

Abstract
Rat astrocyte function is changed by diabetes mellitus relative to the nondiabetic state and we believe that altered function contributes to the central nervous system symptoms manifested by individuals with diabetes. We report here a comparison of astrocyte glutamate uptake and GFAP expression in streptozotocin-induced type 1 diabetic rats and insulin treated diabetic rats at four and eight weeks following diabetes onset. In glial plasmalemmal vesicle (GPV) preparations from treated rats, insulin prevented the increase observed in untreated, diabetic rats of both sodium-dependent and sodium-independent glutamate uptake. We determined by immunoblotting and immunohistochemistry that insulin treatment prevented the decrease of GFAP expression detected in the cerebral cortex, hippocampus, and cerebellum of untreated, diabetic rats. These observations indicate that insulin effects on astrocyte function are significant in managing diabetes-induced central nervous system pathology.
Keywords: astrocyte, diabetes, insulin, GFAP, glutamate
1. Introduction
Type 1 and type 2 diabetes mellitus result in insulin deficiency and/or insulin resistance, hyperglycemia and altered lipid, carbohydrate and protein metabolism (McNeill, 1999). Both types of diabetes affect the central nervous system (CNS) and are associated with increased incidence of seizure, stroke, dementia, and cognitive impairment (Biessels et al., 2006, 2008; Ott et al., 1999; Tiehuis et al., 2008; Whiting et al., 1997). These pathologies emerge from changes in cerebral metabolism, vascular reactivity and increased oxidative stress (Horani and Mooradian, 2003; Mankovsky et al., 1996; Manschot et al., 2007; Valko et al., 2007). Astrocytes play a role in energy metabolism, maintenance of the blood-brain barrier, vascular reactivity, regulation of extracellular glutamate levels, and protection from reactive oxygen species among other functions (Dringen et al., 2000; Tsacopoulos and Magistretti, 1996; Zonta et al., 2003). These observations led us to hypothesize that altered astrocyte activity contributes to the CNS pathophysiology associated with diabetes.
Our laboratory and others demonstrated a decrease in astrocyte glial fibrillary acidic protein (GFAP) expression in type 1 diabetic rats (Afsari et al., 2008; Barber et al. 2000; Coleman et al., 2004; Dennis et al., 2005; Lechuga-Sancho et al., 2006). Decreases in GFAP expression are associated with detrimental conditions in the CNS (Pekny and Pekna, 2004), several of which occur in individuals with diabetes. Poor white matter vascularization, disruptions in the blood-brain barrier (Bouchard et al., 2002; Chehade et al., 2002; Hawkins et al., 2007; Horani and Mooradian, 2003; Huber, 2008; Liedtke et al., 1996), and changes in long term potentiation (LTP) (Kamal et al., 2000; McCall et al., 1996) are reported in GFAP knock-out mice and in diabetic individuals. These observations suggest a link between decreases in GFAP and diabetes-induced CNS complications.
Changes in astrocyte function may also contribute to oxidative damage associated with diabetes (Bhardwaj, et al., 1999; Mastrocola et al., 2005; Rösen et al., 2001). Astrocytes are an integral component of the antioxidant defense system in the brain through the regulation of extracellular glutamate concentrations and production of antioxidant compounds (Dringen et al., 2000; Hertz and Zielke, 2004; Trendelenburg and Dirnagl, 2005; Wilson, 1997). A significant increase in oxidative stress, which causes tissue damage, is symptomatic of diabetes (Mastrocola et al., 2005; Rösen et al., 2001). Elevated glutamate levels were reported in diabetic individuals and increased glutamate concentrations contribute to the production of reactive oxygen species (Ambati et al., 1997; Hansson et al., 2000; Hudspith, 1997; Lieth et al., 1998).
Astrocyte regulation of extracellular glutamate occurs by both sodium-dependent and sodium-independent uptake mechanisms (Danbolt, 2001). Five high affinity, sodium-dependent glutamate transporters have been cloned: GLAST/EAAT1, GLT-1/EAAT2, EAAC1/EAAT3, EAAT4, and EAAT5 (Danbolt, 2001; Gegelashvili et al., 1998; Pines et al., 1992; Rothstein et al., 1994; Storck et al., 1992). The glutamate transporters, GLAST and GLT-1, are located primarily on astrocytes and are critical in maintaining extracellular glutamate at safe levels (Danbolt, 2001). Sodium-independent glutamate uptake by the xc- transporter of astrocytes is linked directly to a glutamate-cystine exchanger and production of the important antioxidant, glutathione (Dringen et al., 2000; La Bella et al., 2007; McBean 2002; Sato et al., 2002; Shih et al., 2006). Altered activity of the xc- transporter can result in changes in glutathione levels and the antioxidant defense system of the brain (Dringen et al., 2000; Shih et al., 2006).
We report here the effects of insulin treatment on astrocyte GFAP expression and glutamate uptake in rats with type 1 diabetes. Type 1 diabetes was induced by the administration of streptozotocin (STZ), a compound that selectively destroys the islet cells of the pancreas (McNeill, 1999) and is used extensively in diabetes research (Asnaghi et al., 2003; Bouchard et al., 2002; Coleman et al., 2004; McNeill, 1999). We focused on the cerebral cortex, hippocampus, and cerebellum because we observed decreased GFAP expression in these regions after 4 and 8 weeks of diabetes (Coleman et al., 2004). The cerebral cortex and hippocampus play important roles in cognitive behavior, seizure activity and contain high levels of the glutamate transporter, GLT-1 (Lehre et al., 1995). The cerebellum contains high levels of the glutamate transporter, GLAST (Lehre et al., 1995).
2. Results
2.1 Diabetic Model
Over 8 weeks following the initial injection, STZ treated animals lost weight and had elevated blood glucose levels compared to untreated controls (Figure 1). These animals also had lower insulin levels, lower brain weights, and larger brain weight/body weight ratios at 4 and 8 weeks following diabetes induction (Table 1). Insulin treatment prevented the diabetes-induced body weight decrease and, for the most part, blocked the blood glucose increase over the 8 week time course (Figure 1). At three time points, 1, 6, and 7 weeks, the insulin treated animals had glucose levels mildly elevated over those of the control group. Insulin treatment also prevented diabetes-induced decreases in blood insulin, brain weight, and altered brain weight/body weight ratio at 4 and 8 weeks (Table 1). In these animals receiving insulin treatment, insulin levels were higher at 8 weeks of diabetes than those in the untreated control group (Table 1).
Figure 1.
