

Abstract
Atovaquone is a substituted 2-hydroxy-naphthoquinone used therapeutically against Plasmodium falciparum (malaria) and Pneumocystis pathogens. It acts by inhibiting the cytochrome bc1 complex via interactions with the Rieske iron-sulfur protein and cytochrome b in the ubiquinol oxidation pocket. As the targeted pathogens have developed resistance to this drug there is an urgent need for new alternatives. To better understand the determinants of inhibitor binding in the ubiquinol oxidation pocket of the bc1 complex we synthesized a series of hydroxy-naphthoquinones bearing a methyl group on the benzene ring that is predicted to interact with the nuclear encoded Rieske iron-sulfur protein. We have also attempted to overcome the metabolic instability of a potent cytochrome bc1 complex inhibitor, a 2-hydroxy-naphthoquinone with a branched side-chain, by fluorinating the terminal methyl group. We have tested these new 2-hydroxy-naphthoquinones against yeast and bovine cytochrome bc1 complexes to model the interaction with pathogen and human enzymes and determine parameters that affect efficacy of binding of these inhibitors. We identified a hydroxy-naphthoquinone with a trifluoromethyl function that has potential for development as an anti-fungal and anti-parasitic therapeutic.
Keywords: Hydroxy-naphthoquinones, Cytochrome bc1 complex, Malaria, Plasmodium, Pneumocystis, Atovaquone
1. Introduction
Malarone is a combination of atovaquone (2-[trans-4-(4’-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-hydroxynaphthoquinone; atovaquone, Fig. 1A) and proguanil that is used therapeutically against Plasmodium falciparum (malaria) and Pneumocystis pathogens [1]. Atovaquone inhibits parasite and fungal respiration by specifically targeting the cytochrome bc1 complex. It is a competitive inhibitor of ubiquinol oxidation that acts at center P of the bc1 complex and locks the iron-sulfur protein in a conformation proximal to cytochrome b, preventing electron transfer to cytochrome c1 [2, 3]. The recent failure of atovaquone treatment has been linked to mutation of amino acids located in or near the atovaquone-binding site on cytochrome b [1, 4].
Figure 1.

(A) Atovaquone (ATV) (B) S-10576 (C) Novel 2-hydroxy-naphthoquinones: 2-OH-3-(2-Me-trifluorooctyl)-naphthoquinone (NQ1), 2-OH-3-(2-Me-octyl)-8-Me-naphthoquinone (NQ2), 2-OH-3-(2-Me-trifluorooctyl)-8-Me-naphthoquinone (NQ3).
Most of the reported mutations in cytochrome b destabilize important hydrophobic interactions between atovaquone and the binding domain on the protein or change the volume of the pocket [5, 6]. Since these mutations occur in the mitochondrially encoded gene for cytochrome b it would be advantageous to target the nuclear encoded subunit of the binding pocket, the Rieske Fe-S protein [7], since mutations arise spontaneously at a much lower frequency in nuclear compared to mitochondrial genes. Earlier modeling studies have indicated that van der Waals interactions between this subunit and the inhibitor are improved when the benzene ring of the 2-hydroxy-naphthoquinone has an alkyl substituent. In order to probe binding determinants located in the Rieske protein we synthesized a series of NQs featuring a methyl group on the aromatic ring.
S-10576 is a 2-hydroxy-naphthoquinone that is structurally similar to atovaquone (Fig. 1B) and that also targets the cytochrome bc1 complex [7]. However, studies designed to test the therapeutic potential of S-10576 in vivo showed that the terminal two carbons of the side chain are modified by oxidation and the resulting more water soluble compound is rapidly excreted [8, 9]. In the present study we sought to improve the metabolic stability of S-10576 by fluorinating the side chain, an increasingly common strategy for fine-tuning the pharmacological properties of drugs [10]. As atovaquone also inhibits the yeast bc1 complex, we have developed the yeast Saccharomyces cerevisiae as a model to probe binding determinants at center P of the bc1 complex.
We previously designed a model that described the molecular basis for atovaquone binding in the ubiquinol oxidation pocket of cytochrome b [2]. Using this model we have compared the binding affinities of atovaquone, S-10576 and the new hydroxy-naphthoquinones. Comparing the inhibitory activity of these hydroxyl-naphthoquinones and modeling their interactions with the binding site provides insight into the structural parameters that affect inhibitor binding.
