

Abstract
Purpose
Pre-existing antiviral antibodies in cancer patients can quickly neutralize oncolytic measles virus (MV) and decrease its anti-tumor potency. In contrast to `naked' viruses, cell-associated viruses are protected from antibody neutralization. Hence, we hypothesized that measles virotherapy of ovarian cancer in measles immune mice might be superior if MV infected mesenchymal stem cell (MSC) carriers are used.
Experimental Design
Antimeasles antibodies titers in ovarian cancer patients were determined. The protection of MV by MSC from antimeasles antibodies, the in vivo biodistribution profiles and tumor infiltration capability of MSC were determined. Measles naïve or immune tumor-bearing mice were treated with naked virus or MSC-associated virus and mice survivals were compared.
Results
MSC transferred MV infection to target cells via cell-to-cell heterofusion and induced syncytia formation in the presence of high titers of antimeasles antibody; at levels which completely inactivated naked virus. Athymic mice bearing intraperitoneal human SKOV3ip.1 ovarian tumor xenografts passively immunized with measles immune human serum were treated with saline, naked MV or MV infected MSC. Bioluminescent and fluorescent imaging data indicated that intraperitoneally administered MSC localized to peritoneal tumors, infiltrated into the tumor parenchyma and transferred virus infection to tumors in measles naïve and passively immunized mice. Survival of the measles immune mice was significantly enhanced by treatment with MV-infected MSC. In contrast, survivals of passively immunized mice were not prolonged by treatment with naked virus or uninfected MSC.
Conclusions
MSC should be used as carriers of MV for intraperitoneal virotherapy in measles-immune ovarian cancer patients.
Keywords: Mesenchymal stem cells, oncolytic measles virus, ovarian cancer, virus neutralizing antibodies
INTRODUCTION
Epithelial ovarian cancer is the most lethal of all gynecologic malignancies, killing more than 15,000 women in the United States each year (1). Due to the lack of effective screening modalities, the majority of patients present with advanced Stage III disease at the time of diagnosis where the cancer still remains confined within the peritoneal cavity (2). Primary treatment is maximal debulking surgery followed by chemotherapy using carboplatin and paclitaxel or carboplatin alone (3). More than 75% of patients will eventually relapse, and salvage therapies for recurrent disease are not curative. Various novel biological therapeutics are being developed for the treatment of ovarian cancer; these include immunotherapy using tumor vaccines, monoclonal antibody therapy, gene transfer of cytotoxic and anti-angiogenic transgenes and virotherapy using replication-competent tumor selective viruses (4–8).
We have been developing the Edmonston vaccine lineage of measles virus as a tumor selective oncolytic agent for cancer therapy (9). Oncolytic measles virus uses the hemagglutinin (H) envelope glycoprotein to infect cancer cells via the cellular CD46 receptor and the fusion (F) envelope glycoprotein to trigger fusion of the viral-cell membranes for virus entry (10). Expression of these fusogenic H and F proteins on surfaces of virus infected cells results in massive intercellular fusion with uninfected neighboring CD46 positive cells to generate the characteristic MV-induced cytopathic effects (CPE) of syncytia formation (11). We recently demonstrated that overexpression of CD46 on cell surfaces results in the preferential killing of tumor cells (12, 13). Indeed, human ovarian cancer cells overexpress CD46 (14) and are highly susceptible to measles induced CPE and cell killing (10, 12).
A phase I dose escalation clinical trial testing the safety of intraperitoneal administration of 103 to 109 TCID50 of MV-CEA, a recombinant MV genetically modified to express a soluble marker peptide to enable noninvasive monitoring of the profiles of viral gene expression, was recently completed (10, 15). The virus was well tolerated, and no dose-limiting toxicity was observed. There were, however, early indications of biologic activity, especially in patients treated with higher doses of MV-CEA (16). As a possible follow-on trial using measles virus in ovarian cancer patients, we are exploring various strategies to improve delivery of measles virus to the tumor site, especially in patients with pre-existing antimeasles antibodies. We and others have reported that cells can potentially be used as carriers to deliver oncolytic viruses to tumor xenografts in murine models, although only one study has evaluated the therapeutic activity of cell carriers given (intratumorally) to mice with preexisting antiviral antibodies (17–22). Potentially, any cell can be used as a virus carrier; for example, irradiated cell lines (20, 23), cytokine induced killer cells (18), activated T cells (21), MSC (24), and CD14+ monocyte derived dendritic cells (25). Mesenchymal stem cells are attractive as cell carriers because, in addition to their reported ability to home to tumors (26), adipose tissue derived MSC are readily obtained from adipose tissues that are available as surgical wastes from gastric bypass or from fat biopsies. MSC can be expanded to large numbers in cellular therapy laboratories of medical centers under Good Laboratory Practice conditions, and clinical experience with infusion of MSC into humans is available (27). Here, we have chosen to test adipose tissue derived MSC as a measles virus carrier in mice bearing orthotopic human ovarian tumor xenografts, focusing on their potential to overcome antiviral immunity in mice passively immunized with antimeasles antisera.
MATERIALS AND METHODS
Viruses, Lentivectors and Cell Lines
Recombinant Edmonston strain measles virus expressing firefly luciferase (FLuc), red fluorescent protein (RFP), green fluorescent protein (GFP), and sodium iodide symporter (NIS) were generated as described previously (28, 29). Viral titers were determined by TCID50 titration on Vero cells. To generate the lentivectors, 293T cells were cotransfected with gag-pol expression plasmid pCMV8.91, VSV.G envelope expression plasmid pMD-G, and vector plasmid encoding Gaussia luciferase with an IRES linking cyan fluorescent protein (a kind gift from Dr Bakhos A Tannous) (30, 31). Vector supernatant was collected 48 hours later, filtered (0.45 um) and frozen at −80°C. Human ovarian cancer cells, SKOV3ip.1, stably expressing GLuc were maintained in alpha MEM (Lonza, Allendale, NJ) that was supplemented with 20% fetal bovine serum (FBS, Gibco), 100 U mL-1 penicillin-streptomycin, and 2 mM L-glutamine. Human ovarian cancer cell lines A2780 and OVCAR5 (kind gift from Dr Viji Shridhar) were maintained in 10% FBS-DMEM. Adipose derived MSC were maintained in Advanced MEM (Invitrogen, Carlsbad, CA) that was supplemented with 5% platelet lysate, 2 U mL-1 heparin, 100 U mL-1 penicillin-streptomycin, and 2 mM L-glutamine. All media and growth supplements were purchased from Invitrogen.
