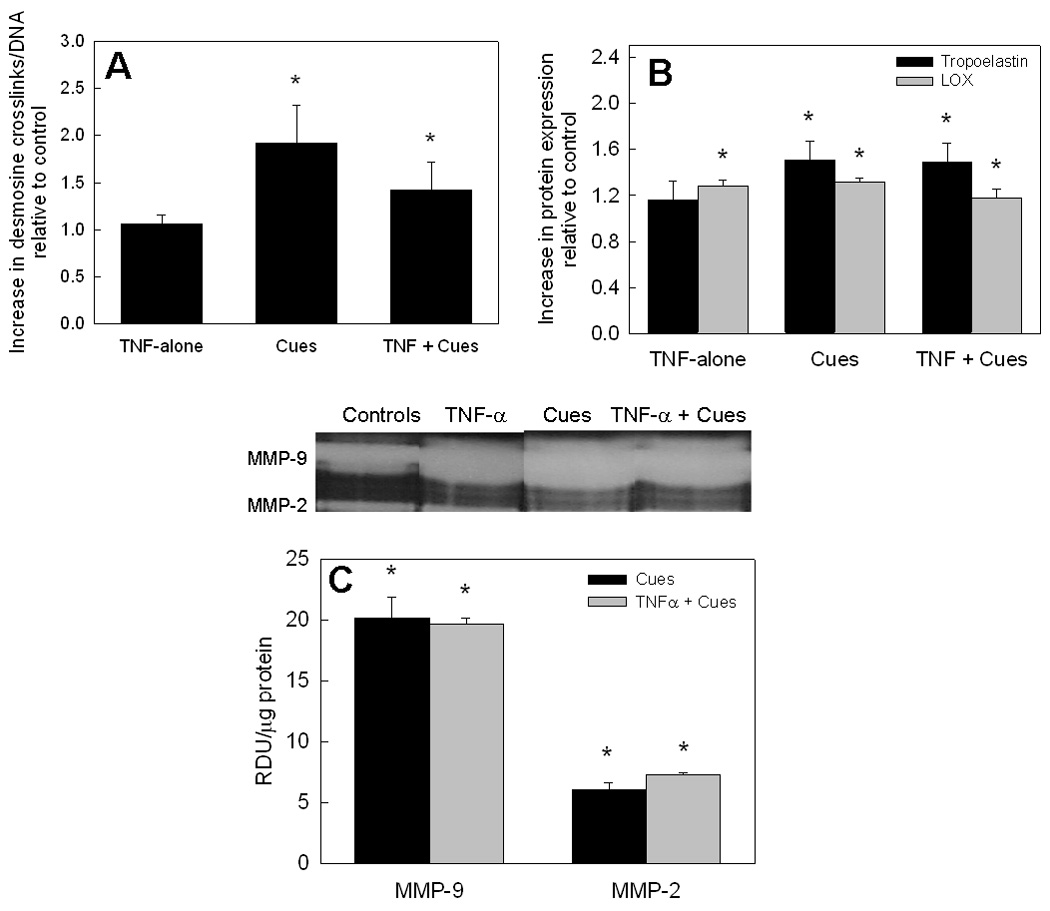
(A) Desmosine amounts measured in test cell layers were normalized to corresponding DNA amounts (ng/ng), and further a similar ratio obtained for the non-additive controls. Comparable trends were observed for the desmosine/DNA density and respective insoluble matrix elastin/DNA for selected cases. (B) SDS-PAGE/ Western blot analysis of tropoelastin and LOX proteins within the pooled medium cultures at the end of 21 days. Data shown represent mean ± SD of 3 repeats/ case and are shown normalized to controls. (C) Gel zymography analysis revealed the presence of MMPs-2 and 9 within cultures containing cues alone or together with TNF-α. Representative zymogram bands are also shown for comparison between the controls and test cases. It can be seen that the amount of MMPs-2, 9 in cultures receiving both the cues and TNF-α were not significantly different from those in TNF-α alone stimulated cultures. P < 0.05 represents significant differences in values relative to non-additive controls (*).
