

Abstract
Several studies have demonstrated that sphingosine kinase 1 (SphK1) translocates to the plasma membrane (PM) upon its activation and further suggested the plasma membrane lipid raft microdomain (PMLRM) as a target for SphK1 relocalization. To date, however, direct evidence of SphK1 localization to the PMLRM has been lacking. In this report, using multiple biochemical and subcellular fractionation techniques we demonstrate that endogenous SphK1 protein and its substrate, D-erythro sphingosine, are present within the PMLRM. Additionally, we demonstrate that the PMA stimulation of SphK1 localized to the PMLRM results in production of sphingosine-1-phosphate as well as induction of cell growth under serum-deprivation conditions. We further report that Ser225Ala and Thr54Cys mutations, reported to abrogate phosphatidylserine binding, block SphK1 targeting to the PMLRM and SphK1 induced cell growth. Together these findings provide direct evidence that the PMLRM is the major site-of-action for SphK1 to overcome serum-deprived cell growth inhibition.
Keywords: Sphingosine-1-phosphate, sphingosine kinase 1, plasma membrane, lipid raft microdomain, phosphatidylserine, subcellular localization
Introduction
In recent years, the sphingolipid metabolic pathway has received increased attention for its role in regulation of critical cell functions including cell growth, apoptosis, differentiation, migration and activation (reviewed in [1-4]. Two of the simplest sphingolipid metabolites, ceramide and sphingosine-1-phosphate (S1P), have garnered the lions share of this attention as key regulators of the “Sphingolipid Rheostat” [5]. These two metabolites have opposing effects on cellular fate and as such their generation and metabolism are tightly regulated by the cell. Ceramide, which is a pro-apoptotic metabolite, can be either synthesized de-novo, originating, in the ER, or can be generated at the plasma membrane (PM) by hydrolysis of sphingomyelin by sphingomyelinase [6]. Ceramide can be further deacylated by ceramidase to produce D-erythro-sphingosine. Sphingosine is subsequently phosphorylated by sphingosine kinase (SphK) to form sphingosine-1-phosphate (S1P), thus generating a sphingolipid metabolite that stimulates cell growth, cell survival or migration [7]. S1P mediates its pro-survival/ pro-growth effects by two separate mechanisms. Secreted S1P (mediated by several ABC transporter family members) binds to its cognate cell surface receptors (S1PR1-5) in an autocrine/paracrine manner [7]. Alternatively, S1P can elicit its effects intracellularly, by an uncharacterized mechanism, resulting in cytoskeletal changes, motility, release of intracellular calcium stores, and protection from apoptosis [8].
Cellular S1P levels have been shown to increase rapidly and transiently in response to activators of SphK1, such as cytokines [9], growth factors [10,11], phorbol esters [12], and fetal bovine serum (FBS) [10]. As SphK is the only known enzyme that converts sphingosine to S1P, considerable effort has been placed on the elucidation of its mechanism of activation. The current model for regulation of SphK1 suggests that subcellular localization is a major regulatory factor controlling the production of S1P. Under basally active conditions SphK1 is predominantly present in the cytoplasm. Upon mitogen exposure, the catalytic activity of SphK1 typically increases 1.5 to 4 fold as it translocates to the PM where it phosphorylates D-erythro-sphingosine to generate S1P. Several interaction partners including anionic phospholipids (phosphatidylserine and phosphatidic acid) and Ca2+/calmodulin have been shown to mediate SphK1 relocalization to the PM [13-15]. Additionally, studies indicate that motifs/amino acid residues such as Thr54, Asn89 and Ser225, within SphK1, are required for the relocalization of SphK1 from the cytoplasm to the PM (extensively reviewed in [16,17]).
The PM, once thought to be very homogenous, has been shown to contain small, heterogeneous, dynamic “microdomains” (i.e. lipid rafts, caveolae) proposed to play important roles in cell signaling, migration, viral entry, and cellular polarization [18-21]. These microdomains are enriched in cholesterol and glycosphingolipids, which are particularly concentrated in the outer leaflet of the lipid raft membrane [18],[22]. Lipid rafts are also enriched in simple sphingolipids such as sphingomyelin and ceramide [23]. The presence of sphingolipid metabolic enzymes such as, acid sphingomyelinase and neutral ceramidase, at the cell surface suggests that the lipid raft is a site for the generation of sphingosine [10,24,25]. Sphingosine could then be phosphorylated, by SphK1, to form S1P which, consistent with its role in cell signaling, would be compartmentalized with cell signaling proteins in the lipid raft. To date, however, there is little evidence to indicate that SphK1 is a lipid raft microdomain resident protein.
Early studies of lipid raft microdomain resident proteins relied on their unique resistance to extraction in cold non-ionic detergent (typically 1% Triton X-100) for their isolation and examination [18,26]. Subsequent studies have demonstrated that resistance to Triton X-100 extraction is not sufficient to conclude that a protein is localized to these PM microdomains. Re-evaluation of these studies lead to the establishment of a definition of lipids rafts in which two criteria must be met to definitively state that a protein resides in these PM microdomains: 1) the presence of a protein in the PM microdomain must be dependent on cholesterol and 2) there should be a change in the levels of a protein within the PM microdomain upon stimulation of the cells [26].
In this report, we demonstrate that endogenous human SphK1 and its substrate, D-erythro-sphingosine, reside in the plasma membrane lipid raft microdomain (PMLRM). We further show that PMA stimulation results in increased SphK1 protein localization to the PMLRM along with a concomitant increase in S1P levels. We also demonstrate that blockage of phosphatidylserine (PS) binding is sufficient to abrogate SphK1 localization to the PMLRM and to block the biological effects of SphK1 membrane localization (i.e. S1P production in response to PMA treatment and increased cell growth/survival). Together, these data provide strong evidence that the PMLRM is an important site of action for SphK1 biological effects. These findings, in part, clarify the effects of SphK1 on the numerous cellular signaling pathways to which SphK1 action has been attributed.
Materials and Methods
Cell Culture
Human embryonic kidney 293 cells (HEK293, ATCC: CRL-1573) and HeLa cells (ATCC: CCL-2) were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin at 37°C in a humidified atmosphere of 5% carbon dioxide. Transfections were performed using Lipofectamine 2000 (Invitrogen). Stable lines were generated by selection of antibiotic resistant cells in the presence of 500 μg/mL G418.
cDNA Cloning of Human SphK1
Human sphingosine kinase 1 (GenBank™ accession number AF200328) was cloned from HEK293 total RNA isolated using the RNeasy kit (Qiagen). First strand cDNA synthesis was accomplished with oligo dT primers and Superscript II (Invitrogen). Primers specific for the 5′ (5′CCCAGGAATTCCACCATGGATCCAGCGGGC) and 3′ (3′ATGATCTGCGGCCGCTCATAAGGGTTCTTCTGG) ends of the short isoform (384 amino acids; 1-384) of the wild-type (WT) SphK1 cDNA were designed. PCR products were obtained using Platinum Pfx high fidelity DNA polymerase (Invitrogen) and were used to directionally clone the SphK1 cDNA into the Eco RI (5′) and Not I (3′) sites of pcDNA 3.1+ HIS B (Invitrogen) for mammalian expression as an NH2-terminally tagged His6×-fusion protein. GFP fusion constructs were created by subcloning SphK1 into the Eco RI (5′) and Xba I (3′) restriction sites in pEGFP-C1 (BD Biosciences). The resulting WT SphK1 clones were sequence verified and used for subsequent mutagenesis studies.
The His6×-SphK1 and GFP-SphK1 mutants were generated by mutagenesis of the WT SphK1 cDNA using the Quikchange Site-Directed Mutagenesis system (Stratagene) according to the manufacturer's recommendations. Primers for the mutagenesis are as follows: Thr54Cys (F-5′ CACGCTGATGCTCTGTGAGCGGCGGAACC 3′, R-5′ GGTTCCGCCGCTCACAGAGCATCAGCGTG 3′); and Ser225Ala (F 5′AAGACACCTGCCGCCCCCGTTGTGGTC 3′, R-5′GACCACAACGGGGGCGGCAGGTGTCTT 3′).
Western Blot Analysis
Samples were separated on a 10% SDS-PAGE gels and transferred to PVDF membranes. Membranes were blocked with 5% milk in TBS-T and then incubated with one of the following primary antibodies: anti-SphK1 (Genetech), anti-caveolin-1 and anti-GAPDH (Santa Cruz), anti-Filamin A (Chemicon), anti-flotilin-1 (BD Transduction Laboratories), and anti-His6× (BD Biosciences), The membranes were incubated with the appropriate secondary antibodies (Jackson Labs) and visualized on X-ray film using Super Signal West Dura reagents (Pierce).
Isolation of Triton X-100 Soluble and Insoluble Fractions
Media was aspirated from 10 cm dishes of native HEK293 cells and HEK293 cells stably expressing a His6× epitope tagged recombinant SphK1 protein (His6×-SphK1). Cells were washed once with 1× PBS at 4°C and were flash frozen in liquid nitrogen. Cell lysates were resuspended in 1 mL of 1× TBS containing a protease inhibitor tablet (Roche) and were sonicated briefly (3 15 sec pulses 50% output). The cytosolic fraction was separated from the total membrane fraction by centrifugation at 100,000 xg for 30 min at 4°C. The cytosolic supernatant was removed and the total membrane pellet was resuspended by brief sonication in 500 μL of 1× TBS, 1% Triton X-100 containing protease inhibitors and solubilization of total membrane proteins was allowed to occur for 30 min on ice. The Triton X-100 solubilized membrane fraction was separated from Triton X-100 insoluble material by centrifugation at 100,000 xg for 30 min at 4°C. The Triton X-100 insoluble membrane fraction was resuspended in 250 μL of 2% SDS by brief sonication.
Subcellular Fractionation
Native HEK293 cells and HEK293 cells stably expressing His6×-SphK1 were lysed by Dounce homogenization in 5 mM Tris-HCl, 1 mM MgCl2, 250 mM Sucrose, pH 7.4 containing a Mini EDTA-Free protease inhibitor cocktail tablet and were centrifuged at 500 xg for 10 min at 4°C to remove unbroken cells and nuclei. The post-nuclear supernatant was removed to a new tube and differential centrifugation was performed. The supernatants were sequentially centrifuged at 3,000 xg, 10,000 xg, and 100,000 xg. All centrifugation were 30 min at 4°C. Each pellet was removed and resuspended in the above buffer. The final supernatant (cytosolic fraction) was collected and 5 μg of total protein from each fraction was analyzed by Western blot analysis.
