

Abstract
Transcriptional regulation of the bcl-2 proto-oncogene is highly complex, with the majority of transcription driven by the P1 promoter site and the interaction of multiple regulatory proteins. A guanine- and cytosine-rich (GC-rich) region directly upstream of the P1 site has been shown to be integral to bcl-2 promoter activity, as deletion or mutation of this region significantly increases transcription. This GC-rich element consists of six contiguous runs of guanines and cytosines that have the potential to adopt DNA secondary structures, the G-quadruplex and i-motif, respectively. Our laboratory has previously demonstrated that the polypurine-rich strand of the bcl-2 promoter can form a mixture of three different G-quadruplex structures. In this current study, we demonstrate that the complementary polypyrimidine-rich strand is capable of forming one major intramolecular i-motif DNA secondary structure with a transition pH of 6.6. Characterization of the i-motif folding pattern using mutational studies coupled with circular dichroic spectra and thermal stability analyses revealed an 8:5:7 loop conformation as the predominant structure at pH 6.1. The folding pattern was further supported by chemical footprinting with bromine. In addition, a novel assay involving the sequential incorporation of a fluorescent thymine analog at each thymine position provided evidence of a capping structure within the top loop region of the i-motif. The potential of the GC-rich element within the bcl-2 promoter region to form DNA secondary structures suggests that the transition from the B-DNA to non-B-DNA conformation may play an important role in bcl-2 transcriptional regulation. Furthermore, the two adjacent large lateral loops in the i-motif structure provide an unexpected opportunity for protein and small molecule recognition.
Introduction
The proto-oncogene bcl-2 (B-cell lymphoma gene 2) belongs to the Bcl-2 family of proteins, which consists of pro- and anti-apoptotic factors that regulate the mitochondrial or intrinsic apoptotic pathway. The bcl-2 gene was originally discovered to be highly constitutively expressed in B-cell lymphomas due to the insertion at the reciprocal chromosomal translocation t(14:18) breakpoint.1,2 Rather than stimulate cellular proliferation similar to other oncogenes, Bcl-2 promotes cell survival through the inhibition of cell death1. Bcl-2 is well known for its anti-apoptotic activity in suppressing the function of pro-apoptotic counterparts such as Bax. Studies that have addressed the manner in which Bcl-2 inactivates Bax suggest that the levels of Bcl-2 and Bax protein present determine the susceptibility or resistance of a cell to undergo apoptosis.3 Consistent with this model, cancer cells that overexpress Bcl-2 shift the balance of the cellular Bcl-2:Bax ratio and exhibit an anti-apoptotic phenotype,1,4–6 while an opposite shift in neuronal cells from neurodegenerative diseases, including Alzheimer's and Parkinson's, results in an increased susceptibility to induced apoptosis.7,8 Despite extensive research, the aberrant bcl-2 expression present in various diseases and the direct effect of this expression on cell survival are poorly understood due to the complexity of bcl-2 transcription.
Transcriptional control of the bcl-2 proto-oncogene is highly complex and involves two promoter sites, P1 and P2, with the majority of the control occurring at the P1 site (Figure 1A). The P1 promoter located 1386–1423 base pairs upstream of the translational start site consists of a TATA-less, GC-rich region with multiple transcription initiation sites.9 To date, the mechanisms of bcl-2 transcriptional regulation are not well characterized; however, several response elements within the P1 and P2 promoter regions have been shown to be involved in the activation or deactivation of bcl-2 transcription. Specifically, A-Myb exhibits potent activation of the P2 promoter in t(14;18) lymphoma cells through interaction with the Cdx binding site,8 and in contrast, p53 has been shown to negatively regulate bcl-2 transcription via the TATA element.9 Similarly, both positive and negative transcription factors have been shown to interact with P1 promoter elements and alter bcl-2 gene expression, including NF-κB and WT-1, respectively.10,11 Although there are no consensus sequences for NF-κB, this transcriptional regulator has been shown to activate bcl-2 expression in lymphoma cell lines through the cAMP response element (CRE) and Sp1 binding sites.11 In addition, the interaction of the cAMP response element binding protein (CREB) with the CRE also leads to the activation of bcl-2 transcription.12,13 Alternatively, WT-1 represses bcl-2 transcription in both HeLa and t(14;18) lymphoma cell lines.11 Of particular interest, transcription factors WT-1, Sp1, and E2F recognize and bind within the GC-rich region upstream of the P1 promoter.9,10,14 This GC-rich region contains potential DNA secondary structure–forming sequences. These sequences have recently been shown to form not only G-quadruplexes15–18 but also i-motif 19 structures, which may act as additional regulators of bcl-2 transcription.
Figure 1.
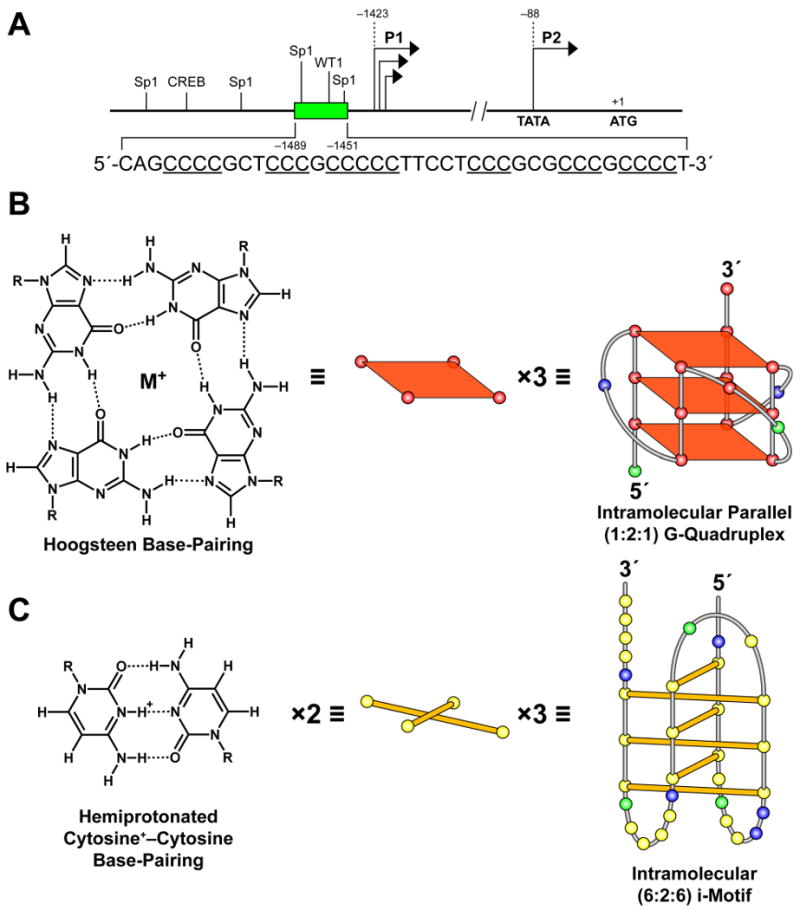
Structure of the human bcl-2 gene promoter region (A), and the guanine–guanine and cytosine+–cytosine base pairing that gives rise to the G-quadruplex (B) and i-motif (C) secondary structures. (A) Locations of the P1 and P2 promoters, the upstream C-rich element (inset), and Sp1, WT1, and CREB transcriptional factor binding sites upstream of the bcl-2 P1 promoter. Three stacked G-tetrads form the G-quadruplex (B) and three sets of two intercalated hemiprotonated cytosine+–cytosine base pairs form the i-motif structure (C). The previously proposed c-myc24 G-quadruplex and i-motif structures formed under conditions of negative supercoiling serve as examples, with the yellow, green, red, and blue circles representing the nucleobases cytosine, adenine, guanine, and thymine, respectively.