Western blot analysis for tropoelastin protein expression (Figure 3B) showed trends similar to that observed from biochemical analysis. TNF-α alone had no effect on tropoelastin protein amounts (1.16 ± 0.16–fold vs. controls), while cues alone and in the presence of TNF-α promoted tropoelastin protein expression by 1.51 ± 0.16 and 1.48 ± 0.17–fold, respectively. Western blot analysis showed LOX protein expression to be enhanced in cultures supplemented with TNF-α or cues alone by 1.27 ± 0.05 and 1.31 ± 0.03–fold, versus controls (p < 0.005; Figure 3B) and less so in cultures that received both the cues and TNF-α (1.17 ± 0.09–fold increase vs. controls; p = 0.03). There were no statistically significant differences in LOX expression between cultures that received TNF-α alone, cues, alone, or both together. Also, there were no significant differences in LOX activities between control cultures and those supplemented with TNF-α either alone or together with cues.
3.4. Cues Repress TNF-α–Induced Activation of RASMCs
Gel zymography analysis revealed that MMP-9 production was increased by addition of cues, alone and together with TNF-α (1.78 ± 0.05 and 1.73 ± 0.05–fold vs. controls, respectively; p < 0.05 vs. controls). However, these increases were not significantly different from that measured in cultures supplemented with TNF-α alone (Figure 1C; p > 0.05 vs. controls). Again, though MMP-2 production levels, both in the presence of cues alone and together with TNF-α were significantly higher than that in controls (Figure 3C; p < 0.05 vs. controls), they were not significantly higher than in cultures treated with TNF-α alone (Figure 1C; p > 0.05 vs. TNF-α alone in both cases). However, elastase activity in the presence of cues alone (0.24 ± 0.01 U/mL) was comparable to that in control cultures (0.23 ± 0.02 U/mL), while the addition of both cues and TNF-α resulted in elastase activity around 0.28 ± 0.01 U/mL, significantly lower than that in TNF-α alone additive cultures (p < 0.01 for TNF-α + cues vs. TNF-α alone).
The release of inflammatory cytokines and interleukins by TNF-α-stimulated RASMCs significantly decreased when cues were added (Figure 1A). It was observed that on average, addition of the cues to TNF-α-stimulated cultures contributed to ∼ 28–46% decrease in the production of interleukins (IL-1, 4, 6, 10), chemokines (FNK, LIX, MCP-1, MIP-3a; 12–40%), and cytokines (TNF-α; 19–53%) and TIMP-1 relative to those conditioned with TNF-α alone (p < 0.05).
Von Kossa staining showed that cultures supplemented with TGF-β (1 ng/mL; Figure 4A) alone contained large black calcific deposits, while further addition of HA oligomers in the absence (Figure 4B) and presence (Figure 4C) of TNF-α completely inhibited calcific deposits.
Figure 4.
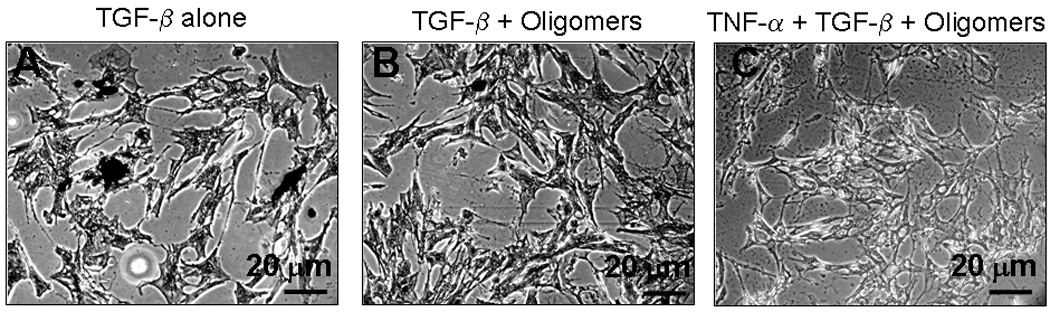
Von kossa staining images of cell layers treated with TGF-β alone, cues alone and cues together with TNF-α. Significant calcific deposits were evident in cultures treated with TGF-β alone, while cultures which received cues alone or together with TNF-α did not contain any calcific deposits.