Generation and characterization of MSC
Adipose tissue derived MSC were generated as described. Briefly, fat tissue was mechanically dissected and disaggregated, cells were pelleted by centrifugation and left to adhere to plastic wells in media supplemented with a 5% solution of lysed human platelets. Typical cell recovery was 1.3 ± 0.68 × 106 cells/gram of tissue (N=7). These cells were phenotyped for typical MSC cell markers, and were negative for Class II, CD14, CD45, CD106, and positive for CD44, CD49d, CD71, CD73, CD90, CD105, CD166 and Class I (A. Dietz, unpublished data). Phenotype of cells remained stable for more than 7 passages and after cryopreservation. Cells were frozen in aliquots in liquid nitrogen and stored until use. Only low passage cells (P5–7) were used for all the experiments of this study.
Virus Infection Assays
MSC were plated overnight in 6 or 12 well plates and MV-RFP at various multiplicities of infection (MOI, ratio of virus to cells) for 2h at 37°C, after which the virus inoculum was removed, and the cells were cultured for 48h in the presence or absence of a fusion inhibitory peptide (FIP, Z-D-Phe-Phe-Gly-OH, Bachem, Torrance, CA). Cells were trypsinized and the percentage of red fluorescent MV-RFP infected cells was determined by flow cytometry. Numbers of viable cells were determined by trypan blue exclusion assay at various time points post infection.
Measles Immune Human Serum
Pooled human AB sera were purchased from Valley Biomedical Inc. (Winchester, VA). The anti-measles antibody (IgG) titers of the sera were determined by the Mayo Clinic Serology Clinical Laboratory (Diamedix enzyme immunoassay, FL). Anti-measles antibody titers are reported as EU/ml. The corresponding full plaque reduction neutralization titers of the sera were also determined (see below).
Virus Neutralization Assays
For the virus neutralization assays, ascites or sera were heat inactivated and diluted in Opti-MEM (2-fold serial dilutions) after which 100–250 TCID50/50 μl MV-GFP was added. The mixture was incubated at 37°C for 30 min after which Vero cells (7×103 cells/well/50 μl) were added and the mixture was plated in 96 well plates. The culture was maintained at 37°C for 2 days. Each dilution was performed in triplicates, and the presence or absence of CPE in the wells was noted. The full plaque reduction neutralization titer is the reciprocal of the highest dilution at which no CPE was noted in any of the replicate wells and before the dilution where CPE was observed in one or more wells.
Immunohistochemical staining for MV-N and CD68 proteins
Tumors were sectioned into halves and frozen immediately in optimal cutting temperature medium. Cryosections (5μm thick) were acetone fixed for 10 min. The cryosections were permeabilized using 0.01% Triton X-100 and 5% horse serum for 15 min and incubated with biotinylated anti-measles virus nucleocapsid (MV-N) protein antibody (Mab 8906, Chemicon, Billerica, MA) for 1 hour. The slides were developed using Vector ABC and alkaline phosphatase substrate kits (Vector Labs, Burlingame, CA) and counterstained with Vector Nuclear Fast Red. For CD68 staining, acetone fixed cryosections were incubated with biotinylated rat antimouse CD68 (Serotec, Raleigh, NC, MCA1957B) for 1 hour. The slides were developed using Vector Elite ABC and DAB substrate kits (Vector Labs, Burlingame, CA) and counterstained with Vector Hematoxylin QS.
In Vivo Experiments
All procedures involving animals were approved by and performed according to guidelines of the Institutional Animal Care and Use Committee of Mayo Foundation. To determine the half-life of human anti-measles antibody post passive transfer into mice, athymic mice were given an IP injection of PBS diluted measles immune human serum. At 3h (day 0), day 1, 2, 3, 4, 7, and 8 post serum infusion, mice were euthanized and bled, and the level of anti-MV neutralizing antibody titer in murine serum was determined by full plaque reduction neutralization assay on Vero cells. The decrease in anti-MV antibody titer was plotted over time.
To establish various orthotopic models of human ovarian cancer, female athymic mice (5–6 weeks of age; Taconic Laboratory, Germantown, NY) were injected IP with 2 × 106 SKOV3ip.1 cells stably expressing Gaussia luciferase-cyan fluorescent protein (SKOV3ip.1-Gluc-CFP) or Fluc (SKOV3ip.1-FLuc), OVCAR5 or A2780 cells. Another cohort of tumor-bearing mice also received CellTracker Red CMTPX dye labeled (red fluorescent) MV infected MSC. Mice were euthanized, and tumors were harvested and examined using a fluorescence microscope.
For the therapy experiments, MSC were pre-infected with MV-NIS (MOI 4.0) for 2h, washed in PBS and exposed to 50 EU of measles immune serum for 30 min, at 37°C, to ensure complete neutralization of any bound virus. An equivalent amount of cell-free MV-NIS viral stock was also exposed to 50 EU of measles immune serum for 30 min, at 37°C. At the end of the incubation period, cells or MV-NIS in measles immune serum were injected IP into mice that had received either 50 EU of measles immune serum (measles immune) or saline (measles naïve) 3 h earlier. Each mouse received 105 TCID50 MV-NIS or 105 infected MSC, diluted in saline or measles immune serum. Mice were imaged to determine tumor burden (FLuc or GLuc). For imaging, mice were given either IP injections of 150 mg/kg D-luciferin (Xenogen) 15 min before imaging for Fluc activity or IP injections of 20 mg/kg colenterazine substrate (Nanolight Technology, Prolume Ltd., Pinetop, AZ) 10 min before imaging for GLuc activity. Whole abdominal bioluminescence signals reflecting tumor burden (GLuc) or viral gene expression (FLuc) were quantitated using the Living Image 2.50 software (Xenogen) according to manufacturer's protocol. Mice were euthanized when they developed ascites, and had lost more than 20% of body weight or could not reach food or water. All remaining mice were euthanized at day 86. Kaplan-Meier survival curves were compared by logrank test in GraphPad Prism (GraphPad Software, San Diego, CA). P values of < 0.05 were considered significant.
RESULTS
Ovarian cancer patients have preexisting antimeasles antibodies
Ascites fluid wastes were collected from ovarian cancer patients who came to Mayo Clinic for drainage of their ascites. All clinical samples were obtained with IRB approval and patient consent. The cellular fraction was removed and the anti-MV IgG titers in ascites fluids were determined. As shown in table 1, all ascites fluids tested positive for anti-measles virus antibodies (anti-MV IgG titers more than 20 EU/ml). The average anti-MV IgG titer was 95 ± 39 EU/ml (mean ± S.D., n=14). The corresponding virus plaque reduction neutralization (PRN) titer of the ascites fluids ranged from 2 to 128 (Table 1).