Sucrose Density Gradient Analysis
Two 15 cm plates of HEK293 or HeLa cells were washed once with 5 mL of 1×PBS and scraped in 2.5 mL PBS+PI. Cells were pelleted at 1,000 xg for 10 minutes at 4°C. Cells were then lysed in 1 mL of MBS (25 mM MES, 150 mM NaCl, pH 6.5) containing 500 mM Na2CO3 final pH 11.5 or in 25 mM MES, 1 M NaCl, pH 6.5 both containing protease inhibitors by 20 strokes with a Dounce homogenizer followed by 10 passages through a 23 gauge needle. Unlysed cells and nuclei were repelleted by centrifugation at 500 xg for 5 minutes at 4°C. The post nuclear supernatant was transferred to a new tube and centrifuged at 100,000 xg for 15 min at 4°C. Cell pellets were resuspended in 1 mL of the appropriate buffer containing protease inhibitors and 3 mL of buffered 60% sucrose were added to adjust final sucrose concentration to 45%. The 4 mL fraction of cell lysate in 45% sucrose was added by glass pipette under an 8 mL discontinuous gradient of 35% (4 mL) and 5% sucrose (4 mL) in the appropriate buffer and centrifuged at 200,000 xg for 16 h at 4°C. One mL fractions were collected from the top of the gradient and 25 μL samples from each fraction were analyzed for the presence of SphK1, and lipid raft markers by Western blot analysis.
Purification of Plasma Membranes
A plasma membrane enriched fraction was prepared from cells essentially as described by Smart et al 1995 [27]. Briefly, four confluent 15 cm dishes were washed twice with 1× PBS and lysed by flash freezing in liquid nitrogen. Cells were resuspended in 5 mL of PBS + 250 mM sucrose containing protease inhibitors, homogenized by 5 passes through a 23 gauge needle, and centrifuged at 500 xg for 10 min at 4°C. The 5 mL post nuclear supernatant (PNS) was centrifuged at 3000 xg for 10 min at 4°C and the supernatant removed. The pellet was resuspended in 1 mL of PBS + 250 mM sucrose containing protease inhibitors and layered on top of 9 mL of 30% Percoll and centrifuged at 84000 xg for 30 min in a 75Ti rotor (Beckman). One mL fractions were removed and analyzed by Western blot for PM markers. The visible plasma membrane band ∼ 4.2 cm from the bottom of the tube (equivalent to 5.7 cm in 25 mL tubes) was removed in 1 mL and centrifuged at 100,000 xg for 15 min to remove Percoll. The plasma membrane layer floating above the pelleted Percoll was removed and diluted to 1 mL with 25 mM MES containing 1 M NaCl and protease inhibitors and resuspended. The samples were then adjusted to 45% sucrose by the addition of 3 mL of 60% sucrose for 1M NaCl containing non-detergent discontinuous sucrose density gradient analysis as described above.
SphK1 Activity Assay and Thin Layer Chromatography
To assess the catalytic activity of endogenous SphK1 in the sucrose density gradient fractions, in vitro SphK1 activity assays were performed. To avoid the inhibitory effects of NaCl on SphK1 activity, 15 μL of each gradient fraction were used in activity assay reactions of 100 μL final volume to yield 150 mM NaCl final concentrations. Briefly, samples were combined with 50 μM D-erythro-sphingosine, 200 μM ATP and 2 μCi [γ-32P] ATP in a 100 μL final reaction volume of SKAAB buffer (20 mM Tris pH 7.4, 1 mM β-mercaptoethanol, 1 mM EDTA, 0.1% Triton X-100, 1 mM Na3VO4, 15 mM NaF, 0.5 mM 4-deoxypyridoxine) for 1 h at 37 °C with shaking. Kinase reactions were terminated by the addition of 10 μL 6N HCl and the radio-labeled lipids were extracted by the addition of 400 μL of Chloroform/MeOH 100:200 v/v and 125 μL Chloroform and 125 μL 1M KCl. The organic phase containing lipids was dried down under nitrogen stream. Samples were then resuspended in 30 μL chloroform and applied to a Silica Gel TLC plate (Whatman, Florham Park, NJ) and the lipids were separated using a butanol:water:acetic acid 3:1:1 v:v:v solvent system. The plates were analyzed by X-ray exposure and the region of the TLC plate corresponding to the Rf value (0.32) of S-1-P was examined [28].
Methyl β cyclodextrin and Filipin Treatment
Confluent 10 cm dishes of HEK293 cells stably expressing His6×-SphK1 were treated with or without either methyl β cyclodextrin (20 mM) or filipin (15 μg/mL) for 30 min. One mL post nuclear supernatants were prepared in 25 mM MES, 150 mM NaCl, 1% Triton X-100, pH 7.5 and the sucrose concentration was adjusted to 45% by the addition of 3 mL of 60% sucrose in the above buffer. Samples were layered under a discontinuous gradient of 5% sucrose (4 mL) and 35% sucrose (4 mL) and separated by ultracentrifugation at 200,000 xg for 16 h at 4 °C. One mL samples were removed and fractions 4 and 5 were combined (raft fraction) as were fractions 9-12 (non-raft fraction).
Cytosolic and Membrane Fraction Isolation (Pool Assay)
Nearly confluent (∼90%) 10 cm plates of His6× epitope tagged WT and mutant SphK1 transfected HEK293 cells were flash frozen and resuspended in 1 mL of 1×TBS containing protease and phosphatase inhibitor cocktails (Roche and Calbiochem respectively). Whole cell lysates were centrifuged at 500 xg for 10 min at 4°C to remove nuclei. Post-nuclear supernatants were separated into cytoplasmic and total membrane fractions by centrifugation at 100,000 xg for 30 min at 4°C. Membranes were subsequently resuspended in 500 μL of 1×TBS, 1M NaCl pH 6.5 buffer. After centrifugation at 100,000 xg for 30 min at 4°C, samples were separated into supernatant and pellet fractions. Pelleted membranes were resuspended in 2% SDS.
PMA treatment
HEK293 cells overexpressing His6×-SphK1 and the His6×-SphK1 mutants were seeded onto 10 cm dishes at 4×105 cells/mL in DMEM containing 2% delipidated FBS (dlFBS). After 24 h, cells were stimulated with 300 nm PMA for 20 minutes. The cells were then washed with cold 1×PBS and snap frozen in liquid nitrogen prior to performing the “pool” assay.
Membrane Raft Labeling and Confocal Microscopy
HEK293 cells stably overexpressing GFP-SphK1 constructs were plated at 2×105 cells/mL into 4 well Lab-Tek chamber slides (Nalge Nunc) coated with poly-L-lysine. After 24 h, membrane rafts were labeled with the Vybrant Lipid Raft Labeling Kit (Molecular Probes). Confocal images were obtained using a Leica TCS SP2 AOBS microscope (Microscopy Imaging Core Facility, Penn State College of Medicine). All pictures were taken using a 10× eyepiece, 63× objective. Excitation wavelengths were 488 nm and 594 nm for GFP and Alexa Fluor respectively. The single scan images, taken at a pixel resolution of 1024 × 1024, were overlain to determine colocalization (yellow) of GFP-SphK1 (green) and membrane rafts (red). All images were collected and processed identically.
Sphingolipid Analysis of Membrane Raft Fractions by LC/MS/MS
HEK293 stably transfected cell lines were plated at 4×105 cells/mL in 10 cm culture dishes in DMEM containing 2% dlFBS. The media was removed, cells were washed with 1×PBS and harvested in 1 mL of 1×MBS + 1M NaCl containing protease and phosphatase inhibitors and sucrose gradient centrifugation was performed under 1M NaCl conditions. Density gradients were divided into 2 mL fractions starting from the top of the gradient. Lipids were separated from the sucrose density material by dilution of each 2 mL gradient fraction 15 fold with 1×PBS. Fractions were centrifuged at 100,000 xg for 30 min to pellet lipids, PBS was decanted, and lipids were dissolved in methanol. The organic phase was dried down under nitrogen stream, and lipids were separated and quantified using LC/MS/MS as described by Sullards et al [29] with minor modifications. Briefly, 10 μL of internal standards (25 μM C17 sphingosine and 25 μM C17 S1P, Avanti Polar Lipids) were added to each sample, resuspended in positive ion MS spray solution (methanol: acetic acid, 99:1 v:v, containing 5 mM ammonium acetate), and filtered through 0.22 μm filters (Millipore, Bedford, MA) into 96 well plates. Quantitation by LC/MS/MS using multiple reaction monitoring was performed as previously described [29] using a 4000 Q trap LC/MS/MS system (Applied Biosystems) with an Aligent 1100 Series binary pump and an HTS PAL autosampler. For the experiments measuring S1P formation after PMA treatment, 24 h after cell plating, the cells were stimulated with 300 nM PMA for 20 min and LC/MS/MS was performed as described.
Growth and survival assays
HEK293 cells stably transfected with His6×SphK1 constructs were growth arrested by overnight incubation in DMEM containing 0.5% FBS. After 24h (day 0), media was changed to DMEM containing 0.5% FBS. After 24h, 48h and 72 h individual 96 well plates were TCA fixed and upon completion of the experiment, all plates were stained with Sulforhodamine B (SRB) for 1 h. Wells were extensively washed with 0.1% acetic acid and the remaining SRB was solubilized with 10 mM Tris and absorbance was determined using a 96-well plate reader at 570 nM [30].
Results
Endogenous SphK1 associates with intracellular membranes and the plasma membrane
The current model of SphK1 activation involves agonist-induced translocation of basally active SphK1 from the cytoplasm to the PM, where it achieves full catalytic activity [31]. Numerous studies have demonstrated by immunofluorescent examination that SphK1 relocalizes from the cytosol to the PM upon stimulation of SphK1 activity by mitogenic factor treatment [9-12]. There are also several reports suggesting that SphK1 associates with subcellular organelle membranes, in particular ER and lysosomes [32,33]. However, most SphK1 localization studies have relied on over-expression model systems raising the possibility that this localization is an artifact of the over-expression system. To clarify the presence of endogenous SphK1 in subcellular organelles and at the PM, we performed a standard subcellular fractionation (Figure 1A) [34]. Endogenous SphK1 is weakly detected in the P2 pellet fraction (3,000 xg) which contains mostly heavy mitochondria. Endogenous SphK1 is also detected in the P3 (10,000 xg) and P4 (100,000 xg) pellets which contains primarily lysomes/peroxisomes/Golgi and ER/PM vesicles respectively. The presence of SphK1 in the P3 and P4 pellet fractions indicates that SphK1 is found in both subcellular organelle membranes and the PM. A similar distribution of a His6× epitope tagged recombinant SphK1 (His6×-SphK1) protein (Figure 1A) was observed demonstrating that over-expression does not aberrantly target SphK1 to non-native membrane locales.