3.5 Structural Analysis of Matrix Elastin
Immunofluorescence imaging showed that elastin (red) was more abundant in cultures that received TGF-β and HA oligomeric cues alone (Figure 5C) or together with TNF-α (Figure 5D), than in controls (Figure 5A) or TNF-α-only cultures (Figure 5B). However, elastin-containing cultures that were not treated with the anti-elastin primary antibody did not exhibit any florescence confirming lack of non-specific binding of the fluorophore.
Figure 5.
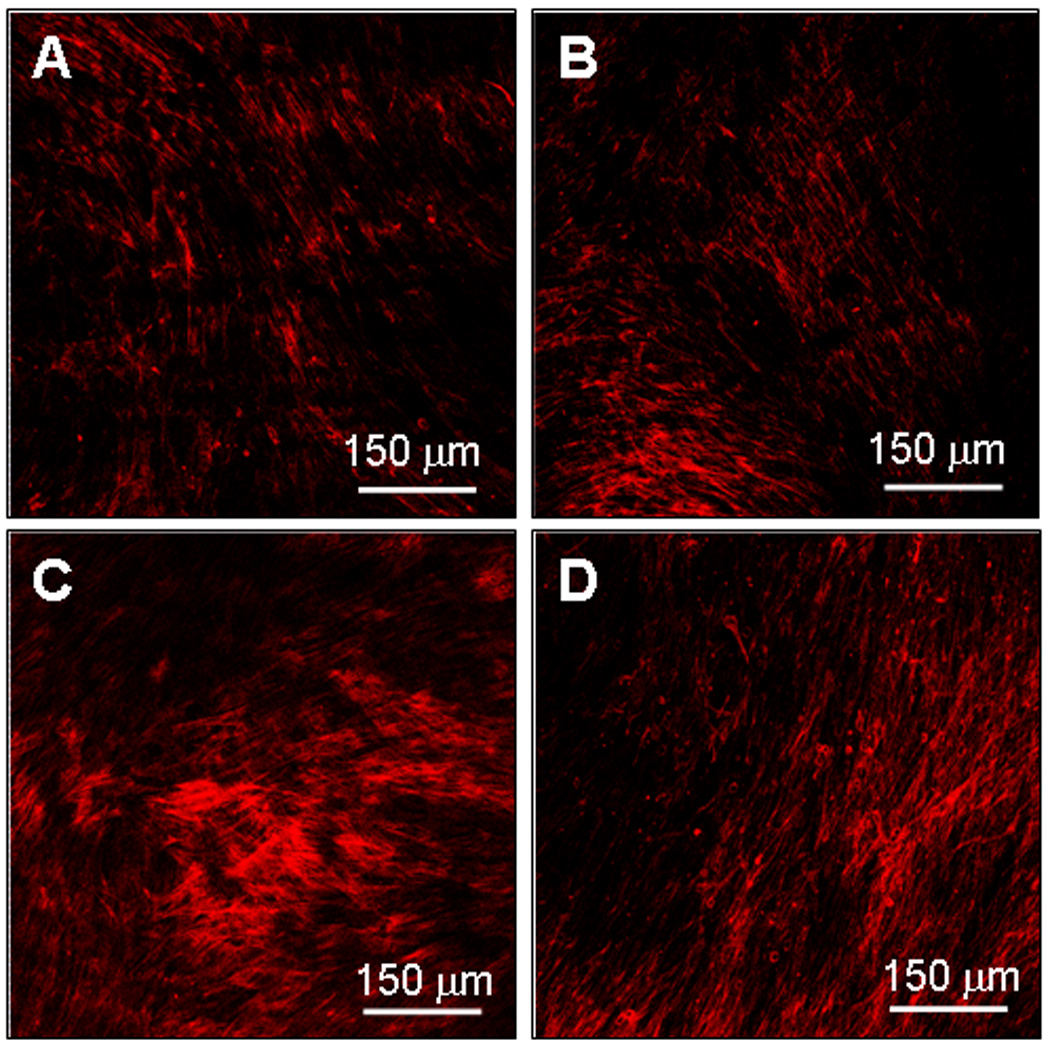
Immunodetection of elastin protein within control and test cell layers after 21 days of culture. An increase in matrix elastin protein (red) is evident in cultures which received cues alone (panel C) or together with TNF-α (panels D), while cultures which received no-additives (panel A) or TNF-α alone (panel B) showed relatively less fluorescence intensity. Scale bar: 150 µm.