Table 1.
Anti-measles IgG levels (EU/ml) and corresponding virus plaque reduction neutralization (PRN) titers in ascites fluids of ovarian cancer patients (n=14). A titer of >20 EU/ml is considered measles seropositive.
| Sample ID | Anti-MV Ab titer | PRN titer |
|---|---|---|
| OvCa12 | 26.10 | 64 |
| OvCa9 | 38.40 | 8 |
| OvCa8 | 54.30 | 2 |
| OvCa2 | 75.70 | n.d. |
| OvCa4 | 85.70 | n.d. |
| OvCa5 | 86.00 | n.d. |
| OvCa1 | 94.50 | n.d. |
| OvCa10 | 102.80 | 32 |
| OvCa3 | 108.00 | n.d. |
| OvCa7 | 108.00 | n.d. |
| OvCa6 | 110.00 | n.d. |
| OvCa14 | 145.80 | 128 |
| OvCa11 | 148.50 | 64 |
| OvCa13 | 154.40 | 64 |
N.d.= not done.
Infection of MSC by measles virus
To determine the susceptibility of MSC to MV infection, MSC were infected with MV-RFP at MOI (0.01, 0.1, 1.0 or 4.0). As shown in figure 1A, there was a corresponding increase in the numbers of infected cells with increase in MOI. To enable quantitation of the percentage of infected cells by flow cytometry, MSC were maintained in media containing a fusion inhibitory peptide (+FIP) that prevents intercellular fusion (syncytia formation). At MOI of 1.0 and 4.0, 20% and 60% of cells were expressing RFP by 48h post infection, respectively (Fig. 1B).
Figure 1.

Adipose tissue derived mesenchymal stem cells (MSC) are susceptible to infection by measles virus. (A) Photographs MV-RFP infected MSC taken at 48h post infection using different multiplicities of infection (MOI). Cells were maintained in the absence (−FIP) or presence (+FIP) of a fusion inhibitory peptide, FIP. (B) Quantitation of MV-RFP infection in MSC by analyzing for numbers of RFP positive cells (+FIP) by flow cytometry at 48h post infection. (C) Viability of infected MSC over time. (D) Progeny produced by virus infected MSC or Vero cells at 48h post infection at MOI 0.2 or 1.0. Amount of cell-associated virus or released virus in the supernatant were titrated on Vero cells. Error bars represent S.D. (n=3 replicates).
Viability of MSC post infection by MV-GFP (MOI 1.0) was determined by trypan blue exclusion assay (Fig. 1C). In the presence of FIP, there was no significant difference in numbers of viable cells in the infected culture over time. The numbers of viable cells decreased by 96h if the infected MSC were allowed to fuse with neighboring cells in the absence of FIP. The amount of viral progeny produced by infected MSC (cell-associated virus) or conditioned media (released virus) was determined by TCID50 titrations. Infected MSC were able to propagate the virus, but to a smaller extent, compared to Vero producer cells (Fig. 1D). The majority of measles virus progeny was cell-associated, requiring freeze/thaw cycles to release the viruses (Fig. 1D).
Intercellular fusion is more resistant to neutralizing anti-MV antibodies than cell-free virus
The anti-MV IgG titers in different lots of human AB pooled sera were determined using an enzyme immunoassay. An individual with a titer of more than 20 EU/ml is measles immune. Full plaque reduction neutralization (PRN) assays using 250 TCID50 MV-GFP were also performed to determine the corresponding virus neutralization titers of the sera. Pooled AB serum lot K61552 (277 EU/ml, PRN=256) and lot C80553 (300 EU/ml, PRN=256) were used in all subsequent experiments.
Vero cells were infected with MV-GFP (103 TCID50) in the absence or presence of increasing concentrations of measles immune serum. The numbers of infectious centers (syncytia) in the Vero monolayer were counted at 48h post infection and represented as a percentage of syncytia count in the absence of anti-MV antibodies. `Naked' MV-GFP was neutralized by measles immune sera; no syncytium was detected at the highest concentration of human serum used (1:4 dilution, 69 EU/ml). It was not until at 1:128 serum dilution (2 EU/ml) that 40% of infectious centers was recovered in the Vero monolayer (Fig. 2A).
Figure 2.
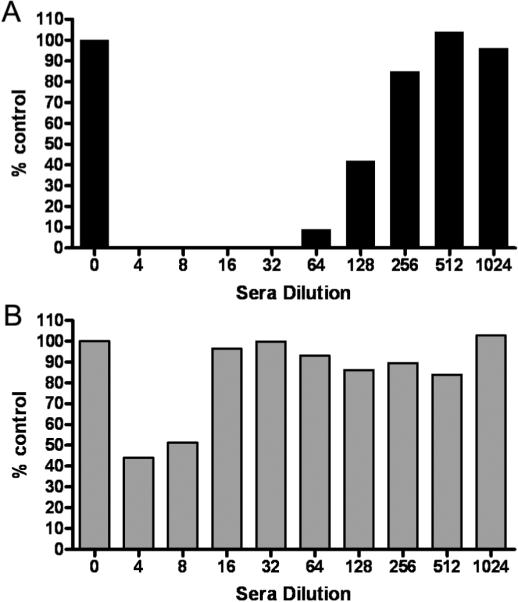
Virus delivered by infected MSC carriers is more resistant to inhibition by antimeasles antibodies than by cell-free `naked' virus. (A) MV-GFP virus was mixed with varying dilutions of human measles immune sera (1:4 to 1:1024 dilution in 5% FBS-DMEM) and added to Vero cells. (B) MSC were infected with MV-GFP (MOI 1.0) and the next day, MSC were mixed with varying dilutions of human measles immune sera and overlaid on Vero cells. The Vero cells were cultured for 2 days and the numbers of syncytia were counted and represented as a percentage of numbers of syncytia found in wells with no serum. A representative experiment from three replicates is shown.
To determine if MSC could deliver MV to target cells in the presence of anti-MV antibodies, MSC were preinfected with MV-GFP at MOI 1.0 and the next day, MSC were overlaid on Vero cells (1 MSC: 100 Vero cells) in the presence of increasing concentrations of measles immune serum. From figure 2B, it is apparent that MSC associated viruses were more resistant to the neutralizing effects of anti-MV antibodies. At 1:4 serum dilution (69 EU/ml), 40% of infectious centers was present and at 1:16 dilution, the numbers of infectious centers were comparable to the control in which no serum was added. Hence, infected MSC were able to fuse with Vero cells and delivered MV infection to the target cells in an environment with a high amount of anti-MV antibodies.