The dynamic nature of DNA topology and the conformational transitions that may occur during transcriptionally induced negative superhelicity as a result of unwinding or resolution of the double helix are becoming increasingly recognized.20–23 Recently it has been demonstrated that G- and C-rich sequences are capable of rearranging from B-DNA conformations into non-B-DNA secondary structures (G-quadruplexes and i-motifs) under negative superhelicity.24 G-rich sequences can give rise to G-quadruplex structures of stacked G-tetrads formed by the Hoogsteen interaction of four guanines stabilized by the presence of a monovalent cation (Figure 1B).27 Similarly, the complementary C-rich strand may fold back on itself to form an i-motif structure comprising two parallel duplexes with intercalated hemiprotonated cytosine+–cytosine base pairs (Figure 1C).28–30 Both secondary structure–forming sequences require adjacent guanine or cytosine tracts separated by one or more bases. These dynamic GC-rich sequences have been discovered within or near numerous promoter regions throughout the human genome, suggesting that these structures may function in the regulation of gene transcription.31–37 Genomic analyses on the prevalence of G-quadruplex-forming sequences within 1 kb upstream of transcriptional start sites have revealed that 43% of promoter regions contain these G-rich elements.32 One such G-rich sequence was identified within the c-myc promoter and has been shown to adopt intramolecular parallel G-quadruplex structures.38–42 In support of the hypothesis that DNA secondary structures serve as modulators of transcription, the c-myc G-quadruplex has been demonstrated to function as a transcriptional repressor in vitro.41 TMPyP4, an established G-quadruplex-interactive compound,41,43 and more recently discovered quindoline derivatives44 and actinomycin D45 bind to and stabilize the c-myc G-quadruplexes and result in the downregulation of c-myc mRNA levels in RAMOS Burkitt's lymphoma41,43,45 and Hep G2 hepatocellular carcinoma44 cell lines. In further support of secondary structure involvement in gene transcription, protein-facilitated unwinding or folding of the c-myc G-quadruplex as observed with NM23-H224,46 and nucleolin47, respectively, resulted in activation or repression of c-myc transcription. Subsequent investigations of G-rich elements within other oncogene promoter regions has led to the discovery of G-quadruplex-forming sequences upstream of the VEGF,48,49 c-kit,50–53 PDGF-A,54 RET,55 KRAS,56 hif-1α,57 bcl-2,15–18 and hTERT58 promoters and downstream of the c-myb59 and Rb60 promoters.
While previous research has focused principally on the characterization of G-quadruplex structures, more recent studies have been expanded to also include characterization of i-motif structures within a variety of promoter regions, such as c-myc,61–63 Rb,60 RET,56 VEGF,49 and EPM1.64 Of interest, the bcl-2 C-rich promoter sequence contains six runs of at least three contiguous cytosines that may serve as the core building block for i-motif formation. Recent experimental data with the bcl-2 C-rich promoter sequence utilizing only the middle four runs of cytosines has illustrated the ability of this sequence to form two intramolecular i-motif structures.19 These bcl-2 i-motif structures were not only present at low pH but persisted at pH 6.1, and approximately 5% of the oligomer remained at pH 7.19 This supports the possibility that i-motif structures may form opposite to the G-quadruplex in vivo and may also play an important role in the transcriptional process.
Since the bcl-2 promoter region consists of six cytosine runs, our studies utilized the full-length sequence rather than the previously studied truncated sequence of four cytosine runs19 to investigate the ability of this C-rich element to form i-motif structures. We demonstrated through CD, FRET, and fluorescence analyses that the full-length C-rich sequence upstream of the bcl-2 P1 promoter forms intramolecular i-motif structures in vitro with a transitional pH of 6.6. Isolation of three separate possible i-motif-forming DNA sequences from the full-length element as well as mutational studies of the full-length sequence confirmed that the full-length sequence is required to form the most stable i-motif structure. Bromine footprinting and fluorescent analog substitution of thymines within the loop regions have provided evidence that the predominant, biologically relevant bcl-2 i-motif species consists of an 8:5:7 loop folding pattern.
Materials and Experimental Methodology
DNA Oligonucleotides
DNA oligonucleotides were synthesized by Biosearch, and strand concentration was calculated using the Beer-Lambert law: A = ε·C·l. The extinction coefficients for each oligonucleotide were determined by the nearest neighbor method. Since these particular oligonucleotides exhibit higher order structures, absorbance at 260 nm was recorded at 95 °C (at least 20 °C above their respective Tm) to ensure the presence of single-stranded DNA. The extinction coefficients (M−1 cm−1) were as follows: Py39WT, 310 640; 5′-IM, 262 040; Mid-IM, 216 260; 3′-IM, 205 220; Py39 mutant, 314 200; Py39MT-5T and 36T, 305 500; Py39MT-12T, 16T, 17T, 18T, 19T, 26T, and 32T, 311 350; Py39MT-15T, 313 150; Py39MT-38T, 306 400. The Py39WT sequence corresponds to the C-rich strand within the bcl-2 promoter region from bases −1489 to −1451. Sequences are provided in Table 1.
Table 1.
Sequences of bcl-2 Oligonucleotides.
| DNA Oligomer | Sequence (5′→3′) |
|---|---|
| Py39WT | CAGCCCCGCTCCCGCCCCCTTCCTCCCGCGCCCGCCCCT |
| 5′-IM | CAGCCCCGCTCCCGCCCCCTTCCTCCCG |
| Mid-IM | GCTCCCGCCCCCTTCCTCCCGCGCCCG |
| 3′-IM | GCCCCCTTCCTCCCGCGCCCGCCCCT |
| Py39 mutant | CAGTTTTGCTCCCGCTTTCTTCCTTTTGCGCCCGCCCCT |
| Py39MT-5T | CAGCTCCGCTCCCGCCCCCTTCCTCCCGCGCCCGCCCCT |
| Py39MT-12T | CAGCCCCGCTCTCGCCCCCTTCCTCCCGCGCCCGCCCCT |
| Py39MT-15T | CAGCCCCGCTCCCGTCCCCTTCCTCCCGCGCCCGCCCCT |
| Py39MT-16T | CAGCCCCGCTCCCGCTCCCTTCCTCCCGCGCCCGCCCCT |
| Py39MT-17T | CAGCCCCGCTCCCGCCTCCTTCCTCCCGCGCCCGCCCCT |
| Py39MT-18T | CAGCCCCGCTCCCGCCCTCTTCCTCCCGCGCCCGCCCCT |
| Py39MT-19T | CAGCCCCGCTCCCGCCCCTTTCCTCCCGCGCCCGCCCCT |
| Py39MT-26T | CAGCCCCGCTCCCGCCCCCTTCCTCTCGCGCCCGCCCCT |
| Py39MT-32T | CAGCCCCGCTCCCGCCCCCTTCCTCCCGCGCTCGCCCCT |
| Py39MT-35T | CAGCCCCGCTCCCGCCCCCTTCCTCCCGCGCCCGTCCCT |
| Py39MT-36T | CAGCCCCGCTCCCGCCCCCTTCCTCCCGCGCCCGCTCCT |
| Py39MT-37T | CAGCCCCGCTCCCGCCCCCTTCCTCCCGCGCCCGCCTCT |
| Py39MT-38T | CAGCCCCGCTCCCGCCCCCTTCCTCCCGCGCCCGCCCTT |
| 10T | CAGCCCCGCNdUCCCGCCCCCTTCCTCCCGCGCCCGCCCCT |
| 20T | CAGCCCCGCTCCCGCCCCCNdUTCCTCCCGCGCCCGCCCCT |
| 21T | CAGCCCCGCTCCCGCCCCCTNdUCCTCCCGCGCCCGCCCCT |
| 24T | CAGCCCCGCTCCCGCCCCCTTCCNdUCCCGCGCCCGCCCCT |
| 39T | CAGCCCCGCTCCCGCCCCCTTCCTCCCGCGCCCGCCCCNdU |
Preparation and End-Labeling of Oligonucleotides
The DNA oligonucleotides were 5′-end-labeled with [γ-32P] ATP upon incubation with T4 polynucleotide kinase for 1 h at 37 °C. Subsequently, T4 kinase was heat inactivated for 5 min at 95 °C, and the labeled oligonucleotides were purified using a Bio-Spin 6 chromatography column (BioRad). The oligonucleotides were further purified by denaturing gel electrophoresis (12% PAGE).