2. Experimental procedures
2.1. Materials
Atovaquone was a gift from GlaxoSmithKline. The 2-hydroxy-naphthoquinones were dissolved in dimethyl sulfoxide at 2 mM concentration and stored at −20 °C.
2.2. Synthesis of naphthoquinones
The hydroxy-naphthoquinones were synthesized as previously reported [7] via radical alkylation of commercial 2-hydroxy-naphthoquinone with alkyl iodides in the presence of tributyltin hydride and 2,2′-azobisisobutyronitrile as radical initiator [11–17]. The synthesis of the 8-methyl and fluorinated derivatives, mass spectrometry, 1H and 19F NMR analysis are described in the Supplementary Data.
2.3. Purification of cytochrome bc1 complexes
Cytochrome bc1 complexes from yeast and bovine heart were isolated from mitochondrial membranes essentially as described previously [18, 19] with some modifications. Membranes were solubilized with dodecylmaltoside and diluted to a salt concentration of 100 mM NaCl with 50 mM Tris (pH 8.0), 0.02% (w/v) dodecylmaltoside before anion exchange chromatography. A salt gradient between 100 and 300 mM NaCl in the above detergent was applied. The bc1 enzyme eluted around 250 mM NaCl and pooled fractions were concentrated to ~0.1–0.3 mM cytochrome bc1 complex by ultrafiltration (Amicon Centricon, exclusion limit 100 kDa).
Quantification of the bc1 complex was performed as reported before [20] by using extinction coefficients of 17.5 mM−1 cm−1 at 553–539 for cytochrome c1 [21] and 25.6 mM−1 cm−1 at 563–579 for the average absorbance of the bH and bL hemes in cytochrome b [22].
2.4. Ubiquinol-cytochrome c reductase activity measurements
Cytochrome c reductase activity of purified cytochrome bc1 complexes were assayed in 50 mM potassium phosphate, pH 7.0, 250 mM sucrose, 0.2 mM EDTA, 1 mM NaN3, 2.5 mM KCN, 0.01% (w/v) dodecylmaltoside and 40 µM cytochrome c at 23 °C. The cytochrome bc1 complex was diluted to 2.5 nM in the assay buffer, inhibitor was added to the assay mixture and allowed to stir with the enzyme for 1 min, after which the reaction was started by adding 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinol, an analogue of ubiquinol. Reduction of cytochrome c was monitored in an Aminco DW-2a spectrophotometer at 550 versus 539 nm in dual wavelength mode. Data were collected and analyzed using an Online Instrument Systems Inc. computer interface and software. IC50 values were calculated as the concentration required to inhibit 50% of the bc1 complex activity compared to untreated enzyme from titration curves of inhibitor concentration versus activity.
2.5. Pre-steady-state reduction of Cytochrome b through Center N and Center P
Pre-steady-state reduction of cytochrome b was followed at 24 °C by stopped-flow rapid scanning spectroscopy using the OLIS rapid scanning monochromator as described previously [20]. The oxidant-induced reduction of cytochrome b through center P was monitored by rapid mixing of 3 µM cytochrome c with 1 µM bc1 complex pre-reduced by incubation for 1 min with 30 µM 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinol before adding 3 µM antimycin and 2 µM NQ1 or 2 µM atovaquone, and further incubating for 1 min. Cytochrome b reduction through center N was obtained by rapid-mixing of 30 µM 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinol with 1 µM oxidized bc1 complex in the presence of 3 µM stigmatellin and in the presence and absence of 2 µM NQ1. For each experiment, 12–19 data sets were averaged. The time course of the absorbance change at 562 and 578 nm was extracted using software from OLIS.
2.6. Molecular modeling
Molecular modeling was carried out on Silicon Graphics O2 and Octane workstations using the commercially available Insight II® software package (Acclerys Inc., San Diego). The starting structure was the energy-minimized atovaquone-liganded yeast cytochrome bc1 complex [2]. Briefly, a minimized conformation of atovaquone was docked into a stigmatellin liganded crystal structure [23] that was modified to include the rotation of Glu-272 and a water mediated hydrogen bond observed in the nHDBT liganded crystal structure [24]. For the structures shown here, the initial docking was achieved by overlay of the hydroxy-naphthoquinone ring with the one from the previously calculated atovaquone structure. The initial position of the linear aliphatic side-chain, prior to the molecular dynamics run, followed the one observed in the nHDBT crystal structure.