Transmission electron micrographs showed deposition of discrete amorphous elastin clumps, and sparse elastin fibers in both control cultures (Figure 6A), and TNF-α-alone stimulated cultures (Figure 6B). When both TGF-β1 and HA oligomers were provided to control cultures (Figure 6C), mature elastin fiber formation was favored, with the matrix containing numerous fully-formed bundles of fibers (100–200 nm diameter), which were not observed in control and TNF-α-only cultures. Fibrillin (immunogold particle-stained, indicated by arrows in Figure 6), which appeared in transverse sections as darkly stained nodules, were located at the periphery of aggregating elastin fiber bundles, signifying normal elastic fiber assembly. The elastogenic cues also promoted fiber formation within TNF-α stimulated cultures (Figure 6D), with less amorphous elastin and more fibrilliar elastin observed when compared to TNF-α-only cultures.
Figure 6.

Representative TEM images of 21-day old RASMC cell layers cultured additive-free (panel A), with TNF-α alone (panel B), cues alone (panel C) or together with TNF-α (panel D). Aggregating amorphous elastin clumps leading to the formation of elastin fibers can be clearly seen in TNF-α –activated cultures treated with cues, while cultures which received no-additives or TNF-α alone showed amorphous elastin deposits with sparse fiber formation. However, in the presence of cues alone, fibrillin-mediated elastin fiber formation is clearly evident (see arrows indicating dark round spots in panel C).
4.Discussion
Literature attests to the contribution of inflammatory mechanisms in cardiovascular disease pathogenesis. Pro-inflammatory cytokines such as TNF-α, IL-1, IL-β1, and IL-6, produced by a variety of cells mediate many biological signaling mechanisms, and facilitate primary host responses and tissue repair26. TNF-α and interleukins have been identified within atherosclerotic and aneurysmal vessel segments27, and appear to enhance elastolytic activity and accumulation of elastin peptides within these tissues, which induce neointima formation28. The importance of TNF-α to initiating aneurysms thus justifies study of TNF-α-stimulated vascular SMC cultures, to investigate activated cell responses and to assess the efficacy of drugs aimed at suppressing these responses. Although pharmacological approaches to attenuate MMP production and activity, and chemical approaches to stabilize elastin against breakdown appear to proffer some promise in halting aneurysm progression, their regression via active regeneration of elastin matrices within has not been possible to date. This is due to poor elastin regeneration by adult vascular cells. In this context, previously, we showed TGF-β1 and HA oligomeric cues to synergistically enhance elastin matrix synthesis and fiber formation by adult vascular SMCs15,20. Currently, we seek to gauge similar utility of these cues for elastogenically-stimulating chronically-activated SMCs, which mimic cells in an aneurysmal environment, while restoring them to a normal phenotype.
In contrast to some prior studies where significant apoptotic cell death was observed upon their exposure to TNF-α29, we observed active cell proliferation. In this context, Wang et al.30 showed that only some sub-populations of vascular SMCs apoptose in response to TNF-α; it is possible that our SMCs represent such non-apoptosing sub-types. Regardless, TNF-a-induced activation of SMC layers was confirmed by (a) increased cellular release of MMPs-2, 9 and other elastases relative to non-additive controls, (b) enhanced endogenous production of interleukins and other cytokines, including TNF-α, and (c) enhanced in vitro calcification of cell layers relative to controls, possibly via TNF-α-mediated increases in intracellular cAMP and osteoblastic differentiation31.