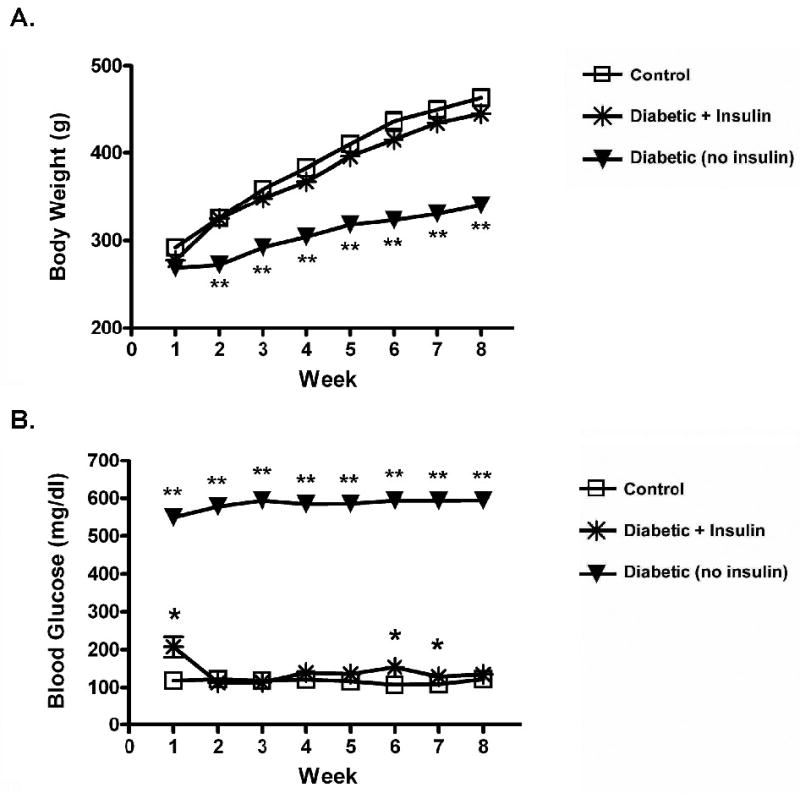
Insulin effects on alterations in body weight (A) and blood glucose (B) for 8 weeks of diabetes. Values represent means ± SEM. Statistical significance is as follows: * p < 0.01 from Control; ** p < 0.001 from Control and Diabetic + Insulin. (Control: n = 24; Diabetic + Insulin: n = 24; Diabetic/no insulin: n = 24). Similar results were seen in animals in the 4 week diabetes group.
Table 1. Diabetic Model.
Effects of insulin treatment on diabetes-induced changes in blood glucose, blood insulin (non-fasted values), body weight, brain weight and brain weight/body weight ratio. Insulin treatment reduced or prevented diabetes-induced changes in blood glucose, blood insulin, body weight, brain weight, and brain weight/body weight ratio at 4 weeks (Control: n = 13; Diabetic + Insulin: n = 13; Diabetic: n = 13) and 8 weeks (Control: n = 13; Diabetic + Insulin: n = 13; Diabetic: n = 13) of diabetes duration.
| Control | Diabetic + Insulin | Diabetic | |
|---|---|---|---|
| Blood Glucose (mg/dl) | |||
| 4 wks | 118.6 ± 1.6 | 137.4 ± 14.5 | 583.0 ± 9.6 a |
| 8 wks | 122.4 ± 2.3 | 128.2 ± 6.6 | 599.1 ± 0.9 a |
| Blood Insulin (ng/ml) | |||
| 4 wks | 2.86 ± 0.26 | 3.09 ± 0.31 | 0.24 ± 0.03 a |
| 8 wks | 2.54 ± 0.21 | 4.17 ± 0.47 b | 0.17 ± 0.02 a |
| Body Weight (g) | |||
| Initial * | 257 ± 5 | 258 ± 5 | 259 ± 6 |
| 4 wks | 405 ± 7 | 387 ± 9 | 299 ± 12 a |
| 8 wks | 463 ± 16 | 449 ± 10 | 332 ± 13 a |
| Brain Weight (g) | |||
| 4 wks | 1.957 ± 0.020 | 1.969 ± 0.016 | 1.836 ± 0.013 a |
| 8 wks | 2.007 ± 0.023 | 2.048 ± 0.014 | 1.825 ± 0.020 a |
| Brain Weight/Body Weight Ratio | |||
| 4 wks | 0.0048 ± 0.00008 | 0.0051 ± 0.0001 | 0.0063 ± 0.0003 a |
| 8 wks | 0.0044 ± 0.0001 | 0.0046 ± 0.0001 | 0.0056 ± 0.0002 a |
1The initial body weight includes the pooled data from both the 4 and 8 week groups (Control: n = 26; Diabetic + Insulin: n = 26; Diabetic: n = 26). Values represent means ± SEM of animals used in studies for protein determination and glutamate uptake
p < 0.01 from Control and Diabetic + Insulin;
p < 0.01 from Control.
2.2 Western blot analysis of GFAP, GLT-1 and GLAST
Immunoblot analysis showed that diabetes induced a decrease in GFAP expression, rescued by insulin treatment, in the cerebral cortex, hippocampus and cerebellum at 4 (graph not shown) and 8 weeks following onset (Figure 2: A, B, C). Compared to controls after 4 weeks of untreated diabetes, GFAP levels were 82.8 ± 5.1% in the cerebral cortex; 69.0 ± 8.1% in the hippocampus; and 73.1 ± 7.5% in the cerebellum. By 8 weeks of untreated diabetes, GFAP levels continued to decrease to 65.3 ± 12.5% in the cerebral cortex and 58.0 ± 6.9% in the cerebellum compared to controls. GFAP expression in the hippocampus at 8 weeks of diabetes remained unchanged at 69.9 ± 9.9% that of the control animals. In contrast, expression of the glutamate transporters GLT-1 and GLAST did not change in the brain regions evaluated (Figure 2: D-F; G-I).
Figure 2.
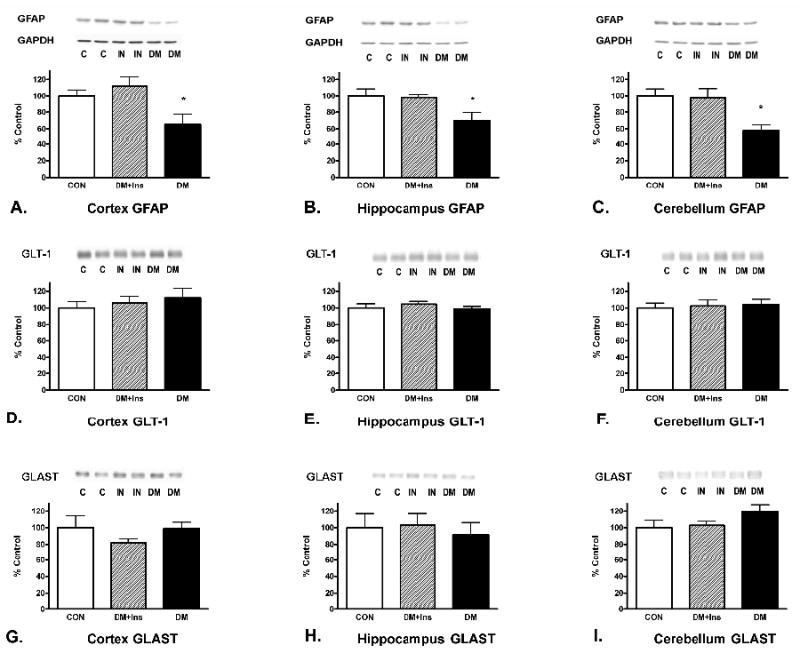
Insulin effects on alterations in protein levels for astrocytic GFAP (A-C), glutamate transporters, GLT-1 (D-F), and GLAST (G-I) after 8 weeks of diabetes. GAPDH, used as a housekeeping protein and to normalize data, was unaltered by the treatments. Values represent means ± SEM (* p < 0.05). Control (CON): n = 8; Diabetic + Insulin (DM+IN): n = 8; Diabetic/no insulin (DM): n = 8. Similar results were seen after 4 weeks of diabetes. Immunoblots from representative samples are shown.