The NQs and cytochrome b residues within 4.0 Å were allowed to be flexible. A surrounding 9.5 Å shell of residues in both cytochrome b and the iron–sulfur protein was fixed, and the most distant residues were excluded from the calculation in order to obtain a manageable simulation speed. A 9.5 Å atom-based cut-off for non-bonding interactions was used during the calculations, with the dielectric constant set at 2.0. Eight simulated annealing runs were performed, each from 800 to 298 K, with five temperature steps and a simulation time of 5000 fs/step. The Nose temperature control method was used with a 0.5 fs/iteration time step. A custom macro was written to select the lowest energy structure from each dynamics run for continued modeling. Between each dynamics run, a minimization of 250 iterations was performed. After the final round of molecular dynamics, the lowest energy structure was minimized to a final convergence criterion of 0.001, using Cauchy's steepest descent method as implemented in the Discover 3® module within the Insight II® software, followed by conjugate gradient and Newton methods in succession. Of the eight minimized results obtained, the three lowest energy structures were chosen for binding energy calculation.
The binding energy calculation was adapted from a previous method [7] and uses a common subset that included the NQ and cytochrome b residues within 4.0 Å of the inhibitor. The reported value for each NQ is an average of the three calculated lowest energy structures and contains non-bonding interactions (van der Waals and electrostatic) as well as internal conformational energies of the ligand and adjacent pocket residues.
3. Results
3.1. Inhibition of yeast bc1 complexes by 2-hydroxy-naphthoquinones
The hydroxy-naphthoquinone S-10576 (Fig. 1B) is a potent inhibitor of the cytochrome bc1 complex [5]. However, although effective against avian malaria, S-10576 was ineffective in humans due to rapid metabolic degradation [25]. To overcome this limitation for the potential use of this compound as a therapeutic we synthesized two derivatives of S-10576 (Fig. 1C) bearing a metabolically stable trifluoromethyl group and six with a methyl moiety on the aromatic ring. The methyl substituent was introduced into the number 8 position on the aromatic ring on the basis of molecular modeling studies that indicated the possibility of non-covalent interactions between a methyl group in this location and the Rieske iron-sulfur protein as shown in Fig. 2. As can be seen from the figure, the lowest energy conformation of the bound ligand positions the 8-methyl group within van der Waals distance of the sulfur atom of Cys180 of the Rieske protein.
Figure 2.

Structure of 2-OH-3-(2-Me-octyl)-8-Me-naphthoquinone (NQ2) docked in the ubiquinol oxidation pocket at center P. Portions of the Rieske protein and cytochrome b are shown as gold and cyan ribbons, respectively. Van der Waals radii depicting the non-covalent interaction between the 8-methyl group of the naphthoquinone ring and Cys-180 (C180) of the Rieske protein are shown. The hydrogen bond between the 2-hydroxy group of NQ2 and His-181 of the Rieske protein and the water-mediated hydrogen bond between the 4-carbonyl oxygen of NQ2 and Glu-272 of cytochrome b are indicated by dashed white lines. Carbon atoms are shown in green, oxygens are red, hydrogens are white, nitrogens are blue, sulfurs are yellow, and iron are purple. The structure was created by substitution of NQ2 into the stigmatellin liganded crystal structure of the yeast bc1 complex [23] and energy minimization as described in the Experimental Procedures.
The titration curves in Fig. 3A show the inhibitory activity of the new agents against cytochrome bc1 complexes purified from yeast, which shares a high degree of sequence identity with parasitic enzymes [7]. Of the S-10576 analogs, the trifluorinated NQ1 was the most active, followed by the 8-methyl substituted NQ2. The S-10576 derivative with both a fluorinated chain and a substituted aromatic ring, NQ3, was the least active.
Figure 3.

Effect of 2-hydroxy-naphthoquinones on bc1 activity of (A) yeast and (B) bovine bc1 complexes. The cytochrome c reductase activity of purified enzyme was measured as described in Materials and Methods in the presence of increasing concentrations of inhibitor. The assays were performed in duplicate and the results are shown as averages. Activities are expressed as a percentage of the turnover number of each strain in the absence of inhibitor.