Figure 1. Subcellular localization of endogenous and untagged over-expressed SphK1.
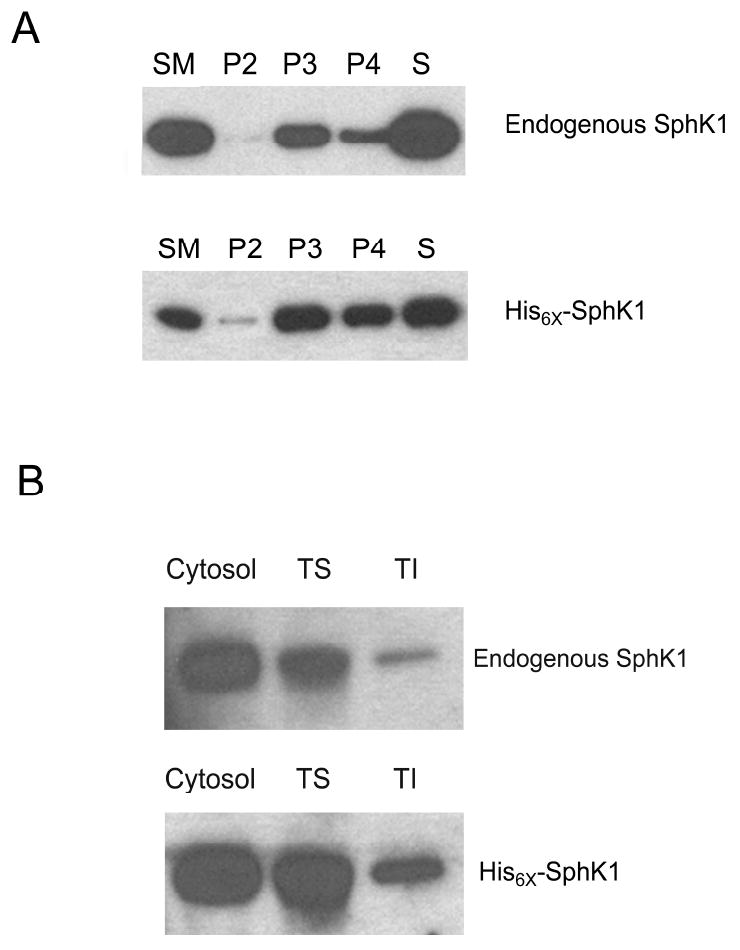
(A) Endogenous and over-expressed His6× epitope tagged SphK1 were separated by subcellular fractionation as detailed in the Materials and Methods section. 5 μg of starting material (SM), heavy mitochondrial fraction (3,000 xg pellet; P2), intracellular membranes including lysomes/peroxisomes/Golgi/ER (10,000 xg pellet; P3), ER/Plasma membranes (100,000 xg pellet; P4) and cytosolic supernatant (S) were separated by SDS-PAGE and analyzed for the expression of SphK1 using anti-SphK1 antibodies (n=3). (B) Endogenous and over-expressed His6×-SphK1 were separated into cytosolic, Triton X-100 soluble (TS) and Triton X-100 insoluble (TI) fractions. 5 μg of total protein were separated by SDS-PAGE and analyzed by Western blot analysis using anti-SphK1 antibodies (n=5).
Endogenous SphK1 protein is localized to the Triton X-100 insoluble total membrane fraction
Many of the recent biochemical studies, that examine SphK1 translocation to the membrane, have focused solely on the “Triton X-100 soluble” (TS) SphK1 total membrane fraction and have not examined whether SphK1 is present in the “Triton X-100 insoluble” (TI) fraction [14,35-37]. Additionally, those reports that have examined the TI fraction have relied on over-expression of recombinant SphK1 for examination of SphK1 membrane association [38]. To determine whether endogenous SphK1 is present within the TS and TI total cellular membrane fractions, we examined the membrane localization of endogenous SphK1 protein in HEK293 cells by Western blot analysis. In addition, we also examined whether His6×-epitope tagged human SphK1 is localized to the TS and TI fractions of stably transfected HEK293 cells. As shown in Figure 1B, both endogenous and recombinant SphK1 proteins are indeed present in the TS and TI fractions of total membrane preparations confirming the presence of endogenous and recombinant SphK1 within the TI total membrane fraction. Importantly, that recombinant SphK1 is present within the TI fraction in approximately the same ratio as endogenous SphK1 further indicates that the relative distribution of SphK1 within its intracellular fractions is not altered by this over-expression system.
Endogenous SphK1 is present within the PMLRM
Our data presented above indicate that both endogenous and recombinant SphK1 are present at the PM and are also present within the TI fraction of membranes. Until recently, the presence of a protein in the Triton X-100 insoluble PM fraction was taken as an indication that the protein was localized to the PMLRM and many studies relied on sucrose density gradient ultracentrifugation in the presence of 1% Triton X-100 to demonstrate localization of the protein to the PMLRM. However, through a series of studies, Triton X-100 has been shown to aberrantly target proteins to the PMLRM and is no longer sufficient to demonstrate PMLRM localization [26]. Therefore, additional methods are required to validate the presence of endogenous SphK1 in the PMLRM fraction.
In this regard, non-detergent sucrose density gradients (ND-SDG) have replaced Triton X-100 containing sucrose density gradients for the study of PMLRM resident proteins [18,26]. One such ND-SDG method employs 500 mM Na2CO3 at pH 11.5, and is therefore of limited use to the study of SphK1 as the high pH of the Na2CO3 buffer abrogates SphK1 activity (data not shown) which has an optimal pH range of 6.5 to 8 [39,40]. One function of Na2CO3 in the ND-SDG is to dissociate peripherally associated membrane proteins, including the actin cytoskeleton, from the PM [41]. We reasoned that 1M NaCl (pH 6.5), due to its ability to dissociate ionic interactions, should have the same effect while maintaining a more physiological pH, thus preserving SphK1 catalytic activity. To validate our NaCl containing ND-SDG method (NaCl method), we compared the distribution of the caveolae/lipid raft microdomain markers caveolin-1 and flotillin-1 obtained from the NaCl method with the distribution obtained from the ND-SDG containing Na2CO3 at pH 11.5 (Na2CO3 method). As shown in Figures 2A and B, the distribution of caveolin-1 and flotillin-1 were very similar between the two methods indicating 1M NaCl can functionally replace Na2CO3 in the ND-SDG preparation. When we performed Western blot analysis of ND-SDG fractions for endogenous SphK1, we detected endogenous SphK1 in the lipid raft fraction by the NaCl method as well as the Na2CO3 method. To further confirm presence of SphK1 in the lipid raft fraction, we performed an in-vitro SphK1 activity assay on SDG fractions isolated by the NaCl method, using thin-layer chromatographic (TLC) separation. Figure 2C clearly shows that the majority of SphK1 catalytic activity is detected in the lipid raft fractions of the SDG (fractions 4-5) while a minor fraction of SphK1 catalytic activity was detected in the non-raft fractions (fractions 9-12). Together, this data demonstrates the presence of endogenous SphK1 protein and activity within the lipid raft of human HEK293 cells.
Figure 2. Non-detergent Sucrose Density Gradient Separation of SphK1.
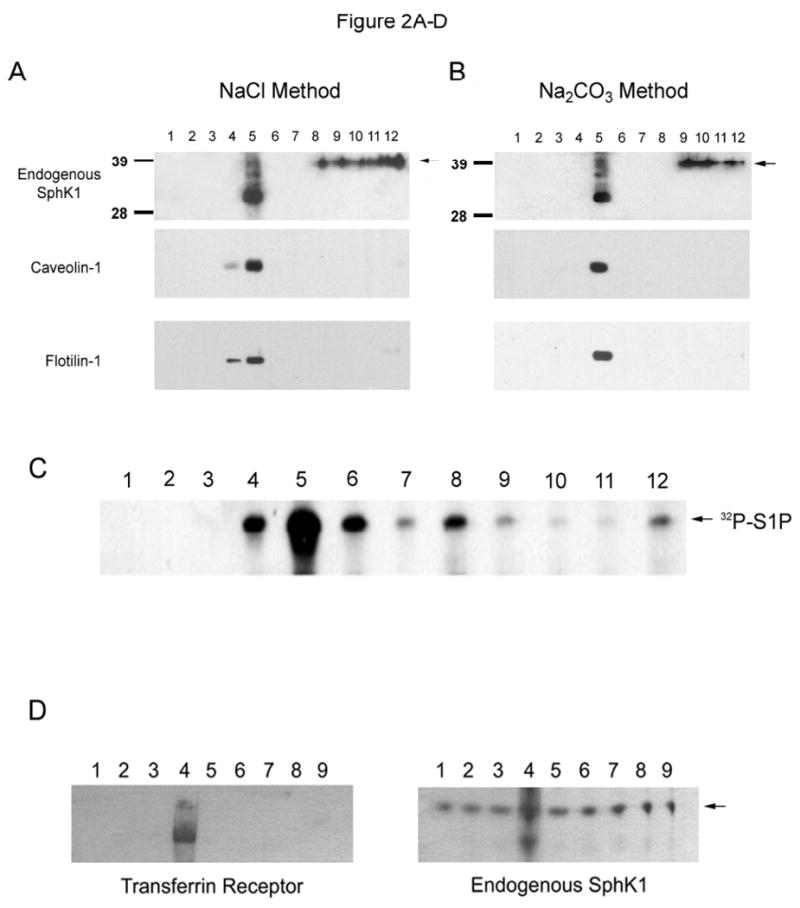
(A) Non-detergent sucrose density gradient separation of SphK1 by the 1M NaCl pH 6.5 method. 25 μL samples of each gradient fraction were separated by SDS-PAGE and analyzed by Western Blot analysis for caveolin-1, flotilin-1 and SphK1 as indicated. Arrow indicates presence of endogenous SphK1 protein. (B) Non-detergent sucrose density gradient separation of SphK1 by the Song et al., 500 mM Na2CO3 pH 11.5, method. 25 μL samples of each gradient fraction were separated by SDS-PAGE and analyzed by Western Blot analysis for caveolin-1, flotilin-1 and SphK1 as indicated. Arrow indicates presence of endogenous SphK1 protein. (C) S1P determination was performed by TLC separation as described in the Materials and Methods section. (D) Plasma membrane enriched fractions were isolated and 25 μL samples of each gradient fraction from the 9 mL 30% Percoll gradient were separated by SDS-PAGE and analyzed for the presence of the plasma membrane marker Transferrin Receptor 1 and endogenous SphK1 by Western blot analysis as indicated. (E and F) HEK293 and HELA cell plasma membrane enriched fractions were separated by 1M NaCl containing sucrose density gradient ultracentrifugation as above. 25 μL samples were separated by SDS-PAGE and analyzed for the presence of endogenous SphK1 and caveolin-1. S1P determination was performed as above. Arrow indicates the presence of endogenous SphK1. (G) HEK293 cells stably expressing His6×-SphK1 were treated with methyl β cyclodextrin or filipin as indicated and separated by sucrose density gradient ultracentrifugation in buffer containing 1% Triton X-100. One mL fractions were removed and fractions 4 and 5 (raft) and 9-12 (non-raft) were pooled. 5 μg samples of each untreated and treated pooled fraction were separated by SDS-PAGE and analyzed for the presence of His6×-SphK1 and caveolin-1 (n=3 for all experiments).