2.3 Immunohistochemistry
We used immunohistochemistry to confirm that insulin treatment prevented the diabetes-induced decrease in GFAP immunoreactivity in the hippocampus, cerebellum, external capsule and corpus callosum regions at 4 weeks (data not shown but similar to 8 week study) and 8 weeks of diabetes (Figure 3). Because cortical astrocyte GFAP immunoreactivity is normally low, differences in cortical expression between the groups were undetectable with immunohistochemistry.
Figure 3.
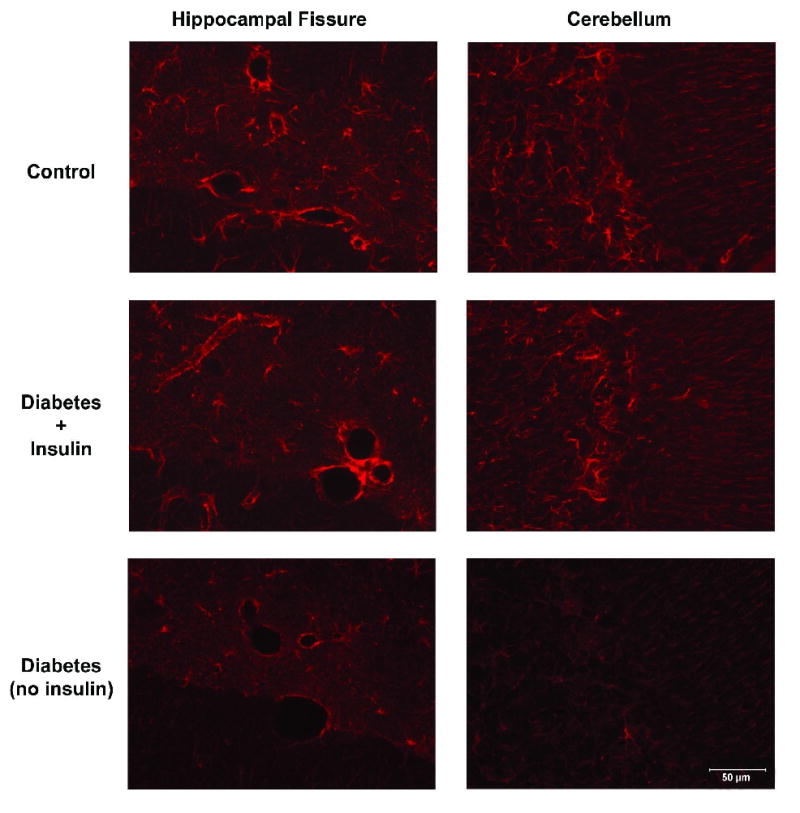
Immunofluorescence of astrocytic GFAP in the hippocampal fissure and cerebellum after 8 weeks of diabetes. Insulin treatment prevented the diabetes-induced attenuation of GFAP immunofluorescence. (Control: n = 8; Diabetic + Insulin: n = 8; Diabetic/no insulin: n = 8). Similar findings were seen after 4 weeks of diabetes. Representative sections are shown (40×).
To determine if the diabetes-induced GFAP expression change was caused by a decrease in astrocyte number, we counted S-100β (+) cells in tissue sections adjacent to GFAP labeled sections. The numbers of S-100β (+) cells were not different in any of the treatment groups (Table 2). No differences in immunoreactivity on tissue sections were detected among the treatment groups for the glutamate transporters GLAST (Figure 4) or GLT-1 (Figure 5) at either time period.
Figure 4. GLAST.
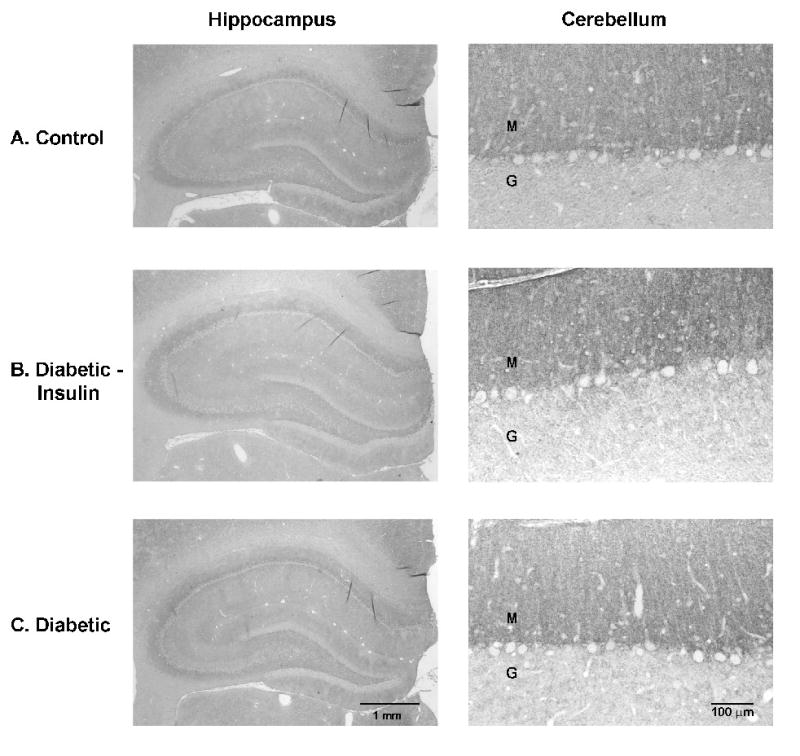
Immunohistochemistry of astrocytic GLAST in the hippocampus and cerebellum after 8 weeks of diabetes. No obvious changes were detectable in the intensity of immunoreactivity among the different treatment groups. (Control: n = 8; Diabetic + Insulin: n = 8; Diabetic/no insulin: n = 8). Similar findings were seen after 4 weeks of diabetes. Representative sections are shown. M: molecular layer; G: granular layer.
Figure 5. GLT-1.
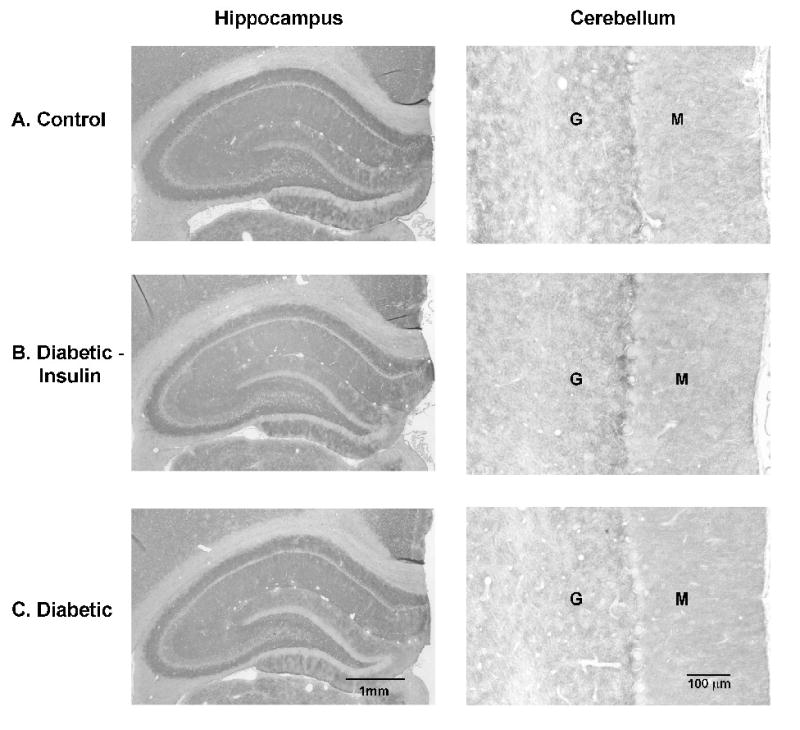
Immunohistochemistry of astrocytic GLT-1 in the hippocampus and cerebellum after 4 weeks of diabetes. No obvious changes were detectable in the intensity of immunoreactivity among the different treatment groups. (Control: n = 8; Diabetic + Insulin: n = 8; Diabetic/no insulin: n = 8). Similar findings were seen after 8 weeks of diabetes. Representative sections are shown. M: molecular layer; G: granular layer.