A series of naphthoquinones (NQ4-7) featuring a methyl group on carbon 8 of the aromatic ring and bearing straight alkyl chains of 8 to 11 carbons were compared with an analogous series that have an unsubstituted carbon 8. The methyl moiety decreased the potency of the naphthoquinones. Comparison of the IC50 values (Fig. 4) shows that the optimal chain length is 8 carbons for the NQ series without the 8-methyl substituent, which is consistent with previous results [4] and 9 carbons for the 8-methyl substituted derivatives. Addition of the 8-methyl group to the NQ with a 9 carbon side-chain increased the IC50 from 180 nM to 550 nM.
Figure 4.

Effect of chain length on inhibition of yeast cytochrome bc1 complexes by 2-hydroxy-naphthoquinones without and with a methyl group on carbon 8 of the naphthoquinone ring. The IC50 values for inhibition of the bc1 complex activity are shown with light gray bars for the 2-hydroxy-3-alkyl-naphthoquinones and dark gray bars for the 2-hydroxy-3-alkyl-8-methylnaphthoquinones.
3.2. Differential binding of 2-hydroxy-naphthoquinones to bovine and yeast bc1 complexes
We also assayed NQ1-3 against the bovine bc1 complex to evaluate the interaction of the inhibitors with the human enzyme, since the bovine and human cytochrome b sequences are 80% identical [7]. As shown in Fig. 3B, the trifluoromethyl substituted NQ1 was the most potent inhibitor of the bovine enzyme followed by atovaquone. The 8-methyl compounds NQ2 and NQ3 were more potent inhibitors of the bovine enzyme than the parent molecule S-10576. This contrasts with the relative efficacies of these three inhibitors with the yeast enzyme, in which case S-10576 was the most potent of these three inhibitors (Fig. 3A).
To compare the specificity of inhibitors with the bovine and yeast bc1 complexes, we defined the ‘Selectivity Coefficient’ of an inhibitor as the IC50 obtained with the bovine enzyme divided by the IC50 obtained with the yeast enzyme. The Selectivity Coefficients for atovaquone, S-10576, NQ1, NQ2 and NQ3 are shown in Fig. 5. S-10576 had the greatest selectivity, which was almost 10 fold greater than that of atovaquone. Although the selectivity of NQ1, NQ2, and NQ3 were less than that of S-10576, all of the derivatives displayed higher selectivity than atovaquone.
Figure 5.

Selectivity of 2-hydroxy-naphthoquinone inhibitors of the bovine and yeast cytochrome bc1 complexes. The bar graphs show the Selectivity Coefficients for inhibition of the yeast versus bovine bc1 complex by atovaquone (ATV), S-10576 and its derivatives 2-OH-3-(2-Me-trifluorooctyl)-naphthoquinone (NQ1), 2-OH-3-(2-Me-octyl)-8-Me- naphthoquinone (NQ2), and 2-OH-3-(2-Me-trifluorooctyl)-8-Me-naphthoquinone (NQ3).
3.3. Molecular modeling of 2-hydroxy-naphthoquinones docked in the yeast cytochrome bc1 complex
Although a crystal structure is not available, atovaquone binding has been modeled using the coordinates of the stigmatellin-liganded yeast enzyme [2]. In a previous study a good correlation was found between relative binding energies of NQs calculated using this model and their inhibitory activity against cytochrome bc1 complexes [7]. In the current study the energy required for binding of the NQs was calculated for each of the modeled structures. The calculated binding energies were then compared with the experimentally measured IC50 values and are shown in Fig. 6. Overall the difference in calculated binding energies of compounds NQ1-3 was small. The methyl-substituted NQ3 had a slightly lower calculated binding energy than S-10576. However, the most potent inhibitor NQ1 was predicted to have a higher binding energy than the parent compound. In particular the angle and torsion components of the binding energy are higher for NQ1 than S-10576.
Figure 6.