Further, we noted that collagen and tropoelastin production in TNF-α-added cultures was not different from control cultures. Some prior studies have also shown TNF-α to suppress collagen gene expression and protein synthesis by fibroblasts32, fat- storing cells33, vascular SMCs and endothelial cells12. One study12 showed that 10 ng/mL TNF-α, identical to that used here, suppressed collagen production by 20% relative to control cultures, only when cell cultures were confluent. However, 10 ng/mL TNF-α did not have any impact on collagen production within sub-confluent SMC cultures12, which agrees with our present findings since our cultures were only ∼70% confluent at 3 weeks. Besides, our culture system closely mimics chronic, long-term TNF-α–induced signaling in aneurysms than a 24-h study, as was done previously12. We also note in our study that TNF-α induces multi-fold increases in total matrix elastin synthesis relative to controls, which we hypothesize to be an indirect outcome of TNF-α–induced increase in LOX production (Figure 3B), an enzyme critical for normal biosynthesis and assembly of elastin matrix. Supporting this hypothesis, we have seen that exogenous LOX stimulates elastin matrix synthesis relative to controls (unpublished work), in a manner similar to that observed in this study. Moreover, our analysis shows that tropoelastin was not more efficiently crosslinked by desmosine, as the crosslink density (i.e., desmosine content/ insoluble elastin matrix ratio) remained unaffected relative to controls. Collectively, these results suggest that TNF-α induces cytokine and LOX release by SMCs that may in turn modulate cellular matrix synthesis.
In agreement with our published observations 15 when elastogenic cues alone were provided to healthy adult SMCs, here too we observed that both in the absence and in the presence of TNF-α, collagen and tropoelastin production were increased, possibly a downstream intracellular signaling outcome34 of interactions of TGF-β1 and HA oligomers with their respective cell-surface receptors35. The enhanced elastin matrix yield in the presence of cues, regardless of the presence of TNF-α also, may be due to the significant increases, in both cases, in production of the elastin crosslinking enzyme LOX, relative to controls. The addition of cues also improved elastin fiber assembly over controls, probably by physical coacervation of tropoelastin precursors by HA oligomers to result in more efficient crosslinking by LOX36. Our finding that the cues do not enhance basal elastase activity of the SMCs or their release of MMPs, and that they suppress calcification of cell layers as induced by TGF-β1 alone, strengthen our claims to the elastogenic utility of TGF-β1 and HA oligomeric cues within aneurysms.
Relative to cultures which received TNF-α alone, production of cytokines/ chemokines/ interleukins was significantly attenuated, and TIMP-1 production suppressed within cultures that received both TNF-α and elastogenic cues. Surprisingly, TIMP-1 production decreased in cultures that received both TNF-α and elastogenic cues by 40 ± 6%, relative to that in controls. We hypothesize that such attenuation may be due to the contradictory downstream effects of simultaneous cellular interactions with TNF-α and TGF-β37. Regardless, the overall observations of suppressed cytokine/chemokine release in presence of both TNF-α and the elastogenic cues is highly significant to treatment of aneurysms. A very important determinant of vascular patency following injury/ disease initiation in vivo is the interaction between recruited leukocytes and ECs. Enhanced production and release of chemokines (e.g., IL-8) and acute inflammatory cytokines (e.g., TNF-α, IL-1) by cells in aneurysmal segments can locally attract T-cells to incite acute inflammation, while chemokines such as MCP-1, MIP-3α can summon chronic inflammatory cell types (e.g., monocytes, macrophages) to effect matrix degradation and remodeling38. Thus, under inflammatory conditions, an attenuated release of leukocyte-recruiting cytokines/ chemokines by elastogenic cues in the presence of TNF-α, is likely to provide a more stable matrix micro-environment.
Vascular calcification is a complex process that frequently involves differentiation of SMCs to an osteoblastic phenotype, which can be induced by both TNF-α and TGF-β25,31. In agreement with this, we observed calcific deposits in SMC cultures exposed to either of these factors alone. While TNF-α induces matrix calcification via increases in intracellular cAMP31, TGF-β appears to enhance bone protein production via increased expression of the elastin-laminin receptor (ELR)25, and by upregulating cGMP39. Here, we found elastogenic cues alone to inhibit calcification in adult SMC cultures. In a previous study20, we showed that HA oligomers suppress ELR activity, which might explain the absence of calcific deposits even when TGF-βl is supplemented. Another finding was that these elastogenic cues also attenuate TNF-α-induced calcification. Though the mechanisms underlying the anti-calcific effects of HA and TGF-βl on TNF-α–activated SMCs is beyond the scope of this study, we believe that our results attest to involvement of these factors via two different mechanisms of calcification that exhibit some downstream opposition to result in a net decrease in matrix calcification.