2.4 GPV isolation and glutamate uptake studies
We used GPV glutamate uptake assays to determine if diabetes affected glutamate uptake. Diabetes resulted in a significant increase in total GPV glutamate uptake, increasing both sodium-dependent and sodium-independent uptake at 4 weeks and 8 weeks of diabetes duration (Figure 6). Glutamate uptake in the presence of dihydrokainate (DHK) was increased in diabetic rats at both time periods. There was no difference in the uptake contributed by the GLT-1 transporter in the different treatment groups. Insulin treatment prevented the increase in glutamate uptake in non-treated diabetic animals at both time periods with the exception of the DHK/insulin treated group at 8 weeks, where uptake was greater than control, but less than untreated diabetes. There were no differences in glutamate uptake between the different treatment groups at either time period in the presence of L-trans-Pyrrollidine-2,4-dicarboxylic acid (PDC). No increases in the protein levels of GLAST, GLT-1, or EAAC1 were detected in the GPV fractions among the treatment groups (data not shown).
Figure 6.
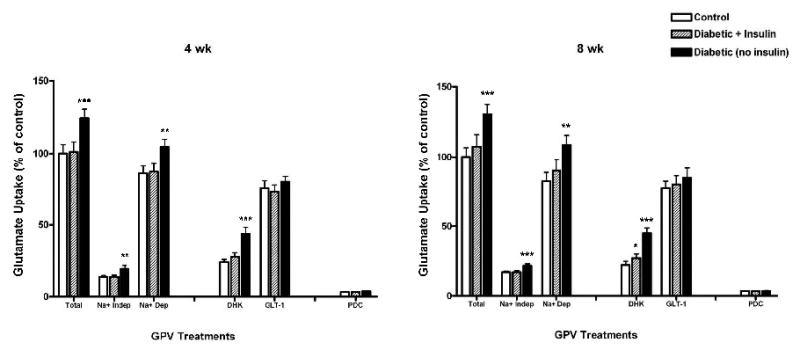
Insulin effects on GPV 3H-glutamate uptake after 4 and 8 weeks of diabetes. Total GPV glutamate uptake (Total); Sodium-independent uptake (Na+ Indep); Sodium-dependent uptake (Na+ Dep; Total uptake minus Na+ Indep uptake); dihydrokainate (DHK; selective GLT-1 blocker, 100 μM); GLT-1 transporter uptake (GLT-1; Na+ Dep uptake minus DHK uptake); L-trans-Pyrrollidine-2,4-dicarboxylic acid (PDC, general glutamate transporter blocker, 100 μM). Statistical significance is as follows: * p < 0.05 from Control; ** p < 0.01 from Control and Diabetic + Insulin; *** p < 0.001 from Control and Diabetic + Insulin. (4 wks: Controls: n = 5; Diabetic + Insulin: n = 5; Diabetic: n = 5; 8 wks: Controls: n = 5; Diabetic + Insulin: n = 5; Diabetic: n = 5).
3. Discussion
In our study, insulin treatment prevented the decrease of astrocyte GFAP levels and the increase in glutamate uptake observed in diabetic animals. Additionally, insulin treatment blocked blood glucose increase and decreases in brain and body weight. These observations are consistent with insulin therapy's overall amelioration of complications associated with diabetes (Nathan et al., 2009).
In the course of this study, we noticed some apparently anomalous blood insulin and glucose values in the insulin treated group. Although markedly reduced in non-treated diabetic animals, insulin levels were normal or slightly elevated in control and insulin-treated animals, respectively. Using the LINCO radioimmunoassay kit, insulin levels in non-diabetic, fasting animals are 0.5 - 2.0 ng/ml. We recorded insulin levels in non-fasted control animals, which yield slightly higher values at 4 (2.86 ± 0.26 ng/ml) and 8 weeks (2.54 ± 0.21 ng/ml). Values for the insulin treated animals at 8 wks were elevated from control (4.17 ± 0.47 ng/ml). We believe these elevated levels stem from the use of exogenous insulin that can result in the production of anti-insulin antibodies. Anti-insulin antibodies can cause falsely elevated measurements when determined by a solid phase radioimmunoassay such as we used in this study (Linshin Canada Inc., manufacture's product information). Additionally, insulin levels may be increased by anesthesia with pentobarbital (Vera, et al., 2002). However, we collected insulin samples within 5 minutes of anesthesia of all animals to minimize the pentobarbital effect.
A second anomalous measurement was that of blood glucose which was elevated mildly at 1, 6, and 7 weeks in the insulin treated group. We believe that these increased blood glucose levels derive from our use of pellets to deliver insulin. One week is usually required to normalize blood glucose using the pellets. The increase in blood glucose at 6-7 weeks was likely a result of declining insulin release from the pellets which occurs at 45 ± 5 days according to the manufacturer (Linshin Canada Inc.). Indeed, implantation of a new pellet normalized blood glucose levels to those of control individuals.
Insulin treatment prevented diabetes-induced decreases in astrocyte GFAP. The decrease in GFAP seen in diabetic animals reflects a decrease in GFAP expression rather than a decrease in astrocyte number. This observation differs from that of Lechuga-Sancho and coworkers who reported a decrease in rat hypothalamic astrocyte numbers 6 weeks after diabetes onset (Lechuga-Sancho et al., 2006). One explanation for the difference is that Lechuga-Sancho et al. counted GFAP (+) cells but a diminution of GFAP immunoreactivity could cause an undercounting. We, on the other hand, counted S-100β (+) cells because S-100β co-localizes with GFAP (Coleman, unpublished; Sorci et al., 1998) and is predominantly expressed by astrocytes (Hertz et al., 2001; Melzer et al., 2001; Mercier and Hatton, 2000). A second possibility that may account for the difference is that not all astrocytes are identical (Hewett, 2009) and astrocytes in different brain regions respond differently to diabetes.
A second effect of insulin treatment on astrocyte function was to modify an increase in glutamate uptake in diabetic rats. Both sodium-dependent and sodium-independent glutamate uptake were 25-30% greater in glial vesicle preparations in diabetic animals compared to non-diabetic control animals but the response in the two systems were not identical. Insulin treatment effects on sodium-dependent glutamate uptake were time sensitive. 1Insulin treatment prevented the increase in sodium-dependent glutamate uptake at 4 weeks of diabetes but by 8 weeks insulin only partially reversed changes in glutamate uptake. This observation suggests a change in sensitivity to insulin signaling or a change induced by diabetes not directly sensitive to insulin signaling.