Localization of MSC post IP administration into mice
Athymic mice were implanted intraperitoneally with SKOV3ip.1 human ovarian cancer cells and 7 days later, mice were injected IP with MV-Luc infected MSC and imaged non-invasively for Fluc gene expression. Firefly luciferase signals were seen both in tumor-free and tumor-bearing mice given MV-Luc infected MSC at day 1 post MSC delivery (Fig. 3A). In the non-tumor bearing mice (top panel), the MV-Luc infected cells were seen at the injection site and at the omental region (highlighted by orange line). By day 3, the luciferase signals had decreased significantly and were undetectable by day 7 after death of the virus infected MSC cells (Fig. 3A, top panel). In contrast, bioluminescent signals remained high and continued to increase over time in tumor-bearing mice given MV-Luc infected MSC. It is evident that in tumor-bearing mice, the infected MSC were able to transfer MV-Luc infection to the peritoneal tumors. The infected tumors continued to propagate the virus, resulting in increase of FLuc bioluminescent signal even after death of the infected MSC (Fig. 3A). As expected, naked MV-Luc was highly efficient at infecting tumors in these measles naïve mice (Fig. 3A, bottom panel).
Figure 3.

In vivo distribution of MSC post IP delivery. (A) Serial bioluminescent imaging of mice post IP injection of MV-Luc infected MSC (MV/MSC) into mice with no tumor or mice bearing intraperitoneal human SKOV3ip.1 ovarian tumors. Another group of tumor-bearing mice received MV-Luc (MV). (B) MV infected MSC were labeled with a fluorescent CellTracker Red CMTPX dye and injected IP into mice bearing peritoneal SKOV3ip.1, A2780 or OVCAR5 human ovarian tumors. Mice were euthanized 24 hours later. Photographs (40X or 100X magnification) of freshly harvested tissues showing red fluorescent MSCs on the omentum or peritoneal wall of mice with no tumor or on the tumor nodules of mice. (C) Confocal microscopy images of tumor cryosections demonstrating presence of red fluorescent MSC on the surface and in the parenchyma of SKOV3ip.1 tumors. Scale bars represent 100 μm unless otherwise indicated.
To determine if MSCs co-localized with the tumor nodules, another cohort of mice bearing intraperitoneal SKOV3ip.1, OVCAR5 or A2780 human ovarian tumors were injected IP with CellTracker Red CMTPX dye labeled MSC (MV infected or uninfected). Tumors were harvested and examined directly under a fluorescence microscope 24h later. Abundant red fluorescent MSC were found at the omental tumor nodules and on smaller peritoneal tumor nodules that have seeded on the mesenteric linings or intestines (Fig. 3B). There was no apparent difference in the biodistribution of uninfected and infected MSC; both types of MSC were able to traffick to and localize on the tumors. Gross examination of the peritoneal cavity indicated that peritoneal tumor nodules in all 3 models of ovarian cancer were covered with red fluorescent MSC. In the OVCAR5 model in which there was significant coverage of the peritoneum wall by a cake of tumor cells, abundant MSC were found on the tumor cake (Fig. 3B). In contrast, the tumor free peritoneal wall next to this tumor cake had minimal numbers of MSC (Fig. 3B). When given IP into control athymic mice with no tumor, MSC accumulated predominantly on the omentum (Fig. 3B). Cryosections of the peritoneal tumors were examined using confocal microscopy to determine the extent of MSC infiltration into the tumor parenchyma. At 4h and 24h post cell infusion, red fluorescent MSC were seen lining the surface of the tumor nodules and in the tumor parenchyma (Fig. 3C).
Passive Immunization of Athymic Mice with Measles Immune Serum
Mice are not susceptible to infection by measles virus because they lack the CD46 receptor required for virus entry. Measles virus is also unable to replicate in murine tissues due to an intracellular block to transcription of viral genes (32, 33). Thus, we used the passive immunization method in which measles immune human serum was injected IP into mice to generate measles immune animals. We validated this method by giving athymic mice an IP injection of measles immune human serum and measuring the level of neutralizing anti-MV antibodies present in the mouse sera at different time points. At 3h post IP injection of human serum (80 EU per mouse), the mice were confirmed to be measles immune and the full plaque reduction neutralization (PRN) titers of the murine sera ranged between 64 and 512 (Fig. 4A). There was a gradual decrease in the PRN titers over the one week time interval. The estimated half-life of neutralizing antimeasles antibody in the mice was 19.2 hours. In all subsequent experiments, virus or cell carrier delivery was performed at 3h post IP infusion of this measles immune pooled human serum.
Figure 4.

MSC protect measles virus from complete neutralization by anti-measles antibody in mice. (A) Timeline for decay of human measles immune serum in sera of athymic mice over time. Mice were euthanized at each time point to obtain serum for virus plaque reduction neutralization (PRN) assays. Each point is the average of PRN assays from 2 to 4 mice. Curve fit R2=0.99. (B) Representative photographs showing correlation between tumor location (indicated by GLuc activity) and MV-Luc gene expression (FLuc activity), delivered by cell-free virus or MSC-associated virus in measles naïve or measles immune mice. Mice were imaged using colenterazine substrate for GLuc expression one day before imaging for FLuc activity using D-luciferin substrate. The photons count (photons/sec) for each mouse is as indicated.
Antitumor efficacy in the presence of neutralizing antibodies
Athymic mice were implanted with SKOV3ip.1-GLuc tumor cells and 8 days later, mice received saline or equal numbers (1×106) of `naked' cell free MV-Luc or MV-Luc infected MSC. Mice were imaged non-invasively using bioluminescent imaging for GLuc expression with colenterazine substrate (tumor burden) and the next day, for Fluc expression using D-luciferin substrate (viral gene expression). Imaging was performed one day apart to ensure that all GLuc bioluminescent signals had disappeared prior to FLuc imaging. We observed a good correlation between the location and intensity of GLuc and FLuc signals (Fig. 4B), indicating robust infection of the tumors by cell-free and MSC-associated virus in measles naïve animals. In measles immune animals, the MV-Luc infected MSC were able to transfer the infection to ovarian tumors, resulting in increase in FLuc signal. In contrast, `naked' MV-Luc was neutralized by the anti-measles antibodies and the peritoneal tumors were not infected (Fig. 4B). Hence, delivery of MV is superior when MSC are used as virus carriers in measles immune animals. The increase in FLuc signal in the tumors at day 7 was not due to presence of infected MSC. A significant portion of virus infected MSC died by day 3 post IP delivery into mice resulting in complete loss of bioluminescent signals (Fig. 3A).