Upregulated collagen matrix production by elastogenic cues was attenuated down to control levels on addition of TNF-α. This might be due to our observed decrease in LOX production in the latter cultures, relative to cultures that received TNF-α or cues alone. On the other hand, tropoelastin and matrix elastin production continued to be enhanced by elastogenic cues, more so in the presence of TNF-α than its absence, indicating synergy in elastin synthesis signaling pathways. Though matrix elastin production was enhanced by TNF-α, both alone and together with cues, dramatic differences in the quality was observed. While elastin was deposited as amorphous, non-fibrillar clumps in TNF-α-only cultures, TNF-α and cues together promoted mature elastin fiber formation with fibrillar microfibrils bordering their periphery. We believe that the observed increase in elastic fiber quality in the latter case (Figure 6D) might be due to the presence of HA oligomers, which may physically coacervate amorphous elastin protein to facilitate fiber formation and crosslinking36.
5.Conclusions
We have shown that TNF-α activates cultured vascular SMCs in a manner that broadly simulates events within inflammatory vascular aneurysms in vivo. Though this in vitro activated cell culture model does not represent the complexity of the in vivo aneurysmal matrix microenvironment, it nevertheless provides evidence as to the efficacy of elastogenic cues in (a) upregulating elastin matrix production by activated vascular SMCs, (b) organizing elastin protein into fibers, and (c) simultaneously stabilizing this matrix by attenuating production of elastolytic enzymes. These results could form a basis for future elastin regenerative therapies for regression/ repair of vascular aneurysms.
Acknowledgements
The authors would like to acknowledge Dr. Samir Ibrahim for help with cytokine array, and Drs. Agneta Simionescu and Dan Simionescu for help with the gel zymography. Funding from the American Heart Association (SDG 0335085N) and the National Institutes of Health (C06RR018823, EB006078-01A1, HL092051-01A1) is also greatly appreciated.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ernst CB. Abdominal aortic aneurysms. N Engl J Med. 1993;328:1167–1172. doi: 10.1056/NEJM199304223281607. [DOI] [PubMed] [Google Scholar]
- 2.Wills A, Thompson MM, Crowther M, Sayers RD, Bell PR. Pathogenesis of abdominal aortic aneurysms-cellular and biochemical mechanisms. Eur J Vasc Endovasc Surg. 1996;12:391–400. doi: 10.1016/s1078-5884(96)80002-5. [DOI] [PubMed] [Google Scholar]
- 3.Mecham RP, Broekelmann TJ, Fliszar CJ, Shapiro SD, Welgus HG, Senior RM. Elastin degradation by matrix metalloproteinases. Cleavage site specificity and mechanisms of elastolysis. J Biol Chem. 1997;272:18071–18076. doi: 10.1074/jbc.272.29.18071. [DOI] [PubMed] [Google Scholar]
- 4.Hinek A, Wrenn DS, Mecham RP, Barondes SH. The elastin receptor: a galactoside-binding protein. Science. 1988;239:1539–1541. doi: 10.1126/science.2832941. [DOI] [PubMed] [Google Scholar]
- 5.Hance KA, Tataria M, Ziporin SJ, Lee JK, Thompson RW. Monocyte chemotactic activity in human abdominal aortic aneurysms: role of elastin degradation peptides and the 67-kD cell surface elastin receptor. J Vasc Surg. 2002;35:254–261. doi: 10.1067/mva.2002.120382. [DOI] [PubMed] [Google Scholar]
- 6.Basalyga DM, Simionescu DT, Xiong W, Baxter BT, Starcher BC, Vyavahare NR. Elastin degradation and calcification in an abdominal aorta injury model: role of matrix metalloproteinases. Circulation. 2004;110:3480–3487. doi: 10.1161/01.CIR.0000148367.08413.E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niederhoffer N, Lartaud-Idjouadiene I, Giummelly P, Duvivier C, Peslin R, Atkinson J. Calcification of medial elastic fibers and aortic elasticity. Hypertension. 1997;29:999–1006. doi: 10.1161/01.hyp.29.4.999. [DOI] [PubMed] [Google Scholar]