The major component of the sodium-dependent glutamate uptake increase was via GLAST (or other non-GLT-1 transporters) since there was no change in the GLT-1 uptake component. We detected no significant increases by western blotting in GLAST or GLT-1 protein levels in the GPV fractions, nor were we able to detect an increase in membrane immunoreactivity for GLAST and GLT-1 in tissue sections. These findings suggest that the increased glutamate uptake resulted from increased affinity of the transporters for glutamate (Duan et al., 1999; Ward et al., 2005). Ward and coworkers reported unchanged GLAST and EAAT4 expression levels after 12 weeks of diabetes in retinal glial cells although glutamate uptake increased by as early as one week (Ward et al., 2005). The increased uptake was attributed to an increased efficiency of uptake. The increase in glutamate uptake in diabetic rats could be a compensatory functional change that protects neurons from the excitotoxic effects of glutamate.
Insulin treatment prevented the diabetes-induced increase in sodium-independent glutamate uptake in the glial preparation at both 4 and 8 weeks. This effect of insulin could prove important in the antioxidant status of the brain in diabetes. An increase in extracellular glutamate uptake by cellular xc- transporters inhibits the uptake of cystine, thereby depleting cells of the antioxidant, glutathione (McBean, 2002; Shih et al., 2006). Glutathione levels decrease in the diabetic brain (Bhardwaj, et al., 1999) but the mechanisms responsible are unknown. We speculate that the diabetes-induced increase in sodium-independent glutamate uptake could lead to reduced cystine transport into astrocytes and result in decreased production of glutathione. Bhardwaj and colleagues showed that insulin treatment in the diabetic rat results in partial or full recovery of a number of antioxidant compounds in the brain, including glutathione (Bhardwaj, et al., 1999). Additional studies are needed to determine the underlying mechanism(s) of action of diabetes and insulin treatment on system xc- transporters and their role in the antioxidant status of the brain.
In summary, the present study demonstrates a significant effect of insulin treatment on diabetes-induced changes in astrocyte GFAP levels and glutamate uptake. Whether the effects of diabetes on astrocyte function are a result of hyperglycemia, insulin deficiency, or both is not known. Insulin's prevention of diabetes-induced alterations in astrocyte function is an avenue of study of both normal astrocyte function and insulin function in the CNS of both non-affected and diabetic individuals.
4. Experimental Procedure
4.1 Experimental groups
Male Wistar rats received an intravenous injection into the tail vein of STZ (40 mg/kg) with control rats receiving vehicle (citrate buffer) (Coleman et al., 2004; Lin et al., 2002; McNeill, 1999). Diabetes was verified by blood glucose levels greater than 350 mg/dl, 24 hr post injection. Blood glucose samples were collected by inserting a 27 gauge needle into the tail vein and collecting a drop of blood. The drop of blood was immediately placed onto an ACCU-CHEK glucose test strip and evaluated with the ACCU-CHEK glucose meter (Roche Diagnostic Corporation, Indianapolis, IN). Two days following the induction of diabetes, animals were briefly anesthetized with isoflurane and implanted with a slow-release insulin pellet (Linshin Canada Inc., Ontario, Canada) into the diabetes/insulin group, or with a control pellet into the control group and diabetes/no insulin group. Insulin pellets (2 mm in diameter; 7 mm long) contain insulin in microcrystallized palmitic acid and release approximately 2 U/24 hour/implant for > 40 days (per manufacturer). The control pellets (2 mm in diameter; 7 mm long) contain microcrystallized palmitic acid only. The pellets were placed in the subcutaneous tissue under the abdominal skin according to the manufacture's recommendation using the supplied 12G needle with trocar. Blood glucose (non-fasted) levels and body weight were determined weekly. Additional insulin pellets were implanted based upon blood glucose values as needed. Corresponding control pellets were implanted at similar time frames. Insulin levels (non-fasted) were determined by radioimmunoassay according to the manufacture's recommendations (LINCO Research, St. Charles, MO) at the end of the study for each group. Blood samples for insulin assay were collected at the time of sacrifice following decapitation from trunk blood within 5 minutes of anesthesia with pentobarbital (50 mg/kg intraperitoneal, IP). Intra- and interassay coefficients of variation were 5.2% and 8.2% respectively.
The number of animals in each treatment group for each time period is indicated below for specific studies and tissue processing. All studies were approved by the Auburn University Institutional Animal Care and Use Committee.
4.2 Protein extraction and Western blot analysis
Animals and tissues for protein determination/immunoblotting were handled as previously described (Coleman et al. 2004). Briefly, animals were anesthetized with pentobarbital (50 mg/kg, IP), decapitated and brains rapidly removed (4 week studies: Control, n=8; Diabetic + Insulin, n=8; Diabetic/no Insulin, n=8; 8 week studies: Control, n=8; Diabetic + Insulin, n=8; Diabetic/no Insulin, n=8). The brains were weighed, dissected into cerebral cortex, hippocampus, and cerebellum, and immediately frozen (stored at -80°C until analysis). Tissue samples were homogenized in 1% sodium dodecyl sulfate (SDS; 85-90°C). Protein concentrations were determined using BCA protein assay determination according to the manufacture's recommendations (Pierce, Rockford, IL). Equal amounts of protein (10 μg) in Laemmli sample buffer (50 mM Tris-HCL, pH 6.8, containing 2% SDS, 0.1% 2-mercaptoethanol, and 20% glycerol) were electrophoresed on a 10% SDS-polyacrylamide gel, transferred to nitrocellulose, and membranes stained with Ponceau stain (0.5%) to assure equal loading of proteins and labeling of molecular weight markers. Membranes were blocked with phosphate buffered saline (PBS; 0.01 M phosphate buffer, 0.0027 M potassium chloride, 0.137 M sodium chloride, pH 7.4) containing 0.1% Triton ×, 0.1% Tween 20, plus 7% nonfat dry milk for 1 hr at 37°C, followed by incubation with antibody to GFAP (1:500, mouse IgG1, clone 52, carboxy terminus of human GFAP, Transduction Laboratory, Lexington, KY), GLT-1 (1:1000, guinea pig polyclonal, carboxy terminus of rat GLT-1, Chemicon, Temecula, CA), GLAST (1:1000, guinea pig polyclonal, carboxy terminus of rat GLAST, Chemicon, Temecula, CA), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a housekeeping protein (1:1000, mouse IgG, clone 6C5, Chemicon, Temecula, CA) at 4°C overnight. Immunodetection of protein bands was carried out by incubation with a peroxidase linked secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) specific for the given species of origin of the primary antibody, followed by chemiluminescence and Hyperfilm ECL (Amersham Biosciences, Piscataway, NJ). Differences in band intensities were determined by Un-Scan-It (Silk Scientific, Orem, Utah). Levels of GFAP, GLT-1, and GLAST were normalized to GAPDH (housekeeping protein) and compared between treatment groups. Results are reported as percent of control for the respective proteins.
4.3 Immunohistochemistry
Animals and tissues for immunohistochemistry were handled as previously described (Coleman et al., 2004). Briefly, animals were anesthetized with pentobarbital (50 mg/kg, IP), an aortic catheter was placed by transcardiac puncture, flushed with PBS followed by 4% paraformaldehyde in PBS (4 week studies: Control, n=8; Diabetic + Insulin, n=8; Diabetic/no Insulin, n=8; 8 week studies: Control, n=8; Diabetic + Insulin, n=8; Diabetic/no Insulin, n=8). Brains were removed, blocked, post-fixed in 4% paraformaldehyde in PBS (24 hr, 4°C) and stored at 4°C in 1% paraformaldehyde in PBS until processing. Coronal sections were paraffin-embedded and cut in serial section at 7 μm (-3.10 to -3.70 mm from bregma for cortex/hippocampus; -10.30 to −10.90 mm from bregma for cerebellum) (Paxinos and Watson, 1998).