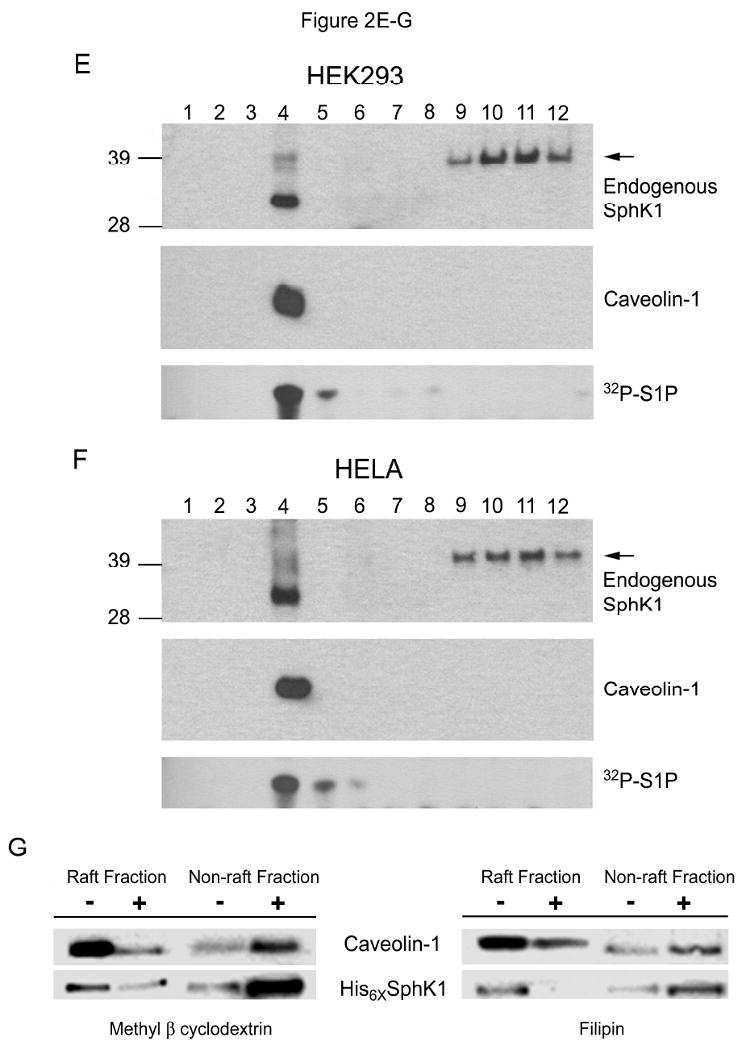
We next wanted to determine whether SphK1 is present within the lipid raft fraction of the plasma membrane. As shown in Figure 2D, when a PM enriched fraction was prepared [27], both the PM marker protein Transferrin Receptor 1 and endogenous SphK1 are enriched in fraction 4. The PM enriched fraction 4 was collected and subjected to our NaCl method to determine whether SphK1 is present within the lipid raft microdomain of the PM. As shown in Figures 2E and F, endogenous SphK1 protein is detected in the PMLRM fraction (fraction 4) isolated from both HEK293 and HELA cells using the NaCl method. Importantly, the presence of SphK1 in the PMLRM was further confirmed by detection of strong SphK1 activity in the PMLRM (fraction 4) of both cell lines by TLC (Figures 2E and 2F).
To provide further evidence supporting the PMLRM localization of human SphK1, we next examined whether the PMLRM localization of SphK1 was dependent on cholesterol. Cholesterol chelating agents, such as methyl-β-cyclodextrin (MβCD) and filipin, disrupt lipid rafts by binding and removing cholesterol from the PMLRM and make a typically Triton X-100 resistant membrane protein (i.e. SphK1) amenable to solubilization [18]. To determine the effects of these agents on the PMLRM targeting of SphK1, we treated HEK293 cells with or without MβCD and filipin and isolated both lipid raft fractions (as indicated by the presence of caveolin-1) and non-raft fractions. As shown in Figure 2G, pretreatment of HEK293 cells stably expressing WT His6×-SphK1 with either MβCD or filipin allowed the solubilization and hence redistribution of WT His6×-SphK1 from the raft fractions to the non-raft fractions. Importantly, this data demonstrates that the presence of SphK1 within the PMLRM is dependent on cholesterol and further confirms that SphK1 is specifically localized to the PMLRM.
Comparison of the SphK1 fraction resistant to salt, pH and detergent
The data presented above indicate that SphK1 protein exists in at least three separate intracellular locales. Clearly, by all methods employed, the vast majority of both endogenous and over-expressed recombinant SphK1 is localized to the cytosolic fraction. The results from the subcellular fractionation methods imply that SphK1 also associates with intracellular membranes and the PM, while the Triton X-100 solubility data indicates that SphK1 resides in both Triton X-100 soluble and insoluble fractions. Furthermore, we have shown that PM localized SphK1 can be subdivided by ND-SDG into PMLRM fractions and non-raft fractions.
We next examined whether the SphK1 fractions resistant to extraction from the membrane by 1M NaCl and Na2CO3 pH 11.5 are equivalent in nature to the TI fraction of SphK1. If these three fractions are equivalent, it may indicate that they represent a single intracellular locale. Whereas if each fraction were separately isolated by each treatment, it could indicate that they each represent a distinct intracellular locale. Using the method detailed in Figure 3A, we isolated the post-nuclear supernatant (PNS) from His6×-SphK1 stably expressed in HEK293 cells and separated the cytosolic fraction (Sample 1) from the total membrane pellet by ultracentrifugation. The total membrane pellet was then divided and resuspended by sonication in either high salt (25 mM MES, 1M NaCl, pH 6.5) or high pH (25 mM MES, 500 mM Na2CO3, pH 11.5) buffer. The SphK1 fractions dislodged from the membrane by high salt and high pH (Samples 2 and 3 respectively) were again separated from the total membrane pellets by ultracentrifugation. To determine whether the SphK1 dislodged from the total membrane fractions by 1M NaCl or high pH (Samples 2 and 3) are equivalent to or distinct from the TS fraction (Figure 1B), we next resuspended the total membrane pellets in 1% Triton X-100 (at 4°C for 30 min) and once again isolated the total membrane pellet by ultracentrifugation. Triton X-100 treatment of the high salt and high pH treated membrane fractions did not appreciably solubilize any SphK1 protein (Samples 4 and 5). There is some SphK1 present in Sample 4 which is likely 1M NaCl dislodged SphK1 protein (Sample 2) that is trapped in outside-in vesicles until these vesicles are disrupted by the 1% Triton X-100 treatment. We did not observe a “trapped” fraction of SphK1 in the Na2CO3 treated sample (Sample 5) due to the propensity of high pH to create membrane sheets rather than membrane vesicles [42]. After Triton X-100 treatment, equal amounts of SphK1 protein remained stably associated with both the high salt and high pH treated total membranes (Samples 6 and 7) indicating that the SphK1 fractions resistant to 1% Triton X-100, 1M NaCl and pH 11.5 treatment are equivalent. This data demonstrates that, by all three methods tested, membrane localized SphK1 can be subdivided into two distinct SphK1 fractions and that, after all three treatments, a fraction of SphK1 remains stably localized to the membrane.
Figure 3. Fractionation of SphK1 by salt, ph and detergent.
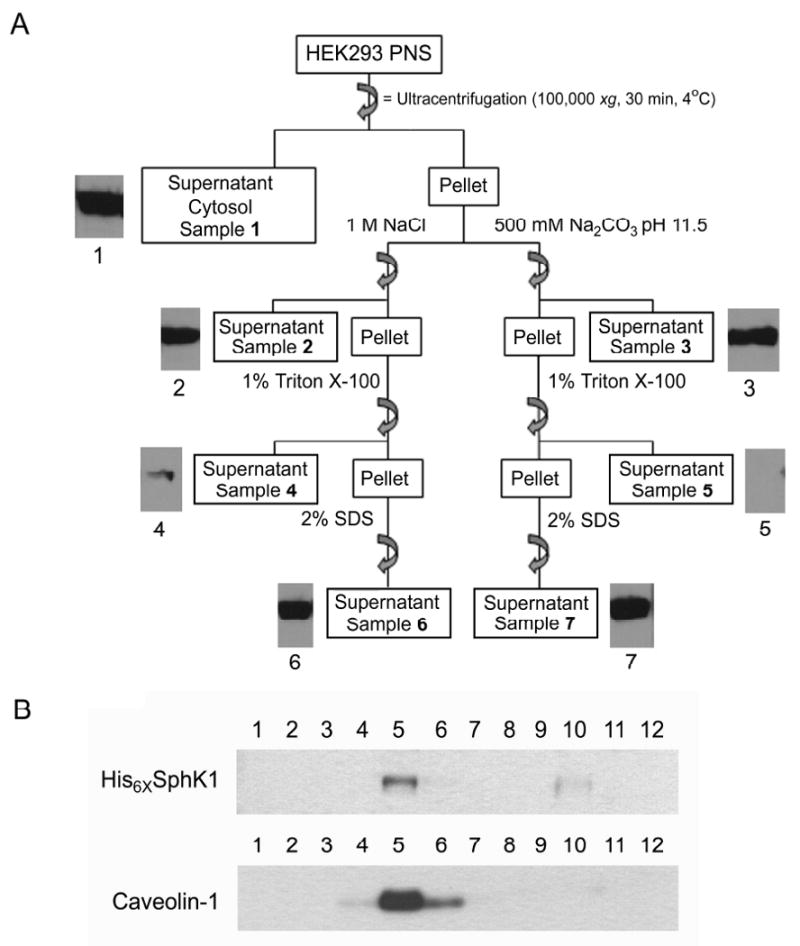
(A) Schematic representation and Western blot analysis of separation of SphK1 into its intracellular populations. His6×SphK1 protein, detected by Western blot analysis using anti-His6× antibodies, is present in the cytosolic (Sample 1), membrane associated (Samples 2 and 3) and membrane integrated (Samples 6 and 7) fractions of HEK293 cells. Both 1M NaCl and Na2CO3 pH 11.5 (Samples 2 and 3 respectively) were able to dislodge the cellular membrane associated fraction and 1% Triton X-100 did not further release SphK1 protein from cellular membranes (Samples 4 and 5; n=4). (B) A HEK293 cell post-nuclear supernatant stably expressing His6×-SphK1 was prepared in the presence 1M NaCl to dislodge the membrane associated fraction of SphK1 from the membrane. Total cell membranes were isolated by centrifugation at 100,000 xg for 30 min at 4°C and the cytoplasmic and membrane associated fractions (supernatant) were discarded. The resulting membrane pellet was resuspended in 25 mM MES + 1M NaCl pH 6.5, was adjusted to 45% sucrose as described in the Materials and Methods and applied to a 1M NaCl containing sucrose density gradient. The presence of His6×-SphK1 and caveolin-1 in 25 μL samples of each fraction of the 1M NaCl containing ND-SDG were analyzed by Western blot (n=3).