- 8.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 9.Dollery CM, Libby P. Atherosclerosis and proteinase activation. Cardiovasc Res. 2006;69:625–635. doi: 10.1016/j.cardiores.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Kahari VM, Chen YQ, Bashir M, Rosenbloom J, Uitto J. Tumor necrosis factor-a down-regulates human elastin gene expression. J Biol Chem. 1992;267:26134–26141. [PubMed] [Google Scholar]
- 11.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–262. [PubMed] [Google Scholar]
- 12.Hiraga S, Kaji T, Ueda Y, Zisaki F, Iwata I, Koizumi F, Okada Y, Katsuda S, Nakanishi I. Modulation of collagen synthesis by tumor necrosis factor alpha in cultured vascular smooth muscle cells. Life Sciences. 2000;66:235–244. doi: 10.1016/s0024-3205(99)00586-x. [DOI] [PubMed] [Google Scholar]
- 13.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, Esato K, Hamano K, Matsuzaki M. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med. 2005;11:1330–1338. doi: 10.1038/nm1335. [DOI] [PubMed] [Google Scholar]
- 14.Isenburg JC, Simionescu DT, Starcher BC, Vyavahare NR. Elastin stabilization for treatment of abdominal aortic aneurysms. Circulation. 2007;115:1729–1737. doi: 10.1161/CIRCULATIONAHA.106.672873. [DOI] [PubMed] [Google Scholar]
- 15.Kothapalli CR, Taylor PM, Smolenski RT, Yacoub MH, Ramamurthi A. TGF-β1 and hyaluronan oligomers synergistically enhance elastin matrix regeneration by vascular smooth muscle cells. Tissue Eng Part A. 2009;15(3):501–511. doi: 10.1089/ten.tea.2008.0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kothapalli CR, Ramamurthi A. Benefits of concurrent delivery of hyaluronan and IGF-1 cues to regeneration of crosslinked elastin matrices by adult rat vascular cells. J Tissue Eng Regen Med. 2008;2:106–116. doi: 10.1002/term.70. [DOI] [PubMed] [Google Scholar]
- 17.Wight TN. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol. 2002;14:617–623. doi: 10.1016/s0955-0674(02)00375-7. [DOI] [PubMed] [Google Scholar]
- 18.Davidson JM, Zoia O, Liu JM. Modulation of transforming growth factor-beta 1 stimulated elastin and collagen production and proliferation in porcine vascular smooth muscle cells and skin fibroblasts by basic fibroblast growth factor, transforming growth factor-alpha, and insulin-like growth factor-I. J Cell Physiol. 1993;155:149–156. doi: 10.1002/jcp.1041550119. [DOI] [PubMed] [Google Scholar]
- 19.Solis-Herruzo JA, Brenner DA, Chojkier M. Tumor necrosis factor alpha inhibits collagen gene transcription and collagen synthesis in cultured human fibroblasts. J Biol Chem. 1988;263:5841–5845. [PubMed] [Google Scholar]
- 20.Joddar B, Ramamurthi A. Elastogenic effects of exogenous hyaluronan oligosaccharides on vascular smooth muscle cells. Biomaterials. 2006;27:5698–5707. doi: 10.1016/j.biomaterials.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 21.Henness S, Johnson CK, Ge Q, Armour CL, Hughes JM, Ammit AJ. IL-17A augments TNF-α–induced IL-6 expression in airway smooth muscle by enhancing mRNA stability. J Allergy Clin Immunol. 2004;114:958–964. doi: 10.1016/j.jaci.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 22.Ammit AJ, Hoffman RK, Amrani Y, Lazaar AL, Hay DWP, Torphy TJ, Penn RB, Panettieri RA. TNFα-induced secretion of RANTES and IL-6 from human airway smooth muscle cells: modulation by cAMP. Am J Respir Cell Mol Biol. 2000;23:794–802. doi: 10.1165/ajrcmb.23.6.4184. [DOI] [PubMed] [Google Scholar]