Sections were deparaffinized and immunohistochemistry carried out as previously described (Coleman et al., 2004). Serial sections were processed for GFAP, S-100β, GLT-1 and GLAST (sections from control rats, diabetic + insulin rats, and diabetic/no insulin rats were processed at the same time under the same conditions for a given protein). All antibodies were tested to determine optimal dilutions and staining parameters prior to the study.
Sections for GFAP immunohistochemistry were labeled by immunofluorescence with anti-GFAP primary antisera (1:1000, rabbit anti-cow, Z334, DAKO, Carpinteria, CA) followed by fluorescent secondary antibody (Alexa FluorR 594 donkey anti-rabbit, Molecular Probes, Eugene, OR) and mounted with Vectashield (Vector Laboratories, Burlingame, CA). Slides were evaluated through an epi-fluorescence-equipped Nikon Eclipse E600W microscope and images captured with a RT-Slider Spot digital camera (Diagnostic Instruments, Sterling Heights, MI).
Sections for S-100β protein (for astrocyte counting) and the astrocyte glutamate transporters, GLT-1 and GLAST, were labeled by chromogenic immunohistochemistry. Primary antibodies were as follows: anti-S-100β (1:40,000, mouse monoclonal clone SH-B1, beta chain, Sigma, St. Louis, MO), anti-GLT-1 (1:24,000, guinea pig polyclonal, carboxy terminus of rat GLT-1, Chemicon, Temecula), or anti-GLAST (1:12,000, guinea pig polyclonal, carboxy terminus of rat GLAST, Chemicon, Temecula, CA). Sections were incubated with biotinylated secondary antibodies (Jackson ImmunoResearch, West Grove, PA), incubated with ABC Elite (Vector Laboratories, Burlingame, CA), and reacted with diaminobenzidine (DAB)/nickel (Vector Laboratories, Burlingame, CA). Slides were dehydrated and cover slipped with Vecta Mount (Vector Laboratories, Burlingame, CA). Control sections (sections lacking primary antibody and/or preabsorption of the primary antibody with antigen) showed no immunoreactivity.
Astrocyte cell counts were performed using immunoreactivity to S-100β on the adjacent serial section to those used for GFAP immunohistochemistry (4 week studies: Control, n=6; Diabetic + Insulin, n=6; Diabetic/no Insulin, n=6; 8 week studies: Control, n=5; Diabetic + Insulin, n=5; Diabetic/no Insulin, n=5). For each animal, S-100β immunoreactive astrocytes from the cerebral cortex (2 regions), hippocampus (5 regions), corpus callosum, external capsule, and cerebellum (3 regions) were counted at 20× magnification using NIH Image J (National Institutes of Health, Bethesda, MD). Cell counts for the cerebral cortex, hippocampus, corpus callosum and external capsule were reported as the number of immunoreactive cells in 0.263 mm2 (20× field). Cell counts for cerebellar astrocytes were made at the margin between the molecular and granular cell layers over a given length and were reported as S-100β immunoreactive cells per 100 mm.
4.4 Glial plasmalemmal vesicle (GPV) isolation
Glial plasmalemmal vesicle isolation was carried-out according to the technique of Nakamura (Nakamura et al., 1993) with modification by Hirst (Hirst et al., 1998). Due to low fractional recovery of the GPV, determination of glutamate uptake from isolated regions of the brain could not be performed (Coleman, unpublished observations; Strużyñska et al., 2001). GPV isolation was therefore performed on total brain homogenate. Brain tissue was collected from control, diabetic + insulin, and diabetic/no insulin rats at 4 and 8 weeks of diabetes duration (4 week studies: Control, n=5; Diabetic + Insulin, n=5; Diabetic/no Insulin, n=5; 8 week studies: Control, n=5; Diabetic + Insulin, n=5; Diabetic/no Insulin, n=5). Rats were anesthetized with pentobarbital and decapitated. Brains were quickly removed, weighed and placed in 10 volumes of ice-cold solution containing 0.32 M sucrose and 1 mM EDTA (pH 7.4). Tissues were homogenized using a Potter's teflon/glass homogenizer. The homogenate was centrifuged at 1000 g for 10 min at 4°C. The supernatant was diluted with SEDH solution (0.32 M sucrose, 1 mM EDTA, 0.25 mM dithiothreitol and 20 mM HEPES, pH 7.4), and centrifuged at 5000 g for 15 min. The supernatant was then centrifuged at 33,500 g for 20 min. The resultant pellet was suspended in SEDH, layered on a discontinuous gradient composed of 20, 10, 6 and 2% Percoll (Sigma, St. Louis, MO) in SEDH, and tubes centrifuged at 33,500 g for 5 min at 4°C. The turbid layer between 2% and 6% Percoll was collected and washed in 10 volumes of SEDH. The pellet was suspended in SEDH, and layered onto fresh gradients for a second purification. The fractions were washed, kept on ice and used within 2 hrs. Aliquots of GPV were taken for protein determination by the Bradford method (Bio-Rad, Hercules, CA). Purity of the GPV prep was evaluated by western blotting (WB) with astrocyte marker proteins GFAP, glutamate transporters, GLT-1 and GLAST, and for neuronal contamination by the predominantly neuronal glutamate transporter, EAAC1, and the synaptic terminal protein synaptobrevin (Daniels and Vickroy, 1998; Hirst et al., 1998; Nakamura et al., 1993).
4.5 GPV glutamate uptake
Glutamate uptake by GPV was measured using the filtration method of Divac (Divac et al., 1977) with modification by Daniels (Daniels and Vickroy, 1998). Aliquots of 2.5 μg of GPV (5 μl; 0.5 μg/μl protein) were added to duplicate tubes in a 25°C water bath and incubated for 2 min. The uptake reaction for was initiated by the addition of 200 μl of L-[2,3,4-3H]glutamic acid (final concentration of 400 nM) in 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 1.2 mM NaH2PO4, 10 mM D-glucose, and 20 mM HEPES. Tubes were rapidly mixed and returned to the water bath for 3 min. The reaction was terminated by the addition of ice cold phosphate buffer. This uptake was considered “total glutamate uptake”. Na+-independent uptake was measured using a buffer with 140 mM choline chloride in place of NaCl. Na+-dependent uptake was determined by subtracting the Na+-independent uptake from the total uptake. The specific GLT-1 blocker, dihydrokainate (DHK, 100 μM) and a non-selective glutamate transporter blocker, L-trans-Pyrrollidine-2,4-dicarboxylic acid (PDC, 100 μM) were evaluated. The GLT-1 uptake portion was determined by subtracting the DHK uptake from the Na+-dependent uptake. Uptake was determined by vacuum filtration on a Millipore 1225 Sampling Vacuum Manifold (Millipore, Bedford, MA) with Whatman GF/B filter discs. Filters were washed in cold phosphate buffer. Radioactivity was measured in a Beckman liquid scintillation counter.