The salt resistant fraction of SphK1 exclusively localizes to the PMLRM
We next wanted to determine whether this 1% Triton X-100, 1M NaCl and pH 11.5 resistant fraction of SphK1 was equivalent to the PMLRM localized fraction of SphK1. To clarify this point, a post-nuclear supernatant of HEK293 cells stably expressing His6×-SphK1 was treated with 1M NaCl and the total membrane fraction was isolated by ultracentrifugation after this treatment. This treatment would effectively remove the cytosolic and 1M NaCl dissociated SphK1 protein from the total membrane fraction leaving only the 1M NaCl resistant SphK1 fraction present in the membrane preparation. We isolated the PM fraction from this salt treated total membrane fraction, fractionated it by our NaCl method and analyzed the resulting fractions for the presence of His6×-SphK1 protein. As shown in Figure 3B, both SphK1 protein and the marker caveolin-1 are detected in the lipid fractions (fraction 5) of the SDG indicating that the 1M NaCl resistant SphK1 fraction is localized to the PMLRM. There is a small amount of SphK1 detected in fraction 10 which likely represents SphK1 protein dislodged by 1M NaCl that is trapped in outside-in vesicles as was observed in Figure 3A above.
Relocalization of His6×SphK1 in response to PMA treatment
Taken together, the data presented above indicate that the PMLRM localization of SphK1 can be examined without the need to employ ND-SDG centrifugation techniques. We, therefore, developed a simplified “pool” assay (Figure 4A) whereby SphK1 is separated into the cytosolic fraction (Pool 1), 1M NaCl dissociated membrane fraction (Pool 2) and the PMLRM fraction (Pool 3). This “pool” assay method was subsequently employed to examine the elements of SphK1 responsible for localization to the PMLRM and the effect of PMA stimulation on the localization of SphK1 to the PMLRM.
Figure 4. Site-directed mutagenesis of the phosphatidylserine binding residues of His6×SphK1 blocks lipid raft localization.
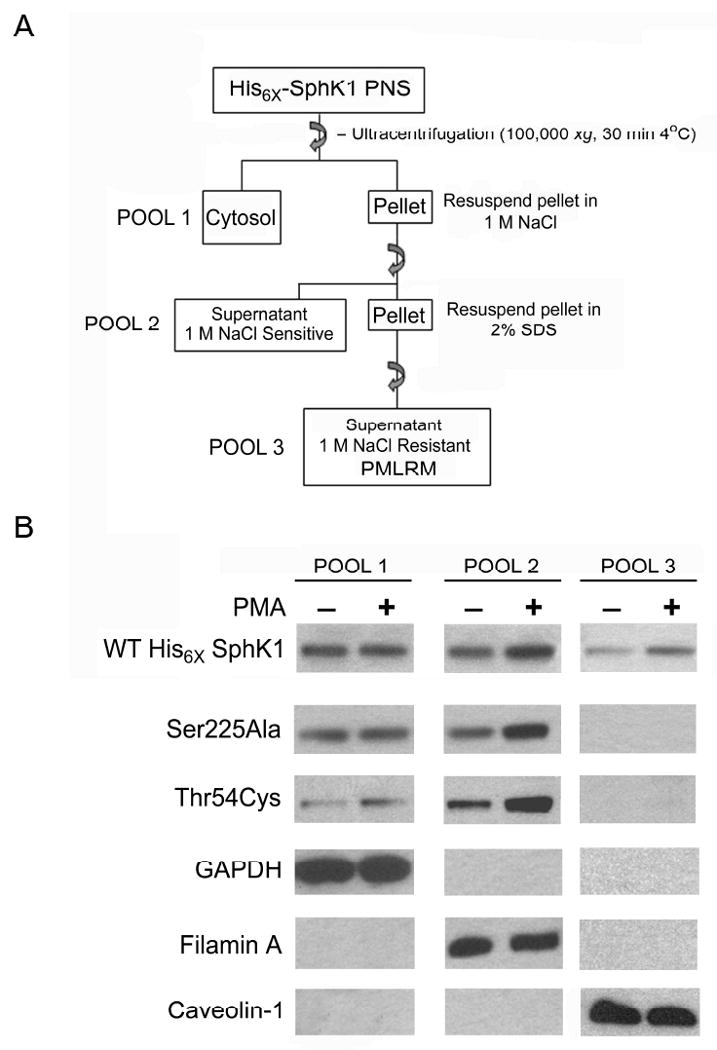
Pool assays were performed on WT His6×SphK1 and the indicated site-directed mutants of SphK1 that were previously shown to block phosphatidylserine binding [51]. Western blot analyses, using anti-His6×, anti-GAPDH, anti-Filamin A, and anti caveolin-1 antibodies, were performed on 5 μg of total protein from each subcellular pool in the absence (-) or presence (+) of PMA treatment (n=3).
By definition, lipid rafts are small heterogeneous domains that can be stabilized to form larger signaling platforms in response to agonists. As stated earlier, the current model for SphK1 activation involves agonist-induced translocation of basally active SphK1 from the cytoplasm to the PM, where it achieves full catalytic activity. One such agonist, PMA, has been shown by both biochemical and immunofluorescent techniques, to induce the relocalization of SphK1 from the cytosol to the PM [38,43]. Having established that SphK1 localizes to two distinct membrane fractions, we next examined the intracellular distribution of WT His6×SphK1 before and after PMA stimulation using our “pool” assay method as outlined in Figure 4A. As shown in Figure 4B, PMA stimulation induces the relocalization of WT His6×SphK1 to both Pool 2 and Pool 3. The above data validates the use of the “pool” assay system to assess the PMLRM localization of SphK1 in response to PMA stimulation.
Previous studies have demonstrated that mutation of amino acids (Thr54, Ser225) shown to mediate the SphK1-phosphatidylserine (PS) interaction affected membrane localization of SphK1 [36,43]. We next examined whether mutation of these residues affected localization of SphK1 to either Pool 2 or Pool 3 using the “pool” assay method. As shown in Figure 4B, Ser 225 mutated to Ala (Ser225Ala) and Thr 54 mutated to Cys (Thr54Cys) abrogated relocalization of the mutant SphK1 proteins to Pool 3, whereas these mutations had no effect on localization to Pool 2 in the absence of PMA treatment. In fact, PMA stimulation induced the accumulation of SphK1 protein in Pool 2 for both the Ser225Ala, and Thr54Cys mutants. Based on the distribution of marker proteins for each pool (Pool 1: GAPDH; Pool 2: Filamin A; Pool 3: Caveolin-1), these results indicate that agonist stimulation increases SphK1 protein localization to the PMLRM in a PS binding dependent manner, whereas agonist enhanced association of SphK1 protein with Pool 2 is independent of PS binding.
Confocal microscopic localization of SphK1 to the plasma membrane lipid raft microdomain
To further examine the PMLRM localization of WT SphK1 and the SphK1 mutants, we generated GFP-tagged fusion constructs and examined the subcellular localization of the GFP-SphK1 fusion proteins using confocal microscopy. In Figure 5, we clearly demonstrate that WT GFP-SphK1 colocalizes with the PMLRM marker, GM-1 (as shown by regions of yellow colocalization), further indicating that SphK1 is localized to the PMLRM. Mutation of PS binding residues (Thr54Cys and Ser225Ala) blocked translocation to the PMLRM as evidenced by the lack of colocalization with the PMLRM marker, GM-1. These data are consistent with previously published results for a Thr54Ala mutant, and the Ser225Ala mutant [43].
Figure 5. Representative confocal images of SphK1 protein and its mutants expressed in HEK293 cells.
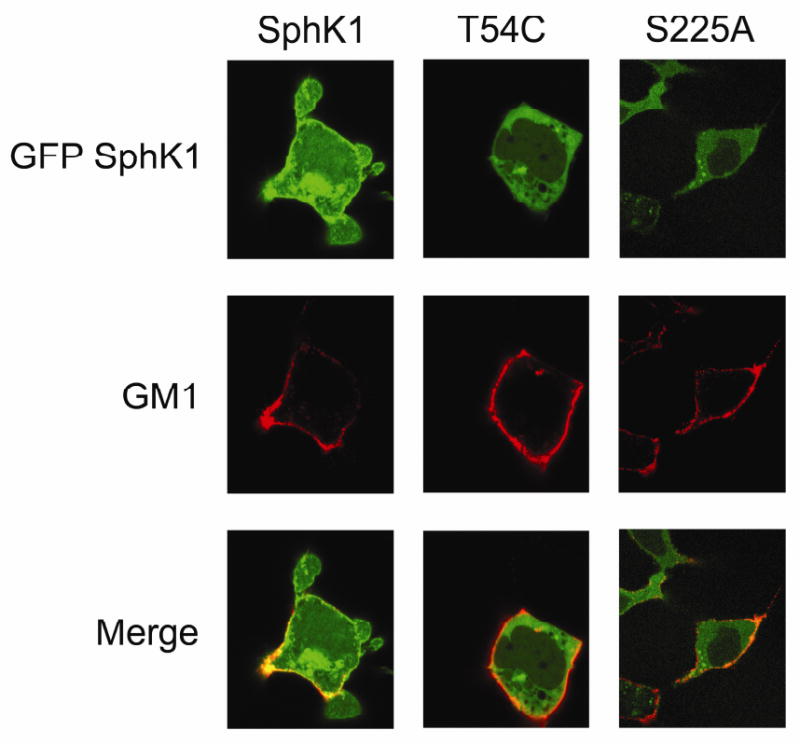
Confocal imaging of GFP-fused WT SphK1, and the site-directed mutants T54C, and S225A was performed. Colocalization of GFP-SphK1 constructs (green) and the lipid raft marker GM-1 (red) are indicated as yellow regions along the PM. All images were collected and processed identically.