Experimentally measured IC50 values compared to calculated binding energies of S-10576, 2-OH-3-(2-Me-trifluorooctyl)-naphthoquinone (NQ1), and 2-OH-3-(2-Metrifluorooctyl)-8-Me-naphthoquinone (NQ3) in yeast cytochrome bc1 complex. The IC50 values for the inhibition of the yeast bc1 complex activity are shown in light gray bars. The dark gray bars show the calculated binding energies.
3.4 Specific binding of 2-hydroxy-naphthoquinone inhibitors to center P of the yeast cytochrome bc1 complex
In order to determine whether NQs bind to center P or N, we monitored cytochrome b reduction in the presence of a center N inhibitor antimycin and a center P inhibitor stigmatellin. As shown in Fig. 7A, NQ1 completely inhibited the additional oxidant-induced reduction of cytochrome b through center P in bc1 complex that had been incubated with quinol to reduce the Rieske protein (resulting also in partial cytochrome b reduction) before addition of antimycin and NQs. In contrast, NQ1 had no effect on cytochrome b reduction through center N in the presence of stigmatellin (Fig. 7B). If NQ1 or atovaquone were preincubated with the fully oxidized enzyme before reduction with quinol, the first turnover at center P was not inhibited (results not shown). This effect of NQs is due to the fact that these inhibitors, like stigmatellin, bind more tightly to center P when the Rieske protein is reduced, and thus differ from inhibitors like myxothiazol that do not interact with the Rieske protein and are therefore insensitive to the redox state of the FeS cluster (See Ref. 26 and references therein). The higher potency of NQ1 relative to atovaquone in inhibiting center P under pre-steady state conditions (Fig. 7A) agrees with the results obtained under multi-turnover conditions (see Fig. 3).
Figure 7.
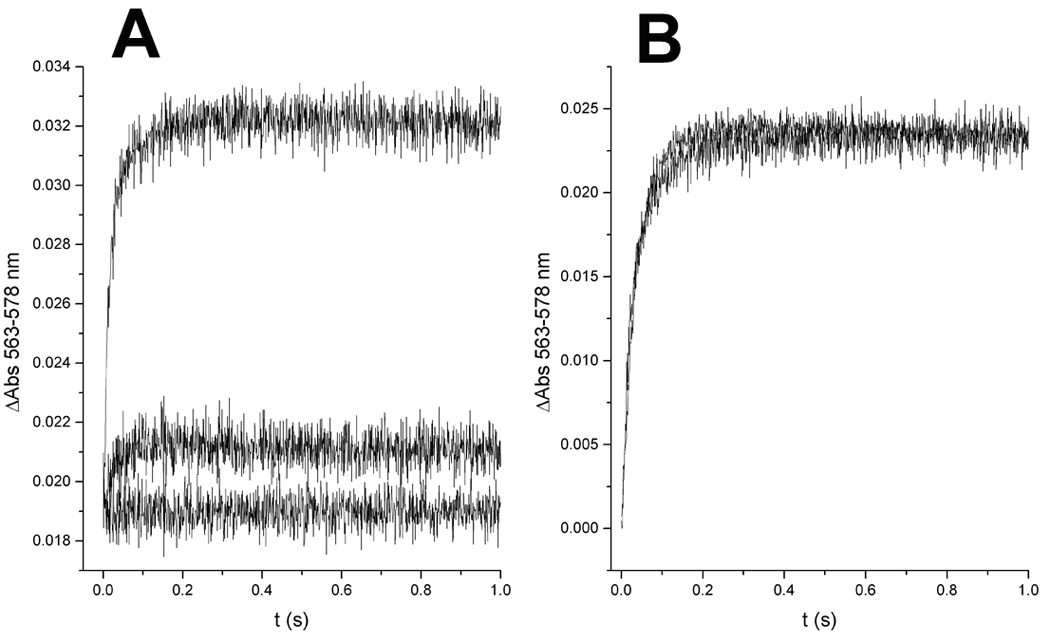
Specific binding of 2-hydroxy-naphthoquinone inhibitors to center P of the yeast cytochrome bc1 complex. Upon rapid mixing of cytochrome c with bc1 complex that had been partially reduced by incubation with quinol before the addition of antimycin and NQs (yielding the initial absorbance shown at t = 0), the additional oxidant-induced reduction of cytochrome b through center P (A) was greatly inhibited by atovaquone (middle trace), and completely by 2-OH-3-(2-Me-trifluorooctyl)-naphthoquinone (NQ1, lower trace). In contrast, cytochrome b reduction through center N (B) obtained by rapid-mixing of 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinol with oxidized bc1 complex in the presence of stigmatellin yielded identical, superimposed kinetic traces in the presence and absence of 2-OH-3-(2-Me-trifluorooctyl)-naphthoquinone (NQ1).