To investigate the antitumor activity of the various treatments, athymic mice were implanted IP with 2 × 106 SKOV3ip.1-GLuc tumor cells and 8 days later, mice were passively immunized by IP administration of measles immune human serum (50 EU) or given saline. Three hours later, mice were given equal numbers (1×105) of cell-free MV-NIS or MV-infected MSC IP. The MV/MSC had been loaded with MV-NIS by incubation with virus at MOI 4.0 for 2 hours (approximately 60% infection, Fig. 1) and washed once in PBS. In the measles immune groups, MV-NIS and MV/MSC were subsequently incubated in vitro with measles immune serum (50 EU) for 30 min at 37°C prior to injection into the animals to ensure neutralization of any surface bound virus. Thus, each measles immune mouse received a total of 100 EU of anti-MV IgG Ab.
The Kaplan Meier survival curves of the mice were plotted (Fig. 5). The median survival for saline control was 31 days (n=5 mice), uninfected MSC was 31 days (n=5 mice), MV (−Ab) was 64 days (n=12 mice), MV (+Ab) was 31 days (n=12 mice), MV/MSC (−Ab) was 62 days (n=10 mice) and MV/MSC (+Ab) was 66 days (n=11 mice). All mice in the saline, uninfected MSC, and MV (+Ab) groups (22 out of 22) were euthanized because they developed bloody ascites (3–4.5 mL), with extensive dissemination of ovarian tumors in the peritoneal cavity, perigastric area and on the peritoneal side of the diaphragm. In contrast, only a few of the mice in the MV (−Ab) or MV/MSC groups (+ Ab and − Ab) developed ascites (6 out of 33). Several of the mice appeared jaundiced and were euthanized (12 out of 33). Examination at necropsy indicated tumor obstruction/constriction around the gall bladder or bile duct. The rest of the animals were euthanized due to weight loss of more than 20%, or development of an ulcerated subcutaneous tumor at the injection site towards the end of the study.
Figure 5.

MSC mediated delivery of measles virus enhanced survival of measles immune mice. (A) Kaplan Meier survival curves of mice given different treatments; saline, uninfected MSC, 105 TCID50 `naked' MV-NIS or 105 MV-NIS infected MSC (MOI 4.0, ~ 60% infection) in measles naïve mice (−Ab) or measles immune mice (+Ab, 100 EU/mouse). (B) Immunohistochemical staining for measles-nucleocapsid (MV-N) protein (blue staining=site of viral gene expression) and CD68 positive macrophages (brown/red staining). Arrows in the last panel point to corresponding necrotic area in the tumor with minimal MV-N staining but strong CD68 staining. Scale bars indicate 500 μm.
The survival curves of the respective treatment groups were compared against the saline control and the p values were determined (Fig. 5A). Uninfected MSC did not have any antitumor activity against human ovarian cancer, survival of these mice was not significantly different from the saline treated group (p=0.92). As expected, MV-NIS (−Ab) significantly extended the survival of mice compared to saline control (p<0.0001). However, the antitumor activity of MV-NIS was negated in passively immunized mice. Thus, there was a significant difference in survival outcome using `naked' MV-NIS in measles naïve mice versus measles immune mice (p<0.0001). In contrast to therapy using cell-free virus, treatment of passively immunized mice with MV-infected MSC significantly extended the survival of mice (p<0.0001), doubling the median survival from 31 days (MV-NIS +Ab) to 66 days for MV/MSC +Ab. Furthermore, there was no significant difference in therapy using MV/MSC in measles naïve or measles immune animals (p=0.93). The presence of pre-existing antimeasles antibodies did not diminish the delivery and transfer of MV from infected MSC. We did not observe a significant difference in survival of measles naïve mice treated with MSC-associated MV compared to cell-free virus (p=0.67).
Cryosections of omental tumors harvested at necropsy were immunostained with an antibody specific for the measles nucleocapsid (MV-N) protein and CD68, a macrophage marker. As shown in figure 5B, MV-N positive areas were found in MV-NIS (−Ab), MV/MSC (−Ab) and MV/MSC (+Ab) tumors but not in MV-NIS (+Ab) tumors. Staining for CD68+ macrophages indicated abundant infiltration of phagocytic macrophages into the MV infected tumors, either surrounding or in necrotic areas or co-localizing with the MV-N positive areas. It is likely that the macrophages are phagocytic in nature, as suggested by the abundance of CD68 staining in the necrotic areas of the tumors (see black arrow in MV/MSC +Ab tumor). There were also areas of MV-N staining with no corresponding CD68 staining (eg. last panel, MV/MSC +Ab tumor).
DISCUSSION
The Edmonston vaccine strain of measles virus is capable of inducing tumor selective destruction of ovarian cancer cells while sparing normal cells in the peritoneal cavity (10, 11). However, many ovarian cancer patients have pre-existing anti-measles antibodies. As a strategy to protect oncolytic MV from neutralizing antiviral antibodies and improve delivery of the virus to peritoneal tumors, we evaluated the potential of adipose tissue derived MSC for delivery of MV to orthotopic human ovarian tumors post IP administration in passively immunized mice.
A major potential of advantage of using cells as carriers for delivery of viruses to tumors is protection of the oncolytic virus from antiviral antibodies. In addition, carrier cells can also interact dynamically with the host by responding to chemokines secreted by tumors and may preferentially accumulate at tumor sites. In contrast, systemically administered viruses are sequestered rapidly by the reticuloendothelial system of the liver and spleen and become unavailable for circulation to the tumor. Virus infected cells may also serve as in situ virus production factories, thereby increasing the numbers of infectious virus available in the tumor microenvironment. The `ideal' cell carrier should be susceptible to virus infection or be able to `carry' the virus on/in the cell, remain viable for a sufficient period of time post infection to transfer the virus and be able to support virus replication/progeny production (19, 21, 34). However, there are also disadvantages associated with cell carriers. In the case of MV, infection of a non-transformed cell like MSC required larger amounts of virus (high multiplicity of infection, MOI 4.0) to obtain significant numbers (60%) of infected cells. Also, the clinical protocol incorporating cell carriers becomes more complex as it may involve harvest of autologous cells from the patients, expansion of the cells and pre-loading of the cells with virus in a GLP cell processing facility. In addition, the trafficking profile of the cell carrier needs to be taken into consideration. Ex vivo expanded cells such as T cells, MSC and dendritic cells tend to arrest in the lungs, liver and spleen post intravascular infusion in experimental animals and humans (21, 25, 35, 36) although Power and Bell recently determined that leukemic cell carriers were not so readily trapped in the lung vasculature and could deliver oncolytic vesicular stomatitis virus to tumors (37).