LC/MS/MS analysis of sphingolipid profiles in SphK1 overexpressing HEK293 cells
Several studies have demonstrated that agonist induced relocalization of SphK1 to the PM correlates with increased production of S1P [9-12]. To determine whether the PMLRM is the site of action for S1P generation mediated by SphK1, we next examined whether the PMLRM contains the substrate for SphK1 (D-erythro sphingosine), whether PMA induces S1P formation, and whether blockage of SphK1 localization to the PMLRM affects the generation of S1P in response to PMA. To specifically address these questions, we examined the ND-SDG fractions of HEK293 cells for the presence of D-erythro-sphingosine and S1P using LC/MS/MS analyses. A 12 mL ND-SDG was divided into 6 - 2 mL fractions, such that fractions 2 and 3 represent the PMLRM, and LC/MS/MS analysis of the fractions was conducted. As shown in Figure 6A, both D-erythro-sphingosine (Sph) and S1P are predominantly detected in fractions 2 and 3 of the ND-SDG. Since relatively low levels of S1P are generated by SphK1 at the expense of Sph, we normalized the formation of S1P to the amount of Sph present in each fraction (S1P/Sph). We next examined whether PMLRM localization was required for the formation of S1P, in response to PMA. S1P formation was calculated for fractions 2 and 3 since these fractions represent the PMLRM fractions. Upon PMA treatment, we observed a significant increase in the formation of S1P within the PMLRM in WT SphK1 overexpressing HEK293 cells (p<0.05; Figure 6B). In contrast, no increase in S1P formation upon PMA treatment was observed in cells expressing the Thr54Cys mutant and the Ser225Ala mutant. These results demonstrate that SphK1 localizes to the PMLRM and generates S1P in response to PMA treatment in HEK293 cells indicating that the PMLRM is one of the functional active sites of SphK1.
Figure 6. D-erythro-sphingosine and S1P distribution in the lipid raft fraction and changes in sphingolipid levels in response to PMA.
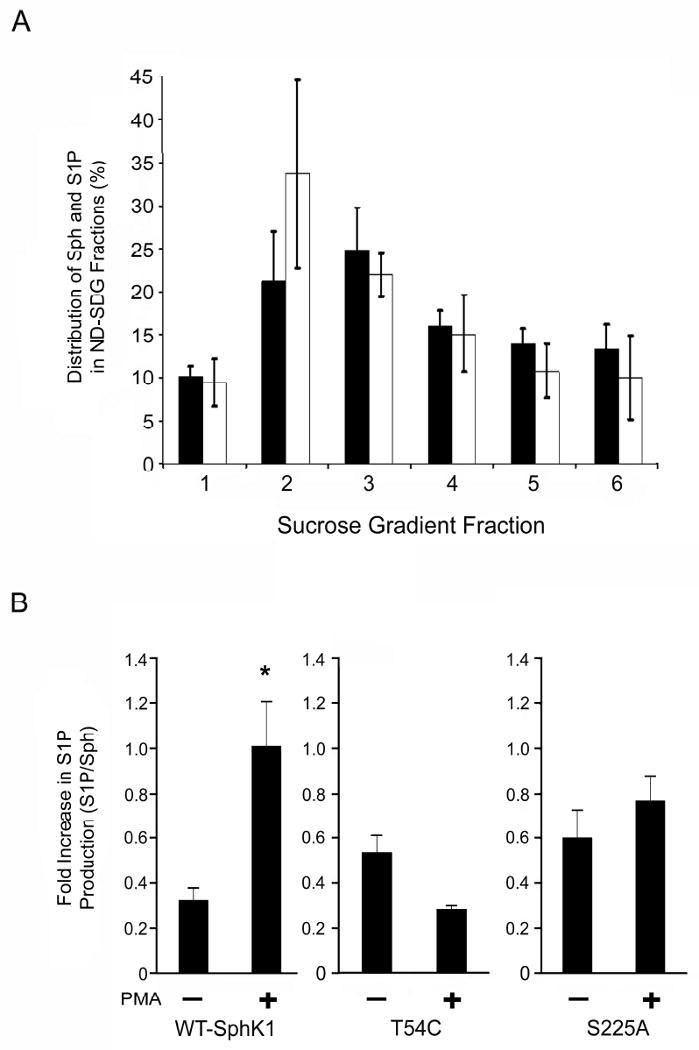
(A) The majority of the intracellular D-erythro-sphingosine and S1P are present in the PMLRM fractions of the ND-SSDG prepared from HEK293 cells. The percent distribution of sphingosine (black bars) and S1P (white bars) were determined by LC/MS/MS analysis (n=3). (B) S1P accumulates in the PMLRM upon PMA stimulation. The S1P/Sph ratio of fractions 2 and 3 were determined by LC/MS/MS analysis in the absence (-) and presence (+) of PMA as indicated (p<0.05; n=3).
Biological function of SphK1 targeting to Pool 3 in HEK293 cells
Our data indicate that one important function of SphK1 localization to the PMLRM is the generation of the pro-growth/pro-survival sphingolipid metabolite, S1P. Previous studies demonstrate that, among many, one important biological function of SphK1 is its ability to overcome serum deprivation induced growth inhibition. To determine whether localization of SphK1 to the PMLRM is required for the biologically functional role of overcoming cell growth inhibition under serum-deprived conditions (i.e. 0.5% FBS), we performed cell growth assays. HEK293 cells stably transfected with vector as well as all SphK1 expressing cell lines grew equally well in complete medium (5.0% FBS; data not shown). However, as shown in Figure 7, under serum deprived conditions (0.5% FBS) vector transfected cells as well as the Thr54Cys and Ser225Ala mutant cell lines did not grow over the 72 h time-course of the experiment. In contrast, WT SphK1 overexpressing HEK293 cells significantly grew over 48 h and 72 h (p<0.001). Together, this indicates that PMLRM localization of SphK1 is required for the biological role of SphK1 in overcoming serum deprivation induced cell growth inhibition.
Figure 7. Lipid raft localization increases survival under reduced serum conditions.
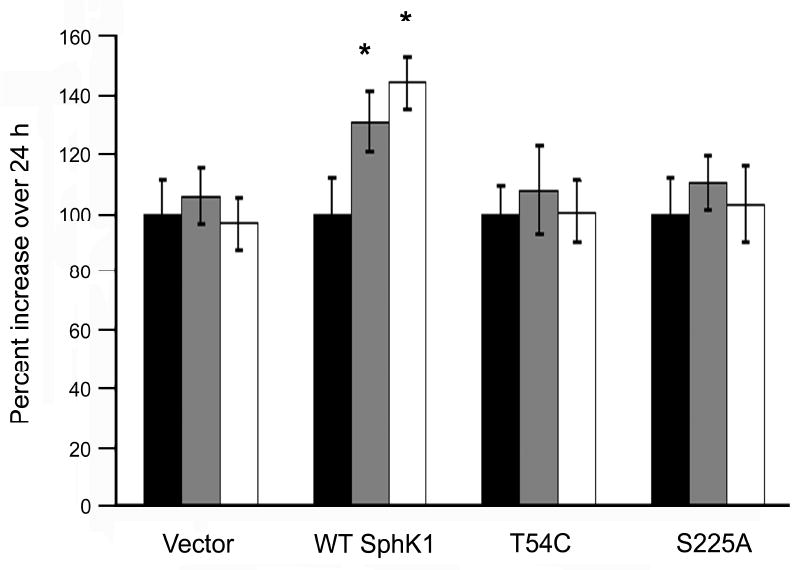
HEK293 cells were growth arrested for 24h in DMEM containing 0.5% FBS, medium was changed to fresh 0.5% FBS containing DMEM and grown for the times indicated. Survival of SphK1 transfected cells was calculated as the percent increase in SRB intensity over the 24 h time points: 24 h (black bars), 48 h (gray bars), and 72 h (white bars) (p<0.001; n=4).
Discussion
Numerous studies have examined the membrane localization of SphK1, by a number of different methods. In this report, we sought to unite these various reports to provide a better understanding of the membrane locales to which SphK1 translocates upon stimulation. Herein, we demonstrate that SphK1 exists as three distinct intracellular populations: a cytosolic fraction, a second population that is dissociated from the membrane by 1M NaCl that possibly associates with organellar or bulk plasma membranes hereafter referred to as the “membrane associated” fraction, and a third population that associates with the PMLRM. Furthermore, we provide evidence that PS binding is required for localization to the PMLRM. We also demonstrate that PMLRM association is required for S1P generation in response to PMA stimulation and for the growth/survival advantage under serum deprived conditions that is provided to cells over-expressing SphK1.
Clearly, SphK1 is predominantly a cytosolic protein accounting for >70% of the total SphK1 present in cells, while membrane localized SphK1 accounts for <30%. Many of the studies that have focused on the translocation of SphK1 from the cytosol to the membrane have employed Triton X-100 to solubilize SphK1 from the total membrane preparation and did not examine the localization of SphK1 to the TI membrane fraction [14,35-37]. In the few reports that examined the TI membrane fraction, SphK1 protein was detected, however these reports relied on epitope tagged over-expressed SphK1 raising the possibility that this localization was an artifact of the over-expression system [38]. We have for the first time detected endogenous SphK1 protein in the TI membrane fraction. We further demonstrate that, the ratio of cytosol:membrane associated:lipid raft associated SphK1 protein is similar in cells expressing endogenous SphK1 only and cells over-expressing recombinant SphK1 protein. While the levels of SphK1 protein are elevated in each fraction with over-expression, the fact that the ratios remain the same indicates that the binding sites for SphK1 in the membrane associated fraction and/or the PMLRM are not saturated under endogenous conditions.