4.6 Statistical analysis
Data were analyzed by one-way Analysis of Variance (ANOVA) using the statistical program Graph Pad Prism version 4.01 for Windows (Graph Pad Software, San Diego, CA). Differences between treatment groups within a given time period were assessed by the Tukey-Kramer Multiple comparison test (Graph Pad Software, San Diego, CA). Data are reported as means ± SEM. Statistical significance was set at p < 0.05.
Acknowledgments
Supported by the National Institutes of Health (KO1 NS44011) and AU FAHDR
Abbreviations
- GPV
glial plasmalemmal vesicle
Footnotes
Section: 8. Disease-Related Neuroscience
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afsari ZH, Renno WM, Abd-El-Basset E. Alteration of glial fibrillary acidic proteins immunoreactivity in astrocytes of the spinal cord diabetic rats. Anat Rec. 2008;291:390–399. doi: 10.1002/ar.20678. [DOI] [PubMed] [Google Scholar]
- Ambati J, Chalam KV, Chawla DK, D'Angio CT, Guillet EG, Rose SJ, Vanderlinde RE, Ambati BK. Elevated gamma-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol. 1997;115:1161–1166. doi: 10.1001/archopht.1997.01100160331011. [DOI] [PubMed] [Google Scholar]
- Asnaghi V, Gerhardinger C, Hoehn T, Adeboje A, Lorenzi M. A role for the polyol pathway in the early neuroretinal apoptosis and glial changes induced by diabetes in the rat. Diabetes. 2003;52:506–511. doi: 10.2337/diabetes.52.2.506. [DOI] [PubMed] [Google Scholar]
- Barber AJ, Antonetti DA, Gardner TW. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. Invest Ophthalmol Vis Sci. 2000;41:3561–3568. [PubMed] [Google Scholar]
- Bhardwaj SK, Sharma P, Kaur G. Alterations in free radical scavenger system profile of Type I diabetic rat brain. Mol Chem Neuropathol. 1998;35:187–202. doi: 10.1007/BF02815124. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Koffeman A, Scheltens P. Diabetes and cognitive impairment. Clinical diagnosis and brain imaging in patients attending a memory clinic. J Neurol. 2006;253:477–482. doi: 10.1007/s00415-005-0036-4. [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a lifespan perspective. Lancet Neurol. 2008;7:184–190. doi: 10.1016/S1474-4422(08)70021-8. [DOI] [PubMed] [Google Scholar]
- Bouchard P, Ghitescu LD, Bendayan M. Morpho-functional studies of the blood-brain barrier in streptozotocin-induced diabetic rats. Diabetologia. 2002;45:1017–1025. doi: 10.1007/s00125-002-0853-2. [DOI] [PubMed] [Google Scholar]
- Chehade JM, Haas MJ, Mooradian AD. Diabetes-related changes in rat cerebral occludin and zonula occludens-1 (ZO-1) expression. Neurochem Res. 2002;27:249–252. doi: 10.1023/a:1014892706696. [DOI] [PubMed] [Google Scholar]
- Coleman ES, Judd R, Hoe L, Dennis J, Posner P. Effects of diabetes on astrocyte GFAP and glutamate transporters in the CNS. Glia. 2004;48:166–178. doi: 10.1002/glia.20068. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Daniels KK, Vickroy TW. Simultaneous isolation of glial and neuronal fractions from rat brain homogenates: comparison of high-affinity L-glutamate transport properties. Neurochem Res. 1998;23:103–113. doi: 10.1023/a:1022413823183. [DOI] [PubMed] [Google Scholar]
- Dennis JC, Coleman ES, Swyers SE, Moody SW, Wright JC, Judd R, Morrison EE. Changes in mitotic rate and GFAP expression in the primary olfactory axis of streptozotocin-induced diabetic rats. J Neurocytology. 2005;34:3–10. doi: 10.1007/s11068-005-5044-x. [DOI] [PubMed] [Google Scholar]
- Divac I, Fonnum F, Storm-Mathisen J. High affinity uptake of glutamate in terminals of corticostriatal axons. Nature. 1977;266:377–378. doi: 10.1038/266377a0. [DOI] [PubMed] [Google Scholar]
- Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in the brain. Metabolic interactions between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- Duan S, Anderson CM, Stein BA, Swanson RA. Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci. 1999;19:10193–10200. doi: 10.1523/JNEUROSCI.19-23-10193.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegelashvili G, Schousboe A. Cellular distribution and kinetic properties of high affinity glutamate transporters. Brain Res Bull. 1998;45:233–238. doi: 10.1016/s0361-9230(97)00417-6. [DOI] [PubMed] [Google Scholar]
- Hansson E, Muyderman H, Leonova J, Allansson L, Sinclair J, Blomstrand F, Thorlin T, Nilsson M, Rönnbäck L. Astroglia and glutamate in physiology and pathology: aspects on glutamate transport, glutamate-induced cell swelling and gap junction communication. Neurochem Int. 2000;37:317–329. doi: 10.1016/s0197-0186(00)00033-4. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50:202–211. doi: 10.1007/s00125-006-0485-z. [DOI] [PubMed] [Google Scholar]
- Hertz L, Hansson E, Rönnbäck L. Signaling and gene expression in the neuron-glia unit during brain function and dysfunction: Holger HydNn in memoriam. Neurochem Int. 2001;39:227–252. doi: 10.1016/s0197-0186(01)00017-1. [DOI] [PubMed] [Google Scholar]
- Hertz L, Zielke HR. Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends in Neurosci. 2004;27:736–743. doi: 10.1016/j.tins.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Hewett JA. Determinants of regional and local diversity within the astroglial lineage of the normal central nervous system. J Neurochem. 2009 July 17; doi: 10.1111/j.1471-4159.2009.06288.x. Epub. [DOI] [PubMed] [Google Scholar]
- Hirst WD, Price GW, Rattray M, Wilkin GP. Serotonin transporters in adult rat brain astrocytes revealed by [3H]5-HT uptake into glial plasmalemmal vesicles. Neurochem Int. 1998;33:11–22. doi: 10.1016/s0197-0186(05)80003-8. [DOI] [PubMed] [Google Scholar]
- Horani MH, Mooradian AD. Effects of diabetes on the blood brain barrier. Curr Pharm Des. 2003;9:833–840. doi: 10.2174/1381612033455314. [DOI] [PubMed] [Google Scholar]
- Huber JD. Diabetes, cognitive function, and the blood-brain barrier. Curr Pharm Des. 2008;14:1594–1600. doi: 10.2174/138161208784705441. [DOI] [PubMed] [Google Scholar]
- Hudspith MJ. Glutamate: a role in normal brain function, anesthesia, analgesia, and CNS injury. Br J Anaesthesia. 1997;78:731–747. doi: 10.1093/bja/78.6.731. [DOI] [PubMed] [Google Scholar]
- Kamal A, Biessels GJ, Duis SEJ, Gispen WH. Learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: interaction of diabetes and ageing. Diabetologia. 2000;43:500–506. doi: 10.1007/s001250051335. [DOI] [PubMed] [Google Scholar]
- La Bella V, Valentino F, Piccoli T, Piccoli F. Expression and developmental regulation of the cystine/glutamate exchanger (xc-) in the rat. Neurochem Res. 2007;32:1081–1090. doi: 10.1007/s11064-006-9277-6. [DOI] [PubMed] [Google Scholar]
- Lechuga-Sancho AM, Arroba AI, Frago LM, García-Cáceres C, de Célix AD, Argente J, Chowen JA. Reduction in the number of astrocytes and their projections in associated with increased synaptic protein density in the hypothalamus of poorly controlled diabetic rats. Endocrinol. 2006;147:5314–5324. doi: 10.1210/en.2006-0766. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathison J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, Raine CS. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron. 1996;17:607–615. doi: 10.1016/s0896-6273(00)80194-4. [DOI] [PubMed] [Google Scholar]