Autologous T cells, irradiated tumor cells, bone marrow derived MSC, dendritic cells and endothelial progenitor cells have been used to deliver oncolytic viruses to tumor xenografts (38). Coukos et al. first demonstrated the feasibility of using a human ovarian teratocarcinoma line PA-1 cell line for HSV-1716 virotherapy of ovarian cancer (39). PA-1 cells were pre-irradiated at 20 Gy prior to HSV infection after which cells were injected intraperitoneally into athymic mice with peritoneal ovarian tumors. Irradiated PA-1 cells co-localized with tumor xenografts to result in significant enhancement in survival of tumor-bearing mice compared to non-treated controls or virus alone. Hamada et al. were among the first to demonstrate that cell carriers can protect oncolytic viruses from neutralizing antiviral antibodies (40). Tumor cell carriers (A549 lung carcinoma) infected with a conditionally replicating adenovirus were irradiated prior to direct intratumoral injection into subcutaneous ovarian tumors in immunized syngeneic mice. Interestingly, treatment of virus immune mice with cell carriers resulted in superior efficacy and complete tumor regressions compared to nonimmunized mice. Hamada et al. suggested that superior antitumor activity could be due to induction of anti-adenoviral and anti-tumoral cytotoxic T-lymphocyte responses in the pre-immunized animals. Bone marrow derived MSC have been used successfully for delivery of oncolytic adenoviruses for the treatment of ovarian cancer or glioma xenografts (22, 24). Perebova et al. demonstrated that mesenchymal progenitor cells infected by RGD displaying adenoviral vector homed to pre-established ovarian tumor nodules and extended the survival of tumor bearing mice, although the feasibility of MSC to overcome antiviral antibodies was not addressed.
Due to previous debulking surgeries, ovarian cancer patients typically have extensive amounts of adhesions that could significantly hinder intraperitoneal distribution of the virus post IP delivery. In addition, fluids injected into the peritoneal cavity rapidly drain out of the cavity via the diaphragmatic stomata into the lymphatics, and from the mediastinal lymph node into the vascular system (41, 42). In contrast, intraperitoneally administered MSC home to and localize on ovarian nodules in the peritoneal cavity of mice. Indeed, abundant MSC were seen `covering' the omental tumor nodules with minimal MSC in the omentum. In nontumor bearing mice, intraperitoneally injected MSC trafficked to and localized on the omentum instead. The omentum is an extension of the mesothelium that becomes drawn out and folded on itself during development and contains abundant milky spots, which consist predominantly of resident peritoneal macrophages (42, 43). Cancer cells injected intraperitoneally into experimental animals preferentially home to, infiltrate and engraft at the omentum (44, 45). As such, it is ideal that MSC appear to have a tropism for the omentum and tumor nodules, thus improving virus delivery to the tumor cells. The mechanism(s) that determines MSC tropism for tumors have not been fully elucidated, but it is clear that signaling molecules produced by the tumor cells (eg. stromal cell derived growth factor SDF-1, monocyte chemoattractant protein MCP-1, vascular growth factor) and adhesion molecules (eg. ICAM-1, integrins and L-selectin) contribute to MSC tumor homing and engraftment (46–48).
Immunohistochemical staining revealed areas of MV gene expression in tumors from MV-NIS (−Ab), MV/MSC (−Ab) and MV/MSC (+Ab) treated animals but not in tumors from MV-NIS (+Ab) treated mice, confirming that the superior survival of mice required active viral replication and gene expression. Uninfected MSC did not have antitumor activity. Abundant infiltrates of CD68 positive macrophages were found in the measles infected areas/tumors but minimal in uninfected tumors. The role of macrophages in the measles infected tumors remains to be determined. They could be phagocytic macrophages whose role is to clear up nonviable virus infected tumor cells as indicated by necrotic areas with negative/minimal MV-N staining but abundant CD68 staining. On the other hand, there were also areas where MV-N and CD68 staining overlapped. Chiocca and colleagues proposed that CD68 macrophages, as part of the host innate immune response, inhibit virotherapy by restricting the intratumoral spread of oncolytic viruses (49). They have elegantly demonstrated that addition of cyclophosphamide, an immunosuppressive agent, to oncolytic herpes virotherapy resulted in decreased numbers of infiltrating CD68 macrophages, increased intratumoral viral replication and superior survival of mice (50). Certainly, research into optimal ways to combine cyclophosphamide with measles virotherapy in ovarian caner is needed; for example, the timing of drug administration is important as cyclophosphamide (an alkylating agent) may negatively impact the viability of cell carriers.
In contrast to studies using vaccinia or adenoviruses (24, 51), we did not observe an enhancement in the oncolytic activity of measles virus when using cell carriers compared to free virus in measles naïve mice. Measles virus infected MSC did not produce a significant amount of progeny virus; this is expected as measles virus replication is significantly lower in nontransformed cells than in cancer cells (13, 52). Hence, in the case of measles virus, it is likely that virus transfer from the MSC carriers to tumor occurred predominantly via heterofusion between the cell carrier and the tumor cells, rather than release of viral progeny. Data from our study also indicated that H and F induced intercellular fusion is much more resistant (32-fold) to the inhibitory effects of antimeasles antibodies. As such, at 69 EU/ml of measles immune serum, cell-to-cell fusion was able to occur where virus-to-cell fusion was evident only at 2 EU/ml of the serum. This observation bodes well for the use of virus infected cell carriers as a possible means to circumvent antiviral antibodies in measles virotherapy.
TRANSLATIONAL RELEVANCE.