Early studies of SphK biology, prior to the cloning of SphK1 and SphK2, attempted to biochemically determine how many SphK isoforms were present in the cell. Studies of rat tissue concluded that there were cytosolic and two membrane associated SphK activities which primarily colocalized with ER and PM markers [32]. While this study was not able to discriminate between SphK1 and SphK2, it is consistent with the results of our subcellular fractionation of endogenous and over-expressed SphK1. Studies on human platelets were consistent with the findings of Gijbers et al. [32] and also demonstrated that a membrane bound form of SphK1 could be dislodged from the membrane by 1M NaCl extraction [44]. This is again consistent with our observations. Interestingly, subcellular fractionation studies of SphK2 also indicate that this isoform exists in the cytosol, internal membranes (possibly ER) and plasma membrane fractions [40,45] indicating that multiple intracellular localizations may be a general feature of the SphKs. SphK2 has the additional, unique feature of nuclear localization mediated by a nuclear localization sequence while SphK1 is actively excluded from the nucleus by a nuclear export sequence [46,47].
The presence of SphK1 in the TI total membrane fraction was suggestive of a lipid raft microdomain localization. In light of the recent controversy surrounding the use of Triton X-100 in sucrose density gradient ultracentrifugation studies, we chose to employ non-detergent sucrose density gradient (ND-SDG) methods. For reasons stated in the results section above, we combined two ND-SDG methods to combine the advantages of each method. By employing this combined method, we were able to isolate a plasma membrane derived SphK1 fraction that colocalized with caveolae/lipid raft marker proteins and retained SphK1 catalytic activity. This observation is consistent with studies in IgE-Ag stimulated mouse bone marrow derived mast cells (BMMC) which indicate that mouse SphK1 interacts with Lyn and Fyn kinases, and that SphK1 activity co-immunoprecipitates with Lyn kinase isolated from the lipid raft fraction, however the cholesterol dependence of this localization was not examined [48,49]. The detection of SphK1 catalytic activity in the lipid raft fraction of two human cell lines (HEK293 and HELA) indicates that the localization of SphK1 to the lipid raft is not a species specific phenomenon nor is this localization unique to the IgE/Ag stimulation of BMMC cells. Additionally, we demonstrate that this localization occurred under both basal and PMA stimulated states and also demonstrated that this localization was dependent on cholesterol. These findings satisfy the 2 criteria to state with certainty that SphK1 is, indeed, a PMLRM resident protein. Using assay conditions that favored SphK2 catalytic activity while inhibiting SphK1 activity [40], we were also able to detect weak SphK2 activity in the lipid raft fraction of HELA cells (data not shown) indicating that at least a portion of the plasma membrane localized SphK2 associates with the PMLRM fraction.
To facilitate the study of amino acids responsible for localization of SphK1 to the lipid raft microdomain, we developed a rapid assay that effectively separates SphK1 into its three distinct intracellular fractions. Subsequently we demonstrated that after 1M NaCl treatment of total membrane fractions, the remaining membrane bound SphK1 localized to the raft fractions of a ND-SDG indicating that the 1M NaCl resistant SphK1 fraction is indeed the PMLRM associated SphK1 fraction. This assay system allowed us to rapidly assess the membrane localization of SphK1 with or without PMA treatment. It is important to note that this assay does not include Triton X-100 thereby eliminating the chance of mistaking detergent induced relocalization to the “detergent-resistant” membrane fraction for actual lipid raft localization. Additionally, because the over-expressed SphK1, whether epitope-tagged or not, had the same intracellular distribution as endogenous SphK1, we feel that it is appropriate to employ epitope-tagged SphK1 for future studies of the intracellular localization of SphK1.
By our assay method, we demonstrate that SphK1, upon PMA treatment, translocates from the cytosol to both the membrane associated fraction and to the PMLRM. Plasma membrane targeting of SphK1 has been reported to derive from its selectivity for PS, which activates SphK1 by increasing substrate access and membrane affinity [43]. It has been suggested that Ser225, Thr54 and Asn89 in the putative SphK1 membrane binding surface are necessary for lipid selectivity and membrane targeting of SphK1. PS, an allosteric activator of SphK1, is enriched in the inner leaflet of the PMLRM of activated cells [43,50], suggesting that SphK1 may be recruited to these domains through protein/lipid interactions. Consistent with this mechanism, we observed that mutation of Ser225 and Thr54 blocked basal and PMA induced localization to the PMLRM. However, these mutations did not affect the basal nor PMA stimulated relocalization of the “membrane associated” fraction of SphK1.
Whether the relocalization of SphK1 from the cytosol to the “membrane associated” and/or the PMLRM fractions occurs independently or in a processive manner remains to be determined. That the binding sites for membrane and/or PMLRM association are not saturated under endogenous or enforced over-expression conditions may indicate that post-translational modification is required to direct SphK1 to these membrane locales. A processive model for relocalization implies that SphK1 would require multiple post-translational modifications to coordinately regulate the translocation of SphK1 to its ultimate destination, the PMLRM fraction. Whereas independent relocalization to the membrane associated and PMLRM fractions implies independent functions in these two locales. A second possible explanation, for the lack of saturated membrane localization, would be that the increased SphK1 activity (in the SphK1 over-expressing cells) up-regulates or somehow activates the binding partners for SphK1 in these locales (i.e. proteins or phosphatidylserine) to accommodate the increased SphK1 localization. In this case, it is difficult to determine whether SphK1 localization to these membrane locales occurs independently or in a processive manner.
To clarify this issue, we must determine the mechanism by which the 1M NaCl sensitive membrane associated fraction of SphK1 interacts with the membrane and to which membranes this fraction associates. It is possible that this fraction represents an interaction of Sphk1 with the actin cytoskeleton generally, or Filamin A specifically, as previously reported [51] and this investigation is currently ongoing in our lab. It is of note that treatment of isolated membranes with Na2CO3 (pH 11.5) dissociates actin, and presumably Filamin A along with it, from membranes [41]. Our data demonstrating that Filamin A and the membrane associated fraction of SphK1 can be dissociated from the PM upon 1M NaCl treatment supports the hypothesis that this fraction represents an actin cytoskeleton interacting fraction of SphK1.
If this were the case, it supports the independent function model. SphK1 associating with the actin cytoskeleton (our membrane associated fraction) would mediate migration as indicated by Maceyka et al., [52] whereas SphK1 localized to the PMLRM fraction mediates the pro-growth/pro-survival/oncogenic functions of SphK1 as demonstrated by our data and others [35,53]. Independent functions also imply that these localizations are mutually exclusive (i.e. a cell that is migrating is likely not proliferating/surviving and vice versa). That we observed SphK1 in both of these membrane fractions in our pool assays could be a function of the fact that a cell culture dish provides a snap-shot of the steady-state localization of roughly 107 cells at any one time. It is not hard to imagine that some of these cells are migrating (hence membrane associated fraction localization) while others are dividing/proliferating or are under local stress conditions whereby they are forced to survive growth factor withdrawal (hence PMLRM localization).
Furthermore, a number of other mechanisms for membrane association may exist as well. For instance, a recent report has identified a mechanism for SphK1 membrane localization involving Gq family GPCRs, which occurs independently of Ser225 phosphorylation [54]. Additionally, the role of protein: protein interactions (i.e. Ca2+/calmodulin, Lyn and Fyn, etc) must be reconciled to our data. It has been presumed that these interactions provide the necessary anchor to mediate the attachment of SphK1 to the membrane. The fact that PS binding appears to be sufficient to mediate attachment of SphK1 to the PMLRM, suggests instead that the function of these interacting proteins may be to modulate SphK1 catalytic activity. It is also worthy of investigation to determine how PS binding so tightly tethers SphK1 to the PMLRM. While the possibility that additional modification(s) of SphK1 such as myristoylation or palmitoylation of SphK1, as described in a yeast homologue [55], cannot be excluded at this point, these effects would seem to be secondary to the association of SphK1 with PS. One possibility would be that PS binding induces a structural shift in SphK1 exposing sites for additional modification(s) that mediate lipid raft tethering.
Serine 225 phosphorylation, by the MAP kinases ERK1/2, is the only known post-translational modification of SphK1 identified to date [35,36]. Studies indicate that phosphorylation of Ser225 in response to PMA and TNFα leads to the relocalization of SphK1 from the cytosol to the PM, is necessary for SphK1 mediated oncogenic signaling, and increases the membrane penetrance of SphK1 allowing it to more tightly bind PS containing model membrane micelles [35,36,43]. Mutation of this residue to Alanine (Ser225Ala) blocked PM localization abrogating the oncogenic role of SphK1 overexpression [36]. Consistent with these findings, our data demonstrates that the Ser225Ala mutation abrogates PMLRM localization, whereas previous studies have shown that this mutation moderately affected localization to the plasma membrane. One possible explanation of this difference is that the authors only examined the TS membrane fraction and did not examine the TI membrane fraction [36]. Our data also suggests that the bulk of the effects mediated by Ser225 mutation occur via blockage of PMLRM localization. Consistent with this, when the Ser225Ala was recombinantly tagged with an Lck dual myristoylation motif [56], known to target proteins to the PMLRM, the oncogenic effects of the Ser225Ala mutant were restored [35].
The observation that the Ser225Ala mutation blocks the oncogenic effects of SphK1 over-expression further supports PM localization as a key factor for SphK1 function. Consistent with previous reports, we demonstrate that mutation of Ser225 affects agonist induced S1P production and growth/survival of cells under serum deprived conditions [35]. Supporting these observations, we also demonstrate that Thr54Cys mutation abrogates PMA stimulated S1P production and growth/survival under serum deprived conditions. Together, with our data demonstrating that these mutations block PMLRM association, these effects reinforce the hypothesis that localization of SphK1 is critical to its function. Furthermore, our data clearly implicate the PMLRM associated fraction of SphK1 as the key mediator of these functions.
Conclusions
Taken together, our data suggests that upon PMA stimulation, basally active cytosolic SphK1 is activated through PKC signaling [38,57,58] and ERK-dependent phosphorylation of SphK1 [59], which results in redistribution of SphK1 to the membrane associated and PMLRM fractions depending upon the intended fate of the cell (migration or proliferation/survival). Sphingomyelinases and ceramidases may also be activated in concert with SphK1 activation, resulting in generation of additional sphingosine at the membrane rafts [10,24,25]. Once at the PMLRM, SphK1 catalyzes the phosphorylation of sphingosine to form S1P, which functions intracellularly, resulting in growth, differentiation, and proliferative signaling. Alternatively, extracellularly released S1P can signal through S1PRs in an autocrine or paracrine fashion.
Acknowledgments
This study was funded by NIH grant CA 91155 and the Jake Gittlen Research Foundation.