Circular Dichroism (CD)
CD analyses were conducted on a Jasco-810 spectropolarimeter (Jasco, Easton, MD, USA), using a quartz cell of 1 mm optical path length. Specifically, spectra were obtained at an instrument scanning speed of 100 nm/min, with a response time of 1 s, over a wavelength range of 200–350 nm and were recorded three times, averaged, smoothed, and baseline-corrected for signal contributions from buffers. Molar ellipticities for melting curves were recorded at 286 nm (the λ of the maximum molar ellipticity) over a temperature range of 4–100 °C and then plotted against temperatures for Tm determination. Tm values were calculated within ±1 °C error. The i-motif-forming oligonucleotides were prepared at a 5 μM strand concentration in a 50 mM Na cacodylate buffer (pH 4.4, 5.0, 5.4, 5.9, 6.1, 6.5, 7.1, or 8.0). Due to the buffering capacity of cacodylate buffer (pH 5.0–7.4), experiments at pH 4.4 were also performed using Tris-acetate, which has a buffering capacity at pH 4.4. These experiments produced results similar to those with the cacodylate buffer (data not shown), and for experimental consistency, only results using cacodylate buffer were discussed.
Fluorescence Resonance Energy Transfer Assay (FRET)
Biosearch Technologies provided FRET probes of the Bcl-2 Py39WT, mutant, and poly-T sequences modified at the 5′-end with a FAM-fluorophore and at the 3′-end with a black hole quencher. Each probe was placed in a 50 mM Na cacodylate buffer (pH 4.4, 5.0, 5.4, 5.9, 6.1, 6.5, 7.1, or 8.0) at a concentration of 1 μM and incubated at room temperature (25 °C) for 1 h to allow for i-motif formation. Samples were prepared in triplicate, and 100 μL was placed into each well of a black 96-well optiplate (PerkinElmer). Fluorescence measurements were recorded by a Bio-Tek Synergy HT spectrofluorimeter at 25 °C with 20 nm bandwidths. The excitation and emission wavelengths were set at 495 nm and 528 nm, respectively. Endpoint fluorescence or quenching was plotted as the average relative fluorescence units of the triplicate wells after correction for background. The control poly-T probe was used to evaluate the effects of acidic pH on the emission spectra of the FRET probes. The excitation wavelength was fixed at 495 nm, and emission spectra were recorded from 500 to 540 nm for pH 4.4, 5.4, 6.1, 7.1, and 8.0. The maximum fluorescence for emission was observed at 515 nm for probe in pH 4.4 and 5.4, 516 nm for pH 6.1, and 517 nm for pH 8.0 (data not shown).
Naphthodeoxyuridine Fluorescence Assay
A fluorescent thymine analogue, naphthodeoxyuridine (NdU), was prepared as previously reported (Scheme 1).65 Briefly, TMS-mediated activation of benzo[g]quinazoline-2,4-(1 H, 3H)-dione (1)66 in N, N-hexamethyldisilazane followed by condensation with bis-3, 5-O-methylbenzoyl-2-deoxyribofuranosyl chloride (2)67,68 in the presence of CuI afforded an α, β anomeric mixture of NdU derivative 3 in 40% yield over two steps. Removal of the O-3 and O-5 methylbenzoyl groups was accomplished using sodium methoxide in MeOH to afford 4 in 71% yield. 5′-DMTr protection in pyridine and separation of the anomers by chromatography gave 5, as the β-anomer, in 11% yield. Phosphitylation using diisopropylethylamine and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite in CH2Cl2 then afforded NdU phosphoramidite 6 in 41% yield. The NdU was incorporated into five separate oligonucleotides to replace each of the five thymidines present in the Py39WT sequence one at a time. The NdU extinction coefficient of 49 800 M−1 cm−1 was used in calculations for each substituted oligomer.66 The extinction coefficient for each oligomer, 10T, 20T, 21T, 24T, and 39T, was 328 680 M−1 cm−1. Each probe was placed in a 50 mM Na cacodylate buffer (pH 4.4, 5.1, 6.0, 6.3, 6.6, 7.4, or 8.0) at a strand concentration of 10 μM, incubated at 95 °C for 5 min, and allowed to cool to room temperature (25 °C) to allow for i-motif formation. Samples were prepared in triplicate to a final volume of 150 μL, and 50-μL aliquots were placed into each triplicate well of a black 96-well optiplate (PerkinElmer). Fluorescence measurements were recorded using a Bio-Tek Synergy HT spectrofluorimeter at 25 °C with 20 nm bandwidth. The excitation and emission wavelengths were set at 250 nm and 440 nm, respectively. Endpoint fluorescence or quenching was plotted as the average relative fluorescence units of the triplicate wells after correction for background.
Scheme 1.
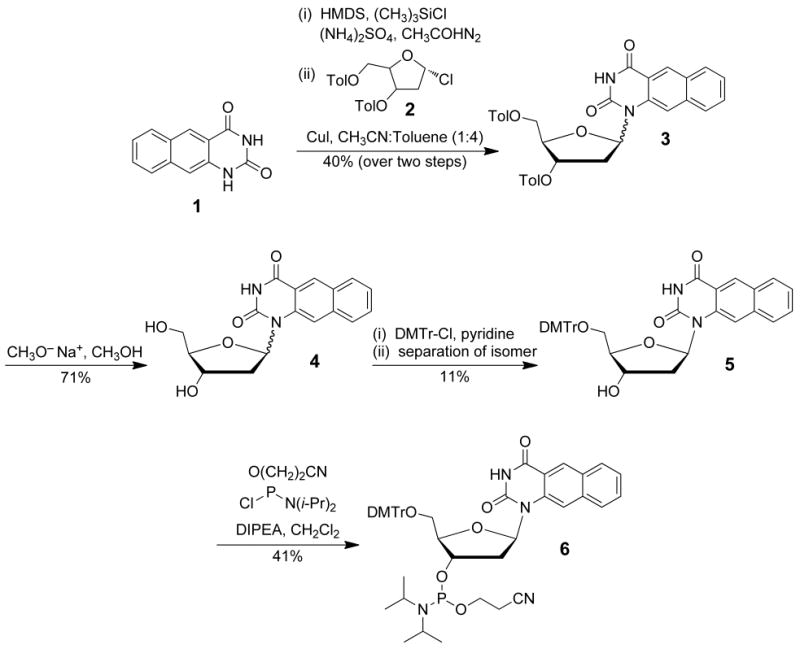
Synthesis of the phosphoramidite of naphthodeoxyuridine.