Recombinant oncolytic measles viruses (MV) derived from the Edmonston vaccine lineage are undergoing phase I clinical testing in cancer patients. While results from a recently completed trial testing intraperitoneal administration of MV-CEA in patients with recurrent ovarian cancer indicated that MV-CEA was well tolerated, it is also apparent that virotherapy was suboptimal in these measles immune patients. Here, we demonstrated that human MSC could protect MV from neutralization by antiviral antibodies and serve as carriers to deliver MV to ovarian tumors. Intraperitoneally administered MSC trafficked to and co-localized with orthotopic OVCAR5, A2780 and SKOV3ip.1 human ovarian tumor xenografts in mice, enhancing contact of virus with the tumors. In addition, virus-loaded MSC transferred MV infection efficiently to ovarian tumor xenografts and significantly extended the survival of mice passively immunized with antimeasles antibodies. Cell carriers should be incorporated in clinical trials using MV in ovarian cancer patients.
ACKNOWLEDGMENTS
We are grateful to the Minnesota Ovarian Cancer Alliance, Andersen Foundation, Alliance for Cancer Gene Therapy and the National Cancer Institute (CA136547 and CA129966) for funding this work. We thank the Mayo Clinic Women's Cancer Program (Drs Kimberly Kalli and Viji Shridhar) for the ascites samples and cell lines and Dr Bakhos A Tannous (Harvard Medical School, Cambridge, MA) for the kind gift of Gaussia luciferase expression plasmid.
REFERENCES
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Badgwell D, Bast RC., Jr. Early detection of ovarian cancer. Dis Markers. 2007;23:397–410. doi: 10.1155/2007/309382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fung-Kee-Fung M, Oliver T, Elit L, et al. Optimal chemotherapy treatment for women with recurrent ovarian cancer. Curr Oncol. 2007;14:195–208. doi: 10.3747/co.2007.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rocconi RP, Numnum TM, Stoff-Khalili M, et al. Targeted gene therapy for ovarian cancer. Curr Gene Ther. 2005;5:643–653. doi: 10.2174/156652305774964668. [DOI] [PubMed] [Google Scholar]
- 5.Kimball KJ, Numnum TM, Rocconi RP, Alvarez RD. Gene therapy for ovarian cancer. Curr Oncol Rep. 2006;8:441–447. doi: 10.1007/s11912-006-0073-x. [DOI] [PubMed] [Google Scholar]
- 6.Agarwal R, Linch M, Kaye SB. Novel therapeutic agents in ovarian cancer. Eur J Surg Oncol. 2006;32:875–886. doi: 10.1016/j.ejso.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 7.Han ES, Monk BJ. Bevacizumab in the treatment of ovarian cancer. Expert Rev Anticancer Ther. 2007;7:1339–1345. doi: 10.1586/14737140.7.10.1339. [DOI] [PubMed] [Google Scholar]
- 8.Chu CS, Kim SH, June CH, Coukos G. Immunotherapy opportunities in ovarian cancer. Expert Rev Anticancer Ther. 2008;8:243–257. doi: 10.1586/14737140.8.2.243. [DOI] [PubMed] [Google Scholar]
- 9.Russell SJ, Peng KW. Viruses as anticancer drugs. Trends Pharmacol Sci. 2007;28:326–333. doi: 10.1016/j.tips.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng KW, TenEyck CJ, Galanis E, et al. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62:4656–4662. [PubMed] [Google Scholar]
- 11.Myers R, Greiner S, Harvey M, et al. Oncolytic activities of approved mumps and measles vaccines for therapy of ovarian cancer. Cancer Gene Ther. 2005;12:593–599. doi: 10.1038/sj.cgt.7700823. [DOI] [PubMed] [Google Scholar]
- 12.Anderson BD, Nakamura T, Russell SJ, Peng KW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64:4919–4926. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- 13.Ong HT, Timm MM, Greipp PR, et al. Oncolytic measles virus targets high CD46 expression on multiple myeloma cells. Exp Hematol. 2006;34:713–720. doi: 10.1016/j.exphem.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Bjorge L, Hakulinen J, Wahlstrom T, Matre R, Meri S. Complement-regulatory proteins in ovarian malignancies. Int J Cancer. 1997;70:14–25. doi: 10.1002/(sici)1097-0215(19970106)70:1<14::aid-ijc3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 15.Committee RDA Recombinant DNA Advisory Committee Meeting, U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health Protocol #0201–523: Phase I trial of intraperitoneal administration of an attenuated strain (Edmonston strain) of measles virus, genetically modified to produce carcinoembryonic antigen, in patients with recurrent ovarian cancer. Hum Gene Ther. 2002 June 20–21;13:2220–2222. 2002. [Google Scholar]
- 16.Galanis E, Hartmann LC, Cliby W, et al. Phase I trial of intraperitoneal (IP) administration of a measles virus (MV) derivative expressing the human carcinoembryonic antigen (CEA) in ovarian cancer patients. Journal of Clinical Oncology. 2006;24:5028. [Google Scholar]
- 17.Raykov Z, Balboni G, Aprahamian M, Rommelaere J. Carrier cell-mediated delivery of oncolytic parvoviruses for targeting metastases. Int J Cancer. 2004;109:742–749. doi: 10.1002/ijc.20013. [DOI] [PubMed] [Google Scholar]
- 18.Thorne SH, Negrin RS, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Science. 2006;311:1780–1784. doi: 10.1126/science.1121411. [DOI] [PubMed] [Google Scholar]
- 19.Power AT, Wang J, Falls TJ, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007;15:123–130. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 20.Iankov ID, Blechacz B, Liu C, et al. Infected cell carriers: a new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol Ther. 2007;15:114–122. doi: 10.1038/sj.mt.6300020. [DOI] [PubMed] [Google Scholar]
- 21.Ong HT, Hasegawa K, Dietz AB, Russell SJ, Peng KW. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther. 2007;14:324–333. doi: 10.1038/sj.gt.3302880. [DOI] [PubMed] [Google Scholar]
- 22.Sonabend AM, Ulasov IV, Tyler MA, et al. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells. 2008;26:831–841. doi: 10.1634/stemcells.2007-0758. [DOI] [PubMed] [Google Scholar]
- 23.Raykov Z, Grekova S, Galabov AS, et al. Combined oncolytic and vaccination activities of parvovirus H-1 in a metastatic tumor model. Oncol Rep. 2007;17:1493–1499. [PubMed] [Google Scholar]
- 24.Komarova S, Kawakami Y, Stoff-Khalili MA, Curiel DT, Pereboeva L. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol Cancer Ther. 2006;5:755–766. doi: 10.1158/1535-7163.MCT-05-0334. [DOI] [PubMed] [Google Scholar]