- 23.Labarca C, Paigen K. A simple, rapid, and sensitive DNA assay. Anal Biochem. 1980;102:344–352. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 24.Joddar B, Ramamurthi A. Fragment size- and dose-specific effects of hyaluronan on matrix synthesis by vascular smooth muscle cells. Biomaterials. 2006;27:2994–3004. doi: 10.1016/j.biomaterials.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 25.Simionescu A, Philips K, Vyavahare N. Elastin-derived peptides and TGF-beta1 induce osteogenic responses in smooth muscle cells. Biochem Biophys Res Commun. 2005;334:524–532. doi: 10.1016/j.bbrc.2005.06.119. [DOI] [PubMed] [Google Scholar]
- 26.Mann DL, Young JB. Basic mechanisms in congestive heart failure: Recognizing the role of proinflammatory cytokines. Chest. 1994;105:897–904. doi: 10.1378/chest.105.3.897. [DOI] [PubMed] [Google Scholar]
- 27.Froon AHM, Greve J-WM, van der Linden CJ, Buurman WA. Increased concentrations of cytokines and adhesion molecules in patients after repair of abdominal aortic aneurysm. Eur J Surg. 1996;162:287–289. [PubMed] [Google Scholar]
- 28.Rabinovitch M. Elastase and cell matrix interactions in the pathobiology of vascular disease. Acta Paediatr Jpn. 1995;36:657–666. doi: 10.1111/j.1442-200x.1995.tb03400.x. [DOI] [PubMed] [Google Scholar]
- 29.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Rao PJ, Castresana MR, Newman WH. TNF-a induces proliferation or apoptosis in human saphenous vein smooth muscle cells depending on phenotype. Am J Physiol Heart Circ Physiol. 2005;288:H293–H301. doi: 10.1152/ajpheart.00165.2004. [DOI] [PubMed] [Google Scholar]
- 31.Tintut Y, Patel J, Parhami F, Demer LL. Tumor Necrosis Factor-a promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation. 2000;102:2636–2642. doi: 10.1161/01.cir.102.21.2636. [DOI] [PubMed] [Google Scholar]
- 32.Solis-Herruzo JA, Brenner DA, Chojkier M. Tumor necrosis factor alpha inhibits collagen gene transcription and collagen synthesis in cultured human fibroblasts. J Biol Chem. 1988;263:5841–5845. [PubMed] [Google Scholar]
- 33.Matsuoka M, Pham NT, Tsukamoto H. Differential effects of interleukin-1 alpha, tumor necrosis factor alpha, and transforming growth factor beta 1 on cell proliferation and collagen formation by cultured fat-storing cells. Liver. 1989;9:71–78. doi: 10.1111/j.1600-0676.1989.tb00382.x. [DOI] [PubMed] [Google Scholar]
- 34.Evanko SP, Angello JC, Wight TN. Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:1004–1013. doi: 10.1161/01.atv.19.4.1004. [DOI] [PubMed] [Google Scholar]
- 35.Marigo V, Voplin D, Vitale G, Bressan GM. Identification of a TGF-b responsive element in the human elastin promoter. Biochem Biophys Res Commun. 1994;99:1049–1056. doi: 10.1006/bbrc.1994.1335. [DOI] [PubMed] [Google Scholar]
- 36.Baccarani-Contri M, Vincenzi D, Cicchetti F, Mori G, Pasquali-Ronchetti I. Immunocytochemical localization of proteoglycans within normal elastin fibers. Eur J Cell Biol. 1990;53:305–312. [PubMed] [Google Scholar]
- 37.Thyberg J. Differentiated properties and proliferation of arterial smooth muscle cells in culture. Int Rev Cytol. 1996;169:183–265. doi: 10.1016/s0074-7696(08)61987-7. [DOI] [PubMed] [Google Scholar]
- 38.Pearce WH, Shively VP. Abdominal aortic aneurysm as a complex multifactorial disease: interactions of polymorphisms of inflammatory genes, features of autoimmunity, and current status of MMPs. Ann N Y Acad Sci. 2006;1085:117–132. doi: 10.1196/annals.1383.025. [DOI] [PubMed] [Google Scholar]
- 39.Kanno Y, Into T, Lowenstein CJ, Matsushita K. Nitric oxide regulates vascular calcification by interfering with TGF-signalling. Cardiovasc Res. 2008;77:221–230. doi: 10.1093/cvr/cvm049. [DOI] [PubMed] [Google Scholar]