Electrophoretic Mobility Shift Assay
The Py39WT, Py39 mutant, and poly-T oligonucleotides were 5′-end-labeled with [γ-32P] ATP as noted above. These oligonucleotides were prepared at 10 000 cpm in 50 mM Na cacodylate buffers, pH 4.4 and 6.1, and subjected to electrophoresis on a 12% non-denaturing PAGE gel. As a control, the same oligonucleotide preparations were also subjected to electrophoresis on a 12% denaturing PAGE gel to demonstrate similar mobility among all three oligonucleotides.
Bromine Footprinting
The bromine footprinting assay protocol was adapted from a previously established procedure.69 Briefly, Py39WT oligonucleotide was 5′-end-labeled with [γ-32P] ATP as previously described. The purified 5′-end-labeled Py39WT oligomer was incubated with molecular bromine formed in situ by mixing an equal molar concentration (50 mM) of KBr with KHSO5 for 20 min and terminated by the addition of 60 μL of a 0.6 M sodium acetate and calf thymus DNA (10 mg/mL) solution. Any unreacted bromine was removed in subsequent ethanol precipitation steps. After ethanol precipitation, the DNA pellet was dried and resuspended in 30 μL of a 100 mM piperidine solution. Samples were heated at 90 °C for 20 min to induce bromination-specific strand cleavage, dried, and resuspended with alkaline sequencing gel loading dye. The bromination-specific strand cleavage was visualized on a sequencing gel (20% PAGE with 7 M urea). A pyrimidine-specific reaction was performed using hydrazine to generate a cytosine sequencing marker.70
Results
The Cytosine-Rich Sequence within the bcl-2 Promoter Region Can Form One or More Intramolecular i-Motif DNA Secondary Structures
The full-length wild-type C-rich sequence from the bcl-2 promoter region (Py39WT, Table 1) was subjected to a pH gradient CD spectral analysis. Previous studies involving i-motif-forming oligonucleotides at acidic pHs have determined that the signature CD spectra specific for an i-motif secondary structure consists of a maximum positive peak within 280–288 nm and a negative peak within 260–267 nm.19,62,64,71
Purified Py39WT oligonucleotide displayed the characteristic i-motif positive peak at 286 nm and a negative peak approximately half the amplitude of the positive peak at 264 nm at pH ≤ 6.5 (Figure 2A, left). These CD spectral features are distinctive for hemiprotonated cytosine+–cytosine base pairs.71 The positive maximum begins to shift toward 277 nm at pH 7.1, and the molar ellipticity decreases by over 50% at pH 8.0, which is consistent with an unordered, random-coiled structure.72 The transitional pH of 6.6 was quantitatively determined from the plot of molar ellipticity at 286 nm versus the corresponding pH (Figure 2A, right). Subsequent CD melt curve experiments were performed to examine the pH dependence of the thermal stability of Py39WT. In agreement with the CD spectra analysis, the melting curves (Figure 2B, right) and calculated Tm (Figure 2B, left) revealed a pH-dependent stability of the i-motif structure. The i-motif structure has a Tm of 69 °C under acidic conditions that decreases with increasing pH to 40 °C near neutral pH. These Tm values are consistent, albeit slightly higher, with previously published data using a Py39WT truncated sequence containing only the middle four runs of cytosines that yielded a Tm of 68 °C at pH 4.5 and 36 °C at pH 6.1.19 Since our thermal stability studies involved the full-length sequence containing all six cytosine runs, it is expected to potentially exhibit greater stability and therefore provide slightly higher Tm values. In addition, a pH gradient experiment was conducted using a FRET assay to further illustrate the potential of the Py39WT sequence to adopt an i-motif structure. A FRET probe consisting of the Py39WT sequence with 5′-end FAM fluorophore and 3′-end black hole quencher labels was assayed for relative fluorescence at various pH levels. In the presence of i-motif formation, the black hole quencher resides in close proximity to the fluorophore and quenches fluorescence. A poly-T probe served as a negative control for the absence of i-motif formation, and a Py39 mutant probe unable to form a stable i-motif was designed as an intermediate control probe. The relative fluorescence units of the Py39WT probe steadily increased with rising pH levels (Figure 2C). At pH 6.5 and higher the Py39WT probe reached similar relative fluorescence units as the mutant and poly-T probes, confirming the loss of i-motif stability that occurs at the transitional pH. The pH-dependent fluorescence of the Py39WT probe exhibited almost 100% quenching under the most stable i-motif structure–forming conditions (pH 4.4), in contrast to the mutant and poly-T probes that displayed significantly more fluorescence (Figure 2C). This trend remains observable at pH 5.0 with a 4- and 21-fold decrease in fluorescence of the wild-type probe compared to the mutant and poly-T probes, respectively, and the decreases at pH 5.4, pH 5.9, and pH 6.1 were 6- and 40-fold, 9- and 23-fold, and 12- and 20-fold, respectively.
Figure 2.
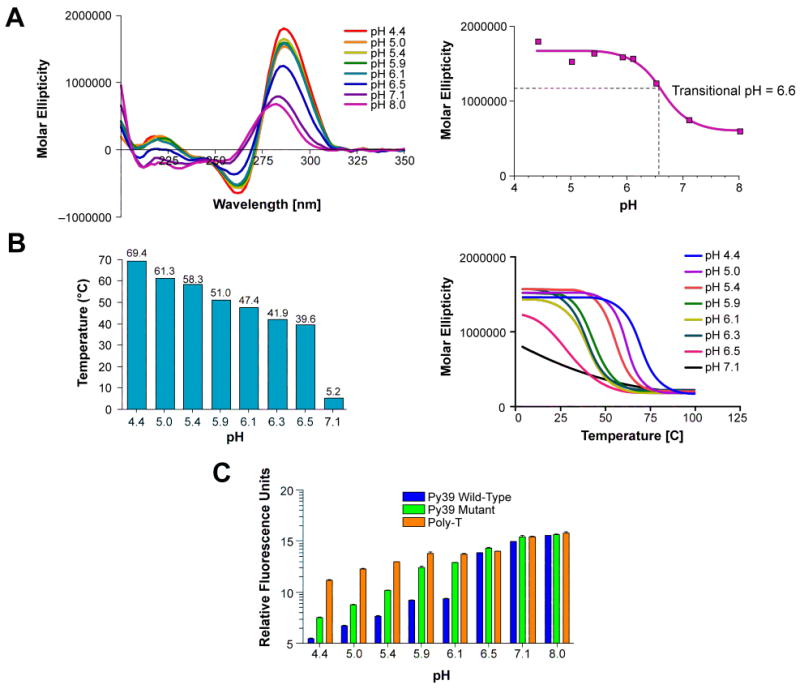
The bcl-2 Py39WT sequence was subjected to a pH gradient for CD spectra analysis (A, left), melt temperature determination (B, left), and the FRET assay (C). The transitional pH was determined from the plot of pH versus the corresponding molar ellipticity at 286 nm (A, right). Melt temperatures were calculated from CD thermal curves (B, right). The bcl-2 Py39 mutant and poly-T sequences were also used in the FRET assay as i-motif unstable and negative controls, respectively. Relative fluorescence was plotted in log scale (C).