- Lieth E, Barber AJ, Xu B, Dice C, Ratz MJ, Tanase D, Strother JM. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Diabetes. 1998;47:815–820. doi: 10.2337/diabetes.47.5.815. [DOI] [PubMed] [Google Scholar]
- Lin CY, Higginbotham DA, Judd RL, White BD. Central leptin increases insulin sensitivity in streptozotocin-induced diabetic rats. Am J Physiol Endocrinol Metab. 2002;282:E1084–1091. doi: 10.1152/ajpendo.00489.2001. [DOI] [PubMed] [Google Scholar]
- Mankovsky BN, Metzger BE, Molitch ME, Biller J. Cerebrovascular disorders in patients with diabetes mellitus. J Diabetes Complications. 1996;10:228–242. doi: 10.1016/s1056-8727(96)90006-9. [DOI] [PubMed] [Google Scholar]
- Manschot SM, Biessels GJ, de Valk H, Algra A, Rutten GE, van der Grond J, Kappelle LJ. Utrecht Diabetic Encephalopathy Study Group. Metabolic and vascular determinants of impaired cognitive performance and abnormalities on brain magnetic resonance imaging in patients with type 2 diabetes. Diabetologia. 2007;50:2388–2397. doi: 10.1007/s00125-007-0792-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrocola R, Restivo F, Vercellinato I, Danni O, Brignardello E, Aragno M, Boccuzzi G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J Endocrinol. 2005;187:37–44. doi: 10.1677/joe.1.06269. [DOI] [PubMed] [Google Scholar]
- McBean GJ. Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci. 2002;23:299–302. doi: 10.1016/s0165-6147(02)02060-6. [DOI] [PubMed] [Google Scholar]
- McCall MA, Gregg RG, Behringer RR, Brenner M, Delaney CL, Galbreath EJ, Zhang CL, Pearce RA, Chiu SY, Messing A. Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc Natl Acad Sci. 1996;93:6361–6366. doi: 10.1073/pnas.93.13.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill JH. Experimental models of diabetes. Boca Raton, FL: CRC Press LLC; 1999. pp. 3–17.pp. 266 [Google Scholar]
- Melzer P, Savchenko V, McKanna JA. Microglia, astrocytes, and macrophages react differently to central and peripheral lesions in the developing and mature rat whisker-to-barrel pathway: a study using immunohistochemistry for lipocortin1, phosphotyrosine, s100 beta, and mannose receptors. Exp Neurol. 2001;168:63–77. doi: 10.1006/exnr.2000.7554. [DOI] [PubMed] [Google Scholar]
- Mercier F, Hatton GI. Immunocytochemical basis for a meningeo-glial network. J Comp Neurol. 2000;420:445–465. doi: 10.1002/(sici)1096-9861(20000515)420:4<445::aid-cne4>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Iga K, Shibata T, Shudo M, Kataoka K. Glial plasmalemmal vesicles: a subcellular fraction from rat hippocampal homogenate distinct from synaptosomes. Glia. 1993;9:48–56. doi: 10.1002/glia.440090107. [DOI] [PubMed] [Google Scholar]
- Nathan DM, Zinman B, Cleary PA, Backlund JY, Genuth S, Miller R, Orchard TJ. Modern-day clinical course of type 1 diabetes mellitus after 30 years' duration: the diabetes control and complications trial/epidemiology of diabetes interventions and complications and Pittsburgh epidemiology of diabetes complications experience (1983-2005) Arch Intern Med. 2009;169:1307–1316. doi: 10.1001/archinternmed.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A, Stolk RP, van Harskamp F, Pols HAP, Hofman A, Breteler MMB. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurol. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic Press; 1998. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M. Astrocyte intermediate filaments in CNS pathologies and regeneration. J Path. 2004;204:428–437. doi: 10.1002/path.1645. [DOI] [PubMed] [Google Scholar]
- Pines G, Danbolt NC, Bjørås M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner BI. Cloning and expression of a rat brain L-glutamate transporter. Nature. 1992;360:464–467. doi: 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- Rösen P, Nawroth PP, King G, Möller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab Res Rev. 2001;17:189–212. doi: 10.1002/dmrr.196. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kunci RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M, Bannai S. Distribution of cystine/glutamate exchange transporter, system xc-, in the mouse brain. J Neurosci. 2002;22:8028–8033. doi: 10.1523/JNEUROSCI.22-18-08028.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26:10514–10523. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorci G, Agneletti AL, Bianchi R, Donato R. Association of S100B with intermediate filaments and microtubules in glial cells. Biochim Biophys Acta. 1998;1448:277–289. doi: 10.1016/s0167-4889(98)00134-7. [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofman K, Stoffel W. Structure, expression, and functional analysis of a Na+-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci. 1992;89:10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strużyñska L, Bubko I, Walski M, Rafalowska U. Astroglial reaction during the early phase of acute lead toxicity in the adult rat brain. Toxicol. 2001;165:121–131. doi: 10.1016/s0300-483x(01)00415-2. [DOI] [PubMed] [Google Scholar]
- Tiehuis AM, van der Graaf Y, Visseren FL, Vincken KL, Biessels GJ, Appelman AP, Kapelle LJ, Mali WP. SMART Study Group. Diabetes increases atrophy and vascular lesions on brain MRI patients with symptomatic arterial disease. Stroke. 2008;39:1600–1603. doi: 10.1161/STROKEAHA.107.506089. [DOI] [PubMed] [Google Scholar]
- Trendelenburg G, Dirnagl U. Neuroprotective role of astrocytes in cerebral ischemia: focus on ischemic preconditioning. Glia. 2005;51:307–320. doi: 10.1002/glia.20204. [DOI] [PubMed] [Google Scholar]
- Tsacopoulos M, Magistretti PJ. Metabolic coupling between glia and neurons. J Neurosci. 1996;16:877–885. doi: 10.1523/JNEUROSCI.16-03-00877.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Vera ER, Battell ML, Bhanot S, McNeill JH. Effects of age and anesthetic on plasma glucose and insulin levels and insulin sensitivity in spontaneously hypertensive and Wistar rats. Can J Physiol Pharmacol. 2002;80:962–970. doi: 10.1139/y02-124. [DOI] [PubMed] [Google Scholar]
- Ward MM, Jobling AI, Kalloniatis M, Fletcher EL. Glutamate uptake in retinal glial cells during diabetes. Diabetologia. 2005;48:351–360. doi: 10.1007/s00125-004-1639-5. [DOI] [PubMed] [Google Scholar]
- Whiting S, Camfield P, Arab D, Salisbury S. Insulin-dependent diabetes mellitus presenting in children as frequent, medically unresponsive, partial seizures. J Child Neurol. 1997;12:178–180. doi: 10.1177/088307389701200305. [DOI] [PubMed] [Google Scholar]
- Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiology Pharmacol. 1997;75:1149–63. [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossman KA, Pozzan T, Carmignoto G. Neuron to astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]