4. Discussion
Atovaquone has broad-spectrum activity against species of Plasmodium, Pneumocystis, Toxoplasma gondii [6] and Babesia spp. [25]. In the current study we synthesized and screened novel NQs that featured straight and branched alkyl chains on the quinoid carbon-carbon double bond and a methyl group on carbon 8 of the aromatic ring. Modeling studies had indicated that a methyl group on carbon 8 of the aromatic ring of NQ may have favorable van der Waals interactions with the Rieske Fe-S protein (Fig. 2). However the 8-methyl derivatives with unbranched alkyl chains are weak cytochrome bc1 complex inhibitors and were not studied further.
Like the hydroxy-naphthoquinone derivatives, stigmatellin binds center P of the bc1 complex via polar and nonpolar interactions [27]. The chromone head group is structurally similar to a naphthoquinone and forms hydrogen bonds with His-161 of the Rieske protein and Glu-272 of cytochrome b. However while the aromatic ring of stigmatellin bears two methoxy substituents, the introduction of a methyl group into the benzene ring of NQ caused a significant loss in inhibitory activity, which indicates the high specificity of the binding pocket for these classes of inhibitors.
The crystal structure of stigmatellin bound to center P identified close (≤4.0 Å) contacts of the protein complex with the ligand [27]. A comparison of the non-bonding interactions at the binding site of stigmatellin and NQ1-3 revealed that there are the same 8 residues close to each inhibitor. These non-bonding contacts are distributed along the alkyl side chains, involving mostly hydrophobic residues of cytochrome b transmembrane helices C and F, and the cd1 and ef helices. The similarity of the interaction of stigmatellin and NQs with these residues highlights the importance of hydrophobic interactions for docking of ligands into the ubiquinol oxidation pocket of the cytochrome bc1 complex.
Previously we found that there was good agreement between the experimentally measure changes in IC50 values for inhibition by NQs and theoretically calculated changes in binding energies [7, 24]. However in the present study the changes in binding energies were small and did not correlate with the IC50 values. This may reflect the fact that IC50 is a free energy (ΔG) term, which includes an entropic parameter, whereas the calculated binding energy is solely enthalphic (ΔH).
Interestingly, Winter et al. discovered that replacement of the hydroxy group in NQ by a trifluoropentyloxy side chain improved the anti-malarial potency of the parent compound by over 50 fold [28]. In addition, haloacridones bearing a terminal CF3 group in the alkoxy substituent exhibited a profound enhancement in anti-malarial action against D6 and Dd2 P. falciparum strains. This suggests that CF3 groups may improve the interactions between the cytochrome bc1 complex and both acridone and NQ inhibitors.
For high species selectivity, the optimal chain length of the NQs consisted of 8 carbons. Similarly, Baramee et al. reported that NQs bearing alkyl chains of 6 to 8 carbons display significant anti-parasitic activity [29]. Fluorination of S-10576 presumably impedes hepatic P450-mediated oxidation, however it decreased the species selectivity of the NQ. This may be due to the larger volume of the trifluoromethyl function, estimated as being slightly larger than an isopropyl moiety [30]. Although the species selectivity of the fluorinated NQ1 was lower than that of the parent compound S-10576, it was still greater than that of atovaquone. Moreover, the IC50 of the trifluorinated NQ1 was significantly lower than that of atovaquone. This suggests that NQ1 is a novel inhibitor of the cytochrome bc1 complex with properties that indicate potential for development as an anti-parasitic and anti-fungal lead compound. Its specificity may be improved by substituting a less bulky side chain such as a monofluorinated 8-carbon chain or a 6 to 7-carbon chain with a terminal trifluoromethyl moiety.