In addition, we conducted a concentration-dependent thermal stability analysis as well as an electrophoretic mobility assay to assess whether the Py39WT oligonucleotide formed an intramolecular or intermolecular secondary structure. Under nondenaturing conditions, the Py39WT sequence displayed faster electrophoretic mobility at both pH 4.4 and 6.1 with respect to the Py39 mutant and poly-T sequences, indicative of an intramolecularly folded structure (Figure 3A). In contrast, denaturing conditions demonstrated that all three sequences exhibited similar mobility and thus consist of the same oligomer length (Figure 3B). CD melt curves of increasing DNA strand concentration (2.5–50 μM) at pH 4.4 and 6.1 revealed a concentration-independent thermal stability of the Py39WT i-motif structure, confirming formation of an intramolecularly folded structure at both acidic and near neutral pHs (data not shown).
Figure 3.
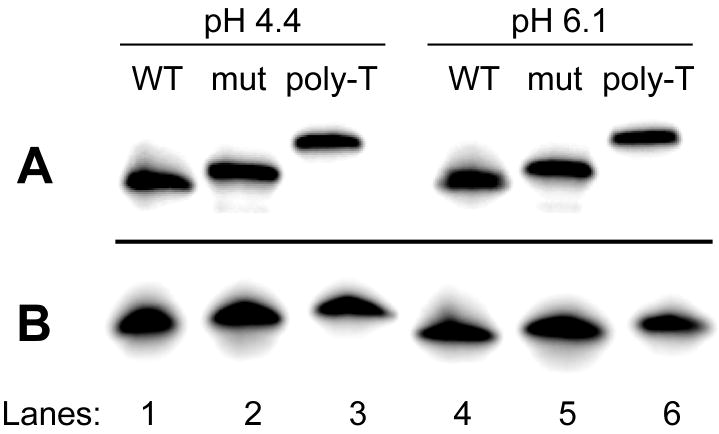
12% non-denaturing (A) and denaturing (B) gel analysis of the bcl-2 Py39WT sequence at pH 4.4 and 6.1 (lanes 1 and 4). For comparison, the Py39 mutant (lanes 2 and 5) and 39-mer poly-T (lanes 3 and 6) sequences were analyzed.
Isolation and CD Analysis of Three Truncated Py39WT Sequences Demonstrate the Ability to Form Different i-Motif Structures within the bcl-2 Promoter
The G- and C-rich strands within the bcl-2 promoter region both display highly complex arrangements of repetitive tracts. Previous studies conducted to elucidate the predominant G-quadruplex structure demonstrated that the 5′- and 3′-end contiguous runs, as well as the middle four guanine runs, formed three different secondary structures.15,18 Therefore, we utilized this approach as the starting point for the determination of the predominant folding pattern of the i-motif. The Py39WT sequence was truncated to isolate the three major i-motif structures that may form from four adjacent 5′-end, middle, or 3′-end runs of cytosines. These i-motif structures are denoted as 5′-IM, Mid-IM, and 3′-IM, and the sequences are provided in Table 1 and Figure 4A. The transitional pH for each truncated sequence was determined following CD spectral analysis of a pH gradient (data not shown). The transitional pHs for 5′-IM (6.4) and Mid-IM (6.2) were comparable to the full-length (6.6), while 3′-IM yielded a lower transitional pH of 5.8. In addition, the CD spectra of all three oligonucleotides at pH 4.4 (Figure 4B) and pH 6.1 (Figure 4C) were compared to spectra obtained from the Py39WT sequence. Although all three truncated sequences displayed spectra specific for an i-motif structure at both pH 4.4 and 6.1, the molar ellipticity decreased by at least 50% and 25%, respectively (Figure 4, B and C). In addition, with the exception of the Mid-IM, which retained the 286 nm positive maximum at pH 4.4, the oligomer fragments of Py39WT displayed a shift in the maximum toward 290 nm (Figure 4, B and C).
Figure 4.
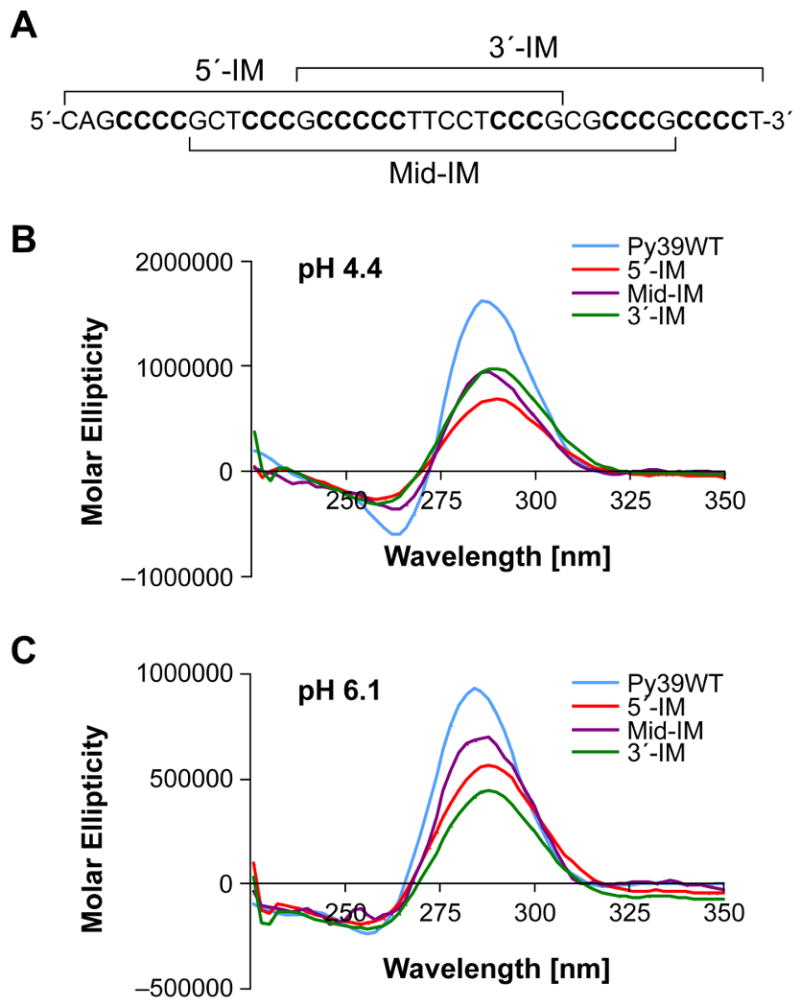
The full-length Py39WT sequence, with each of the truncated sequences bracketed, is shown in panel A. Truncated sequences are also listed in Table 1. Comparison of CD spectra at pH 4.4 (B) and pH 6.1 (C) of the three possible i-motif-forming sequences involving either the 5′-end (5′-IM), middle (Mid-IM), or 3′-end (3′-IM) four runs of cytosines to the full-length (Py39WT).
Characterization of the Predominant bcl-2 i-Motif Species at pH 6.1 Suggests a Major 8:5:7 Loop Folding Pattern of the Full-Length Sequence
A series of mutational, thermal stability, fluorescent thymine substitution, and bromine footprinting experiments were performed to characterize the predominant bcl-2 i-motif conformation from the full-length wild-type sequence. Mutational analysis of the Py39WT sequence was performed in conjunction with thermal stability studies to evaluate which of the major runs of cytosines are most important for i-motif stability. CD melt curves were obtained for each mutant at pH 6.1 and compared to the wild-type sequence. Bromine footprinting analysis was then performed to identify the specific cytosines involved in i-motif formation at pH 6.1 and consequently served as a basis for predicting the loop sizes and folding pattern. Last, each thymine within the Py39WT sequence was substituted with a fluorescent analog, not only to serve as a secondary pH gradient assay, but also to provide information regarding thymine position and stacking environment within the i-motif loop regions.