Abbreviations
- SphK1
Sphingosine Kinase 1
- Sph
Sphingosine
- S1P
Sphingosine-1-phosphate
- PM
plasma membrane
- PS
phosphatidylserine
- TS
Triton X-100 soluble membrane fraction
- TI
Triton X-100 insoluble membrane fraction
- PMLRM
plasma membrane lipid raft microdomain
- ND-SDG
non-detergent sucrose density gradient
- MβCD
methyl-β-cyclodextrin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Argraves KM, Obeid LM, Hannun YA. Adv Exp Med Biol. 2002;507:439–44. doi: 10.1007/978-1-4615-0193-0_68. [DOI] [PubMed] [Google Scholar]
- 2.Cuvillier O. Biochim Biophys Acta. 2002;1585:153–62. doi: 10.1016/s1388-1981(02)00336-0. [DOI] [PubMed] [Google Scholar]
- 3.Maceyka M, Payne SG, Milstien S, Spiegel S. Biochim Biophys Acta. 2002;1585:193–201. doi: 10.1016/s1388-1981(02)00341-4. [DOI] [PubMed] [Google Scholar]
- 4.Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Biochim Biophys Acta. 2006;1758:2016–26. doi: 10.1016/j.bbamem.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Maceyka M, Milstien S, Spiegel S. Prostaglandins Other Lipid Mediat. 2005;77:15–22. doi: 10.1016/j.prostaglandins.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Luberto C, Kraveka JM, Hannun YA. Neurochem Res. 2002;27:609–17. doi: 10.1023/a:1020267831851. [DOI] [PubMed] [Google Scholar]
- 7.Maceyka M, Milstien S, Spiegel S. J Lipid Res. 2009;5(Suppl):S272–6. doi: 10.1194/jlr.R800065-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez SE, Milstien S, Spiegel S. Trends Endocrinol Metab. 2007;18:300–7. doi: 10.1016/j.tem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Xia P, Wang L, Gamble JR, Vadas MA. J Biol Chem. 1999;274:34499–505. doi: 10.1074/jbc.274.48.34499. [DOI] [PubMed] [Google Scholar]
- 10.Olivera A, Spiegel S. Nature. 1993;365:557–60. doi: 10.1038/365557a0. [DOI] [PubMed] [Google Scholar]
- 11.Rius RA, Edsall LC, Spiegel S. FEBS Lett. 1997;417:173–6. doi: 10.1016/s0014-5793(97)01277-5. [DOI] [PubMed] [Google Scholar]
- 12.Mazurek N, Megidish T, Hakomori S, Igarashi Y. Biochem Biophys Res Commun. 1994;198:1–9. doi: 10.1006/bbrc.1994.1001. [DOI] [PubMed] [Google Scholar]
- 13.Delon C, Manifava M, Wood E, Thompson D, Krugmann S, Pyne S, Ktistakis NT. J Biol Chem. 2004;279:44763–74. doi: 10.1074/jbc.M405771200. [DOI] [PubMed] [Google Scholar]
- 14.Sutherland CM, Moretti PA, Hewitt NM, Bagley CJ, Vadas MA, Pitson SM. J Biol Chem. 2006;281:11693–701. doi: 10.1074/jbc.M601042200. [DOI] [PubMed] [Google Scholar]
- 15.Olivera A, Rosenthal J, Spiegel S. J Cell Biochem. 1996;60:529–37. doi: 10.1002/(sici)1097-4644(19960315)60:4<529::aid-jcb9>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 16.Alemany R, van Koppen CJ, Danneberg K, Ter Braak M, Meyer Zu Heringdorf D. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:413–28. doi: 10.1007/s00210-007-0132-3. [DOI] [PubMed] [Google Scholar]
- 17.Taha TA, Hannun YA, Obeid LM. J Biochem Mol Biol. 2006;39:113–31. doi: 10.5483/bmbrep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- 18.Pike LJ. J Lipid Res. 2009:S323–S328. doi: 10.1194/jlr.R800040-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng ZJ, Singh RD, Marks DL, Pagano RE. Mol Membr Biol. 2006;23:101–10. doi: 10.1080/09687860500460041. [DOI] [PubMed] [Google Scholar]
- 20.Pike LJ. Biochem J. 2004;378:281–92. doi: 10.1042/BJ20031672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simons K, Toomre D. Nat Rev Mol Cell Biol. 2000;1:31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 22.Mishra S, Joshi PG. J Neurochem. 2007;103 1:135–42. doi: 10.1111/j.1471-4159.2007.04720.x. [DOI] [PubMed] [Google Scholar]
- 23.Silva LC, Futerman AH, Prieto M. Biophys J. 2009;96:3210–22. doi: 10.1016/j.bpj.2008.12.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auge N, Nikolova-Karakashian M, Carpentier S, Parthasarathy S, Negre-Salvayre A, Salvayre R, Merrill AH, Jr, Levade T. J Biol Chem. 1999;274:21533–8. doi: 10.1074/jbc.274.31.21533. [DOI] [PubMed] [Google Scholar]
- 25.Merrill AH, Jr, et al. Toxicol Appl Pharmacol. 1997;142:208–25. doi: 10.1006/taap.1996.8029. [DOI] [PubMed] [Google Scholar]
- 26.Pike LJ. J Lipid Res. 2006;47:1597–8. doi: 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Smart EJ, Ying YS, Mineo C, Anderson RG. Proc Natl Acad Sci U S A. 1995;92:10104–8. doi: 10.1073/pnas.92.22.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kralik SF, Du X, Patel C, Walsh JP. Anal Biochem. 2001;294:190–3. doi: 10.1006/abio.2001.5166. [DOI] [PubMed] [Google Scholar]
- 29.Sullards MC, Merrill AH., Jr Sci STKE. 2001;2001:PL1. doi: 10.1126/stke.2001.67.pl1. [DOI] [PubMed] [Google Scholar]
- 30.Vichai V, Kirtikara K. Nat Protoc. 2006;1:1112–6. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 31.Safadi-Chamberlain F, et al. Biochem J. 2005;388:827–34. doi: 10.1042/BJ20041726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gijsbers S, Van der Hoeven G, Van Veldhoven PP. Biochim Biophys Acta. 2001;1532:37–50. doi: 10.1016/s1388-1981(01)00111-1. [DOI] [PubMed] [Google Scholar]
- 33.Taha TA, Kitatani K, Bielawski J, Cho W, Hannun YA, Obeid LM. J Biol Chem. 2005;280:17196–202. doi: 10.1074/jbc.M413744200. [DOI] [PubMed] [Google Scholar]
- 34.David L Spector, R.D.G., Leslie A Leinwand (1998), Vol. 1 Cold Spring Harbor Press.
- 35.Pitson SM, Xia P, Leclercq TM, Moretti PA, Zebol JR, Lynn HE, Wattenberg BW, Vadas MA. J Exp Med. 2005;201:49–54. doi: 10.1084/jem.20040559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pitson SM, Moretti PA, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW. Embo J. 2003;22:5491–500. doi: 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kihara A, Anada Y, Igarashi Y. J Biol Chem. 2006;281:4532–9. doi: 10.1074/jbc.M510308200. [DOI] [PubMed] [Google Scholar]
- 38.Johnson KR, Becker KP, Facchinetti MM, Hannun YA, Obeid LM. J Biol Chem. 2002;277:35257–62. doi: 10.1074/jbc.M203033200. [DOI] [PubMed] [Google Scholar]
- 39.Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP. J Biol Chem. 1996;271:9690–7. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- 40.Liu H, et al. J Biol Chem. 2000;275:19513–20. doi: 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- 41.Nebl T, Pestonjamasp KN, Leszyk JD, Crowley JL, Oh SW, Luna EJ. J Biol Chem. 2002;277:43399–409. doi: 10.1074/jbc.M205386200. [DOI] [PubMed] [Google Scholar]
- 42.Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. J Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stahelin RV, Hwang JH, Kim JH, Park ZY, Johnson KR, Obeid LM, Cho W. J Biol Chem. 2005;280:43030–8. doi: 10.1074/jbc.M507574200. [DOI] [PubMed] [Google Scholar]
- 44.Banno Y, Kato M, Hara A, Nozawa Y. Biochem J. 1998;335(Pt 2):301–4. doi: 10.1042/bj3350301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maceyka M, et al. J Biol Chem. 2005;280:37118–29. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- 46.Inagaki Y, Li PY, Wada A, Mitsutake S, Igarashi Y. Biochem Biophys Res Commun. 2003;311:168–73. doi: 10.1016/j.bbrc.2003.09.194. [DOI] [PubMed] [Google Scholar]
- 47.Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S. J Biol Chem. 2003;278:46832–9. doi: 10.1074/jbc.M306577200. [DOI] [PubMed] [Google Scholar]
- 48.Urtz N, et al. Mol Cell Biol. 2004;24:8765–77. doi: 10.1128/MCB.24.19.8765-8777.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olivera A, et al. J Biol Chem. 2006;281:2515–25. doi: 10.1074/jbc.M508931200. [DOI] [PubMed] [Google Scholar]
- 50.Pike LJ, Han X, Chung KN, Gross RW. Biochemistry. 2002;41:2075–88. doi: 10.1021/bi0156557. [DOI] [PubMed] [Google Scholar]
- 51.Kusner DJ, Thompson CR, Melrose NA, Pitson SM, Obeid LM, Iyer SS. J Biol Chem. 2007;282:23147–62. doi: 10.1074/jbc.M700193200. [DOI] [PubMed] [Google Scholar]
- 52.Maceyka M, Alvarez SE, Milstien S, Spiegel S. Mol Cell Biol. 2008;28:5687–97. doi: 10.1128/MCB.00465-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia P, Gamble JR, Wang L, Pitson SM, Moretti PA, Wattenberg BW, D'Andrea RJ, Vadas MA. Curr Biol. 2000;10:1527–30. doi: 10.1016/s0960-9822(00)00834-4. [DOI] [PubMed] [Google Scholar]
- 54.Ter Braak M, et al. Biochim Biophys Acta 2009 [Google Scholar]
- 55.Nagiec MM, Skrzypek M, Nagiec EE, Lester RL, Dickson RC. J Biol Chem. 1998;273:19437–42. doi: 10.1074/jbc.273.31.19437. [DOI] [PubMed] [Google Scholar]
- 56.Zlatkine P, Mehul B, Magee AI. J Cell Sci. 1997;110(Pt 5):673–9. doi: 10.1242/jcs.110.5.673. [DOI] [PubMed] [Google Scholar]
- 57.Shu X, Wu W, Mosteller RD, Broek D. Mol Cell Biol. 2002;22:7758–68. doi: 10.1128/MCB.22.22.7758-7768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jin ZQ, Karliner JS. Cardiovasc Res. 2006;71:725–34. doi: 10.1016/j.cardiores.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 59.Pitson SM, Moretti PA, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW. Embo J. 2003;22:5491–500. doi: 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]