- 25.Peng KW, Dogan A, Vrana J, et al. Tumor-associated macrophages infiltrate plasmacytomas and can serve as cell carriers for oncolytic measles virotherapy of disseminated myeloma. Am J Hematol. 2009;84:401–407. doi: 10.1002/ajh.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kidd S, Spaeth E, Klopp A, et al. The (in) auspicious role of mesenchymal stromal cells in cancer: be it friend or foe. Cytotherapy. 2008;10:657–667. doi: 10.1080/14653240802486517. [DOI] [PubMed] [Google Scholar]
- 27.Giordano A, Galderisi U, Marino IR. From the laboratory bench to the patient's bedside: an update on clinical trials with mesenchymal stem cells. J Cell Physiol. 2007;211:27–35. doi: 10.1002/jcp.20959. [DOI] [PubMed] [Google Scholar]
- 28.Peng KW, Facteau S, Wegman T, O'Kane D, Russell SJ. Non-invasive in vivo monitoring of trackable viruses expressing soluble marker peptides. Nat Med. 2002;8:527–531. doi: 10.1038/nm0502-527. [DOI] [PubMed] [Google Scholar]
- 29.Dingli D, Peng KW, Harvey ME, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103:1641–1646. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- 30.Hewett JW, Tannous B, Niland BP, et al. Mutant torsinA interferes with protein processing through the secretory pathway in DYT1 dystonia cells. Proc Natl Acad Sci U S A. 2007;104:7271–7276. doi: 10.1073/pnas.0701185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tannous BA, Kim DE, Fernandez JL, Weissleder R, Breakefield XO. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol Ther. 2005;11:435–443. doi: 10.1016/j.ymthe.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 32.Niewiesk S, Schneider-Schaulies J, Ohnimus H, et al. CD46 expression does not overcome the intracellular block of measles virus replication in transgenic rats. J Virol. 1997;71:7969–7973. doi: 10.1128/jvi.71.10.7969-7973.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vincent S, Tigaud I, Schneider H, et al. Restriction of measles virus RNA synthesis by a mouse host cell line: trans-complementation by polymerase components or a human cellular factor(s) J Virol. 2002;76:6121–6130. doi: 10.1128/JVI.76.12.6121-6130.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiao J, Wang H, Kottke T, et al. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther. 2008;15:604–616. doi: 10.1038/sj.gt.3303098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Read EJ, Keenan AM, Carter CS, Yolles PS, Davey RJ. In vivo traffic of indium-111-oxine labeled human lymphocytes collected by automated apheresis. J Nucl Med. 1990;31:999–1006. [PubMed] [Google Scholar]
- 36.Hakkarainen T, Sarkioja M, Lehenkari P, et al. Human mesenchymal stem cells lack tumor tropism but enhance the antitumor activity of oncolytic adenoviruses in orthotopic lung and breast tumors. Hum Gene Ther. 2007;18:627–641. doi: 10.1089/hum.2007.034. [DOI] [PubMed] [Google Scholar]
- 37.Power AT, Bell JC. Cell-based delivery of oncolytic viruses: a new strategic alliance for a biological strike against cancer. Mol Ther. 2007;15:660–665. doi: 10.1038/sj.mt.6300098. [DOI] [PubMed] [Google Scholar]
- 38.Russell SJ, Peng KW. The utility of cells as vehicles for oncolytic virus therapies. Curr Opin Mol Ther. 2008;10:380–386. [PubMed] [Google Scholar]
- 39.Coukos G, Makrigiannakis A, Kang EH, et al. Use of carrier cells to deliver a replication-selective herpes simplex virus-1 mutant for the intraperitoneal therapy of epithelial ovarian cancer. Clin Cancer Res. 1999;5:1523–1537. [PubMed] [Google Scholar]
- 40.Hamada K, Desaki J, Nakagawa K, et al. Carrier cell-mediated delivery of a replication-competent adenovirus for cancer gene therapy. Mol Ther. 2007;15:1121–1128. doi: 10.1038/sj.mt.6300128. [DOI] [PubMed] [Google Scholar]
- 41.Abu-Hijleh MF, Habbal OA, Moqattash ST. The role of the diaphragm in lymphatic absorption from the peritoneal cavity. J Anat. 1995;186(Pt 3):453–467. [PMC free article] [PubMed] [Google Scholar]
- 42.Doherty NS, Griffiths RJ, Hakkinen JP, Scampoli DN, Milici AJ. Post-capillary venules in the “milky spots” of the greater omentum are the major site of plasma protein and leukocyte extravasation in rodent models of peritonitis. Inflamm Res. 1995;44:169–177. doi: 10.1007/BF01782815. [DOI] [PubMed] [Google Scholar]
- 43.Krist LF, Eestermans IL, Steenbergen JJ, et al. Cellular composition of milky spots in the human greater omentum: an immunochemical and ultrastructural study. Anat Rec. 1995;241:163–174. doi: 10.1002/ar.1092410204. [DOI] [PubMed] [Google Scholar]
- 44.Tsujimoto H, Hagiwara A, Shimotsuma M, et al. Role of milky spots as selective implantation sites for malignant cells in peritoneal dissemination in mice. J Cancer Res Clin Oncol. 1996;122:590–595. doi: 10.1007/BF01221190. [DOI] [PubMed] [Google Scholar]
- 45.Lopes Cardozo AM, Gupta A, Koppe MJ, et al. Metastatic pattern of CC531 colon carcinoma cells in the abdominal cavity: an experimental model of peritoneal carcinomatosis in rats. Eur J Surg Oncol. 2001;27:359–363. doi: 10.1053/ejso.2001.1117. [DOI] [PubMed] [Google Scholar]
- 46.Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4:206–216. doi: 10.1016/j.stem.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15:730–738. doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 48.Fox JM, Chamberlain G, Ashton BA, Middleton J. Recent advances into the understanding of mesenchymal stem cell trafficking. Br J Haematol. 2007;137:491–502. doi: 10.1111/j.1365-2141.2007.06610.x. [DOI] [PubMed] [Google Scholar]
- 49.Chiocca EA. The host response to cancer virotherapy. Curr Opin Mol Ther. 2008;10:38–45. [PubMed] [Google Scholar]
- 50.Fulci G, Breymann L, Gianni D, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thorne SH, Contag CH. Integrating the biological characteristics of oncolytic viruses and immune cells can optimize therapeutic benefits of cell-based delivery. Gene Ther. 2008;15:753–758. doi: 10.1038/gt.2008.42. [DOI] [PubMed] [Google Scholar]
- 52.Peng KW, Ahmann GJ, Pham L, et al. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood. 2001;98:2002–2007. doi: 10.1182/blood.v98.7.2002. [DOI] [PubMed] [Google Scholar]