A. Mutational studies in combination with thermal stability analysis suggest that the predominant bcl-2 i-motif structure involves non-contiguous runs of cytosines separated by large loops
Sequential mutation of a single cytosine to a thymine within each of the six cytosine tracts was performed to predict which runs of cytosines are involved in the formation of the predominant bcl-2 i-motif (Table 1 and Figure 5A). The Tm of each mutant oligomer at pH 6.1 was then determined by CD thermal analysis (Figure 5A). In addition, each cytosine within tracts III and VI, which consist of more than three cytosines, was mutated in order to determine the specific cytosines involved in the base-pairing of the i-motif (Figure 5A). Overall, mutations of cytosines within runs I, III, IV, and VI most significantly affected the thermal stability of the bcl-2 Py39 i-motif (Figure 5A). The mutation of cytosine C5 within run I produced an 8.5 °C decrease in Tm of the bcl-2 Py39 i-motif. Mutation of each of the central three cytosines in run III (C16, C17, C18) resulted in a 9.5 °C or greater decrease in melting temperature, while mutation of the flanking cytosines, C15 and C19, produced a much smaller ΔTm (−4.9 °C and −6 °C, respectively). A decrease of 5.8 °C in Tm was observed for mutation of C26 within run IV. Mutations of cytosines C35, C36, C37, and C38 within run VI significantly lowered the Tm of Py39WT by 7.5, 7.2, 7.7, and 9.3 °C, respectively. In contrast, mutations of cytosines within runs II (C12) and V (C32) produced minimal effects on the thermal stability of the Py39WT i-motif, with only a 2.1 or 2.8 °C decrease in melt temperature, respectively. These mutational studies implicate the major involvement of runs I, III, IV and VI in providing the stability of the bcl-2 promoter i-motif (Figure 5B) and also provide additional information on which cytosines in runs III and VI are used for i-motif formation.
Figure 5.
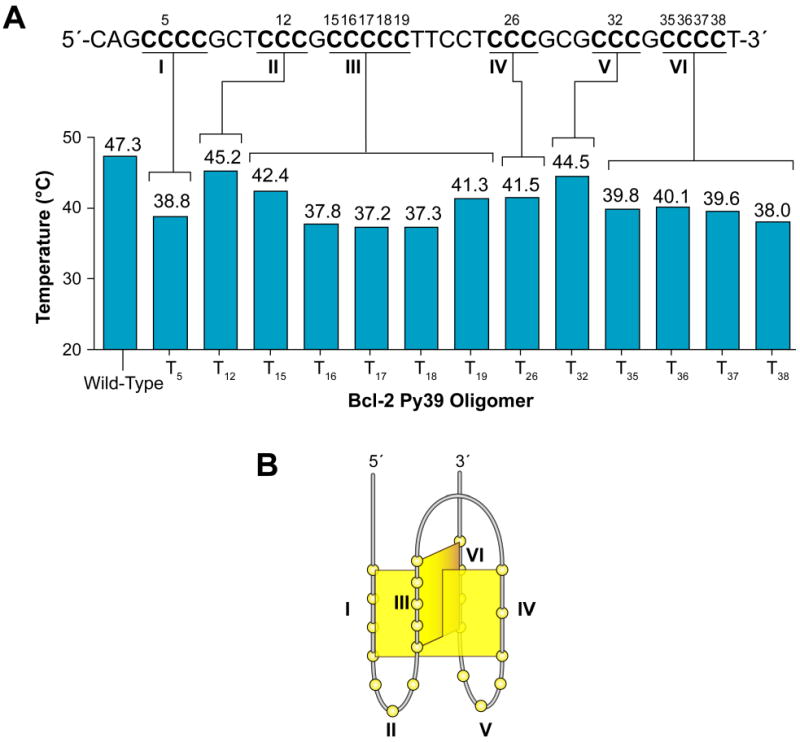
Mutational analysis of the bcl-2 Py39WT sequence. The full-length bcl-2 Py39 oligomer sequence with each of the mutated cytosines labeled is shown in the upper part of panel A. The lower section of panel A displays a graphical representation of the specified bcl-2 Py39 oligomer melting temperatures obtained by CD thermal analysis of each mutant at pH 6.1. A potential folding pattern based on mutant thermal analysis is shown in panel B.
B. The bromine footprinting protection pattern identifies the specific cytosines involved in the bcl-2 promoter i-motif structure
Bromine footprinting can be used to identify the specific cytosines involved in the base pairing and intercalation in the bcl-2 i-motif structure due to preferential bromination of cytosines that lack steric or electrostatic hindrance at the C5 position.24,69 Reactivity of these cytosines with bromine results in their increased susceptibility to cleavage by piperidine, and PAGE can then be used to visualize the resulting cleavage products. Consistent with the result from Tm measurements of mutant oligomers, the overall protection pattern of the Py39WT oligomer at pH 6.1 revealed that cytosines within runs I, III, IV, and VI were consistently protected or partially protected, whereas cytosines within runs II and V displayed strong piperidine cleavage (Figure 6, A and B). The three 3′-end cytosines of run I (C5, C6, and C7) were strongly protected in comparison to the 5′-end cytosine, C4. The 5′-cytosine of run III (C15) showed pronounced cleavage in comparison to the protection of the other four cytosines. Cytosines C17 and C18, were well protected, whereas C16 and C19 were only partially protected. Complete or partial protection from piperidine cleavage was observed for the three cytosines within both runs IV and VI (Figure 6, A and B). In contrast, all of the cytosines within runs II and V displayed significant cleavage (Figure 6A). Significantly, cytosines within the 5′- and 3′-lateral loop regions, C9 and C29, respectively, displayed different cleavage patterns (Figure 6, A and D). Complete protection was observed for C9, which resides between runs I and II, whereas C29, located between runs IV and V, was significantly cleaved. A scan of the band intensities from the 0.5 mM bromine reaction was performed to confirm the protection pattern observed in the PAGE analyses (Figure 6, A and B). A schematic of this protection pattern along with the identification of the cytosine tracts and loop regions is provided in Figure 6C. On the basis of these results, and in conjunction with data obtained from the mutational studies, a folding pattern of the predominant bcl-2 i-motif structure is proposed to consist of seven intercalated cytosine+–cytosine base pairs with loop regions of 8, 5, and 7 bases (Figure 6D).
Figure 6.
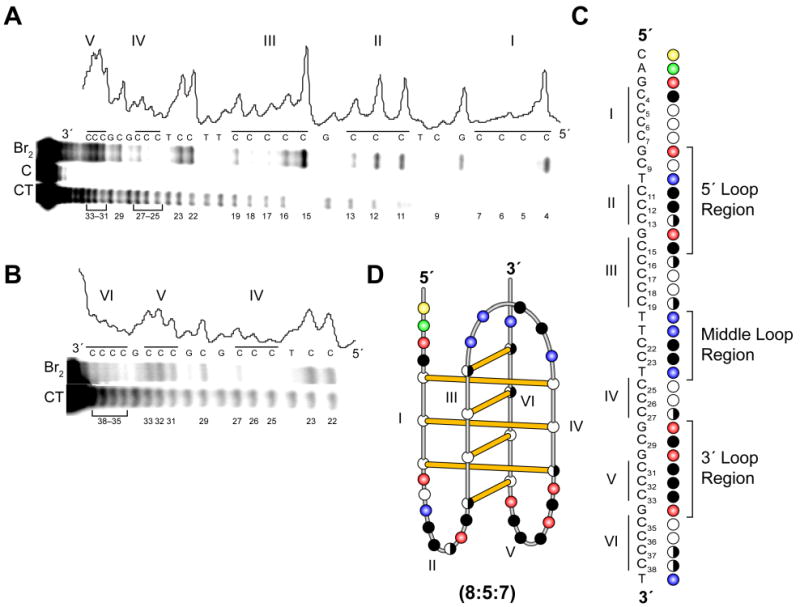
Bromine footprinting pattern obtained from the Py39WT oligomer at pH 6.1. Panels A and B display the autoradiogram and densitometric scans of the PAGE band intensities of the cleavage products from the Py39WT sequence. The CT lanes represent pyrimidine sequencing, while the bromine lanes reflect cytosine cleavage in the presence of 0.5 mM bromine. Lane C in panel A represents the cleavage reaction in the absence of bromine. The scanned band intensities correspond to the 0.5 mM bromine reaction displayed in the bromine lanes. Illustrations of the cytosine protection pattern with the designated loop regions (C) and the proposed folding pattern (D) are based on the bromine footprinting PAGE analysis. The open (white), half open (white/black), and closed (black) circles represent full protection, partial protection, and no protection from cleavage of cytosines, respectively. The yellow, green, red, and blue circles represent the nucleobases cytosine, adenine, guanine, and thymine, respectively.