Supplementary Material
Acknowledgment
This research was supported by NIH grant GM-20379.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vaidya AB, Mather MW. Atovaquone resistance in malaria parasites. Drug Resist. Update. 2000;3:283–287. doi: 10.1054/drup.2000.0157. [DOI] [PubMed] [Google Scholar]
- 2.Kessl JJ, Lange BB, Merbitz-Zahradnick T, Zwicker K, Hill P, Meunier B, Meshnick S, Trumpower BL. Molecular basis for atovaquone binding to the cytochrome bc1 complex. J. Biol. Chem. 2003;278:31312–31318. doi: 10.1074/jbc.M304042200. [DOI] [PubMed] [Google Scholar]
- 3.Mather MW, Darrouzet E, Valkova-Valchanova M, Cooley JW, McIntosh MT, Daldal F, Vaidya AB. Uncovering the molecular mode of action of the antimalarial drug atovaquone using a bacterial system. J. Biol. Chem. 2005;280:27458–27465. doi: 10.1074/jbc.M502319200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srivastava IK, Morrisey JM, Darrouzet E, Daldal F, Vaidya AB. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Microbiol. 1999;33:704–711. doi: 10.1046/j.1365-2958.1999.01515.x. [DOI] [PubMed] [Google Scholar]
- 5.Kessl JJ, Ha KH, Merritt AK, Lange BB, Hill P, Meunier B, Meshnick SR, Trumpower BL. Cytochrome b mutations that modify the ubiquinol-binding pocket of the cytochrome bc1 complex and confer anti-malarial drug resistance in Saccharomyces cerevisiae. J. Biol. Chem. 2005;280:17142–17148. doi: 10.1074/jbc.M500388200. [DOI] [PubMed] [Google Scholar]
- 6.Fisher N, Meunier B. Molecular basis of resistance to cytochrome bc1 complex inhibitors. FEMS Yeast Res. 2008;8:183–192. doi: 10.1111/j.1567-1364.2007.00328.x. [DOI] [PubMed] [Google Scholar]
- 7.Kessl JJ, Moskalev NK, Gribble GW, Nasr M, Meshnick SR, Trumpower BL. Parameters determining the relative efficacy of hydroxy-naphthoquinone inhibitors of the cytochrome bc1 complex. Biochim. Biophys. Acta. 2007;1767:319–326. doi: 10.1016/j.bbabio.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fieser LF, Chang FC, Dauben WG, Heidelberger C, Heymann H, Seligman AM. Naphthoquinone antimalarials. Metabolic oxidation products. J. Pharmacol. Exp. Ther. 1948;94:85–96. [PubMed] [Google Scholar]
- 9.Hudson AT, Dickins M, Ginger CD, Gutteridge WE, Holdich T, Hutchinson DBA, Pudney M, Randall AW, Latter VS. 566C80: a potent broad spectrum anti-infective agent with activity against malaria and opportunistic infections in AIDS patients. Drugs Exptl. Clin. Res. 1991;17:427–435. [PubMed] [Google Scholar]
- 10.Muller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: Looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 11.Moody CJ, Norton CL. Synthesis of 1, 2- fused indoles by radical cyclisation. J. Chem. Soc., Perkin Trans. 1. 1997;17:2639–2643. [Google Scholar]
- 12.Mori K. Revision of the absolute configuration of A-factor. Tetrahedron. 1983;39:3107–3109. [Google Scholar]
- 13.Shirai S, Seki M, Mori K. Pheromone Synthesis, CXCIX. Eur. J. Org. Chem. 1999:3139–3145. [Google Scholar]
- 14.Gallardo H, Merlo AA. Ethyl lactate as a convenient precursor for synthesis of chiral liquid crystals. Synthetic Comm. 1993;23:2159–2169. [Google Scholar]
- 15.Fouquet C, Schlosser M. Improved carbon-carbon linking by controlled copper catalysis. Angew. Chem. 1974;13:83–84. [Google Scholar]
- 16.Parker KA, Spero DM, Koziski KA. Assignment of regiochemistry to substituted naphthoquinones by chemical and spectroscopic method. J. Org. Chem. 1986;52:183–188. [Google Scholar]