C. NdU substitutions indicate the potential involvement of loop thymines in stacking interactions that differ within the 5′ and 3′ loops
To gain further information on the potential for base stacking interaction in the loops of the bcl-2 i-motif, each thymine within the bcl-2 Py39WT sequence was sequentially replaced with the fluorescent thymine analog NdU, which fluoresces in an unquenched environment or becomes quenched in the presence of base–base stacking interactions. Replacement of thymine T39, which resides at the 3′-end of the oligomer and outside the i-motif forming region, served as an internal control and does not exhibit pH-dependent fluorescence (Figure 7A). Significantly, thymines within the loops of the i-motif-forming region of the Py39WT sequence demonstrated different fluorescence patterns. Substituted thymines at positions 20, 21, and 24 displayed quenched fluorescence at low pH levels (pH 4.4–6.3) and fluorescence at higher pH levels (pH 6.6–8.0) (Figure 7A). Incorporation of a fluorescent thymine at positions 20 or 21 resulted in at least 50% less fluorescence, which persisted over the low pH levels (pH 4.4–6.3). Significant quenching was also observed for a fluorescent thymine at position 24 (pH 4.4); however, fluorescence steadily increased with increasing pH. In contrast, substitution of thymine 10 resulted in fluorescence only at acidic pH levels and quenched fluorescence at neutral pH levels (Figure 7A). Figure 7B displays the proposed loop positions of the thymines in a schematic model of the i-motif structure.
Figure 7.
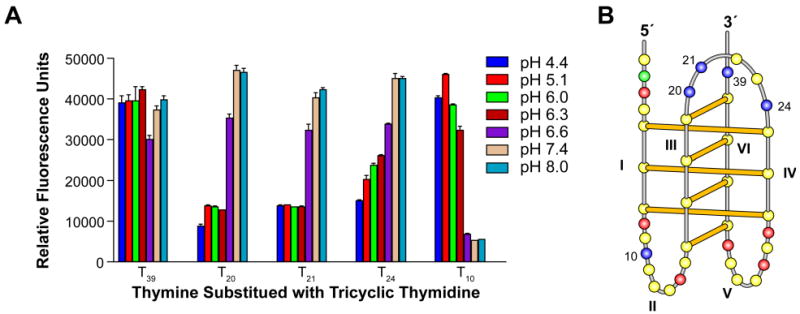
Each of the thymines within the bcl-2 Py39 sequence was substituted with a fluorescent tricyclic naphthodeoxyuridine thymine analog. Fluorescence was plotted for each substituted thymine as relative fluorescence units over a pH gradient (A). Panel B displays the proposed folding pattern of the bcl-2 i-motif with identification of the substituted thymines. The yellow, green, red, and blue circles represent the nucleobases cytosine, adenine, guanine, and thymine, respectively.
Discussion
The bcl-2 proto-oncogene encodes an anti-apoptotic protein involved in the inhibition of the mitochondrial apoptotic pathway. Dysregulation of bcl-2 expression may lead to prolonged cell survival despite apoptotic stimuli, which is hypothesized to result in the chemoresistance observed in a wide variety of cancers that overexpress bcl-2.1,4–6 Alternatively, downregulation of bcl-2 has been associated with extensive neuron apoptosis in a number of neurodegenerative diseases.7,8 The prevalence of defects in bcl-2 expression in the two distinct pathologies makes bcl-2 an attractive target for therapy. The P1 promoter region responsible for the majority of bcl-2 expression consists of multiple transcription factor binding sites, as well as a GC-rich element, that may serve as sites for modulating transcription.
While our previous studies have focused on the G-rich strand within the bcl-2 promoter region, the present study involves the characterization of the dynamic topology of the complementary C-rich strand. The pH-dependent formation of the bcl-2 i-motif within an in vitro system raises the question of the biological relevance of i-motif formation within cellular DNA. Significantly, recent studies have demonstrated that i-motif formation within the c-myc promoter region may occur purely as a result of negative supercoiling.24 This provides a precedent that the C-rich strand of bcl-2 will also exhibit dynamic properties, including an i-motif structure in vivo. Further plasmid chemical probing experiments are required to address the induction of i-motif formation through negative supercoiling within the bcl-2 promoter. These studies are ongoing. There are numerous i-motif folding patterns possible from the six runs of three, four, or five cytosines in the complex runs of cytosines within the bcl-2 C-rich promoter sequence. Consistent with a recent publication in which only the central four cytosine runs were examined,19 our results confirm that intramolecular i-motif formation occurs within the bcl-2 promoter sequence. However, our data suggest the presence of a different conformation that requires all six runs of cytosines of the full-length sequence to form the most stable, biologically relevant i-motif structure. We demonstrate, through mutational studies coupled with bromine footprinting, that the predominant i-motif structure formed by the bcl-2 C-rich promoter sequence consists of an 8:5:7 loop folding pattern (Figures 6 and 7). The discovery of these large lateral loops poses the question, what significance or advantage does the large loop size offer the i-motif structure? Typically, the most stable G-quadruplex structures exhibit smaller loop sizes than those found in i-motifs, which accommodate the characteristic double-chain reversal and lateral loops.73–76 The complementary bcl-2 promoter G-rich strand forms a mixed parallel/antiparallel G-quadruplex that can adopt three different G-quadruplex structures. Although an equilibrium between the three conformations may exist, the middle four runs of guanines were shown to provide the most predominant, stable conformation, consisting of three G-tetrads with a 3:7:1 loop folding pattern, which results in the double-chain reversal and two lateral loops.15 Therefore, the additional non-quadruplex forming guanine tracts within the G-rich strand may provide the necessary number of cytosine tracts for stable i-motif formation. A possible reason for the larger loop sizes in the bcl-2 promoter is that these are needed to provide additional stability, perhaps through the formation of capping structures. Indeed, the mutational studies and the bromine footprinting analyses provided evidence for the involvement of loop cytosines in a capping structure, while thymine involvement was demonstrated through the NdU fluorescence assay. The complete and partial protection observed for the 5′-loop cytosines C9 and C13, respectively (Figure 6A) suggest that these cytosines may be involved in a capping structure formed by the bases within the lateral loop regions. Consistent with the bromine footprinting pattern and the possibility that these cytosines function in stabilizing the i-motif through the formation of capping structures, mutation of C9 to a thymine resulted in a ΔTm of -7.8° (data not shown).