- 17.Guzikowski AP, Cai SX, Espitia SA, Hawkinson JE, Huettner JE, Nogales D, Tran M, Woodward RM, Weber E, Keana JFW. Analogs of 3-hydroxy-1H-1-benzazepine-2,5-dione: Structure-activity relationship at N-methyl-d-aspartate receptor glycine sites. J. Med. Chem. 1996;39:4643–4653. doi: 10.1021/jm960479z. [DOI] [PubMed] [Google Scholar]
- 18.Ljungdahl PO, Pennoyer JD, Robertson D, Trumpower BL. Purification of highly active cytochrome bc1 complexes from phylogenetically diverse species by a single chromatographic procedure. Biochim. Biophys. Acta. 1987;891:227–242. doi: 10.1016/0005-2728(87)90218-0. [DOI] [PubMed] [Google Scholar]
- 19.Snyder CH, Trumpower BL. Ubiquinone at center N is responsible for triphasic reduction of cytochrome b in the cytochrome bc1 complex. J. Biol. Chem. 1999;274:31209–31216. doi: 10.1074/jbc.274.44.31209. [DOI] [PubMed] [Google Scholar]
- 20.Snyder C, Trumpower BL. Mechanism of ubiquinol oxidation by the cytochrome bc1 complex: Pre-steady-state kinetics of cytochrome bc1 complexes containing the site-directed mutants of the Rieske iron-sulfur protein. Biochim. Biophys. Acta. 1998;1365:125–134. doi: 10.1016/s0005-2728(98)00052-8. [DOI] [PubMed] [Google Scholar]
- 21.Yu CA, Yu L, King TE. Preparation and properties of cardiac cytochrome c1. J. Biol. Chem. 1972;247:1012–1019. [PubMed] [Google Scholar]
- 22.Berden JA, Slater EC. The reaction of antimycin with a cytochrome b preparation active in reconstitution of the respiratory chain. Biochim. Biophys. Acta. 1970;216:237–249. doi: 10.1016/0005-2728(70)90215-x. [DOI] [PubMed] [Google Scholar]
- 23.Hunte C, Koepke J, Lange C, Rossmanith T, Mitchel H. Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure. 2000;8:669–684. doi: 10.1016/s0969-2126(00)00152-0. [DOI] [PubMed] [Google Scholar]
- 24.Palsdottir H, Gomez Lojero C, Trumpower BL, Hunte C. Structure of the yeast cytochrome bc1 complex with a hydroxyquinone anion Qo site inhibitor bound. J. Biol. Chem. 2003;278:31303–31311. doi: 10.1074/jbc.M302195200. [DOI] [PubMed] [Google Scholar]
- 25.Krause PJ, Lepore T, Sikand VK, Gadbaw J, Jr, Burke G, Telford SR, 3rd, Brassard P, Pearl D, Azlanzadeh J, Christianson D, McGrath D, Spielman A. Atovaquone and azithromycin for the treatment of babesiosis. N. Engl. J. Med. 2000;343:1454–1458. doi: 10.1056/NEJM200011163432004. [DOI] [PubMed] [Google Scholar]
- 26.Covian R, Pardo JP, Moreno-Sanchez R. Tight binding of inhibitors to bovine bc1 complex is independent of the Rieske protein redox state. Consequences for semiquinone stabilization in the quinol oxidation site. J. Biol. Chem. 2002;277:48449–48455. doi: 10.1074/jbc.M208060200. [DOI] [PubMed] [Google Scholar]
- 27.Lancaster CRD, Hunte C, Kelley J, III, Trumpower BL, Ditchfield R. A comparison of stigmatellin conformations, free and bound to the photosynthetic reaction center and the cytochrome bc1 complex. J. Mol. Biol. 2007;368:197–208. doi: 10.1016/j.jmb.2007.02.013. [DOI] [PubMed] [Google Scholar]
- 28.Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Bagby GC, Rathbun RK, Levin JI, Hinrichs D, Riscoe MK. Evaluation and lead optimization of anti-malarial acridones. Exp. Parasitol. 2006;114:47–56. doi: 10.1016/j.exppara.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 29.Baramee A, Coppin A, Mortuaire M, Pelinski L, Tomavo S, Brocard J. Synthesis and in vitro activities of ferrocenic aminohydroxynaphthoquinones against Toxoplasma gondii and Plasmodium falciparum. Bioorg. Med. Chem. 2006;14:1294–1302. doi: 10.1016/j.bmc.2005.09.054. [DOI] [PubMed] [Google Scholar]
- 30.Hagmann WK. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.