A novel assay was designed based on the unique fluorescence emission properties of a NdU thymine analog that was previously used to detect the formation of triplex DNA.64 For our purposes, the incorporation of NdU into a secondary structure–forming oligomer provided information regarding the local environment of each thymine within the loop regions. The phosphoramidite of NdU was prepared as previously described64 for incorporation into five separate oligonucleotides to sequentially replace each of the five thymines within the Py39WT sequence. Thymine substitution with NdU at positions 20, 21, and 24 within the top (middle) loop resulted in quenched fluorescence in the presence of i-motif formation at pH 6.3 or lower (Figure 7A). Presumably, these thymines are involved in stacking interactions within the loop regions to form a capping structure. The capping structures formed by the large 8:5:7 loop conformation of the bcl-2 i-motif may be responsible for the high transitional pH (6.6) and stability. Similarly, the well-characterized c-myc i-motif also utilizes six cytosine tracts, which results in a 6:2:6 loop conformation, and, as in the bcl-2 i-motif, these large lateral loops most likely contribute to the high transitional pH of 6.5–6.8.60,62 This is in contrast to the previously characterized smaller i-motif loop conformation of 2:(3–4):2 for the VEGF,49 RET,56 and Rb63 structures, which exhibit lower transitional pHs of 5.9, 6.4, and 5.9, respectively. Interestingly, thymine substitution with NdU at position 10 gave a different pH-dependent pattern of fluorescence quenching, suggesting that the local environment of this residue is distinctly different than those at positions 20, 21, and 24.
In addition to the importance of loop size to the overall conformation and stability of the i-motif, perhaps the capping structures formed from these large loops may also serve as recognition scaffolds for specific interaction with nuclear proteins and even small molecules. In support of this hypothesis, loop sizes and capping structures in RNA secondary structures are important for stabilization and protein recognition of hairpins. Single-stranded RNA most often folds back on itself to form a stem-loop structure where the stem adopts a duplex conformation and the loop consists of noncanonical base pairing and stacking of nucleotides.77 In DNA the i-motif structure is also formed from a single-strand that folds into an antiparallel duplex with a loop structure; however, the i-motif consists of a second fold-back that produces additional loop regions.28–30 While the stem of RNA secondary structures primarily consists of Watson-Crick base pairs, there is evidence of non-Watson-Crick regions.77 For example, RNA hairpins have been shown to consist of protonated adenine–cytosine base pairs at neutral pH that contribute to structure stabilization.78,79 In addition, the thermal and structural stability of RNA hairpins also depend on the loop nucleotide sequence and size.80,81 For example, rRNA hairpins appear to be most stable with a tetraloop, whereas stable hairpins formed from tRNA consist of seven nucleotides in the loop region.82 As a consequence, it is not surprising that proteins have been shown to recognize and interact with RNA secondary structures in a sequence-specific fashion. Specifically, the loop regions are important protein recognition sites, as mutations within the loop region disrupt RNA–protein interaction.83 Thus, in addition to providing stability through the formation of capping structures, the two lateral loops of 8 and 7 bases may together offer selectivity in protein recognition and small molecular targeting of the i-motif for modulation of bcl-2 transcription.
Acknowledgments
This research was supported by grants from the Arizona Board of Regents, the National Institutes of Health (GM085585, T32CA09213), and the Leukemia and Lymphoma Society (6225-08). We thank Daekyu Sun for providing experimental guidance with the bromine footprinting protocol and interpretation of data. We also thank David Bishop for his significant contribution in the preparation and editing of the final version of the text and figures displayed in the article.
References
- 1.Vaux DL, Cory S, Adams JM. Nature. 1988;335:440. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 2.Yunis JJ. Science. 1983;221:227. doi: 10.1126/science.6336310. [DOI] [PubMed] [Google Scholar]
- 3.Oltvai ZN, Milliman CL, Korsmeyer SJ. Cell. 1993;74:609. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 4.Baretton GB, Diebold J, Christoforis G, Vogt M, Muller C, Dopfer K, Schneiderbanger K, Schmidt M, Lohrs U. Cancer. 1996;77:255. doi: 10.1002/(SICI)1097-0142(19960115)77:2<255::AID-CNCR6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 5.Tjalma W, De Cuyper E, Weyler J, Van Marck E, De Pooter C, Albertyn G, van Dam P. Am J Obstet Gynecol. 1998;178:113. doi: 10.1016/s0002-9378(98)70636-2. [DOI] [PubMed] [Google Scholar]
- 6.Joensuu H, Pylkkanen L, Toikkanen S. Am J Pathol. 1994;145:1191. [PMC free article] [PubMed] [Google Scholar]
- 7.Berry MD, Boulton AA. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:197. doi: 10.1016/S0278-5846(03)00015-0. [DOI] [PubMed] [Google Scholar]
- 8.Bar-Am O, Weinerb O, Amit T, Youdim MBH. FASEB J. 2005;19:1899. doi: 10.1096/fj.05-3794fje. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y, Mehew JW, Heckman CA, Arcinas M, Boxer LM. Oncogene. 2001;20:240. doi: 10.1038/sj.onc.1204067. [DOI] [PubMed] [Google Scholar]
- 10.Heckman C, Mochon E, Arcinas M, Boxer LM. J Biol Chem. 1997;272:19609. doi: 10.1074/jbc.272.31.19609. [DOI] [PubMed] [Google Scholar]
- 11.Heckman C, Mehew JW, Boxer LM. Oncogene. 2002;21:3898. doi: 10.1038/sj.onc.1205483. [DOI] [PubMed] [Google Scholar]
- 12.Ji L, Mochon E, Arcinas M, Boxer LM. J Biol Chem. 1996;271:22687. doi: 10.1074/jbc.271.37.22687. [DOI] [PubMed] [Google Scholar]
- 13.Wilson BE, Mochon E, Boxer LM. Mol Cell Biol. 1996;16:5546. doi: 10.1128/mcb.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomez-Manzano C, Mitlianga P, Fueyo J, Lee HY, Hu M, Spurgers KB, Glass TL, Koul D, Liu TJ, McDonnell TJ, Yung WK. Cancer Res. 2001;61:6693. [PubMed] [Google Scholar]
- 15.Dexheimer T, Sun D, Hurley LH. J Am Chem Soc. 2006;128:5404. doi: 10.1021/ja0563861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Liu Y, Lin S, Yuan G. Chem Eur J. 2009;15:2445. doi: 10.1002/chem.200801922. [DOI] [PubMed] [Google Scholar]
- 17.Dai J, Chen D, Jones RA, Hurley LH, Yang D. Nucleic Acids Res. 2006;34:5133. doi: 10.1093/nar/gkl610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai J, Dexheimer TS, Chen D, Carver M, Ambrus A, Jones RA, Yang D. J Am Chem Soc. 2006;128:1096. doi: 10.1021/ja055636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan N, Avino A, Tauler R, Gonzalez C, Eritja R, Gargallo R. Biochimie. 2007;89:1562. doi: 10.1016/j.biochi.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 20.Benham CJ. Annu Rev Biophys Biophys Chem. 1985;14:23. doi: 10.1146/annurev.bb.14.060185.000323. [DOI] [PubMed] [Google Scholar]
- 21.Lee JS, Ahsley C, Hample KJ, Bradley R, Scraba DG. J Mol Biol. 1995;252:283. doi: 10.1006/jmbi.1995.0495. [DOI] [PubMed] [Google Scholar]
- 22.Kouzine F, Levens D. Front Biosci. 2007;12:4409. doi: 10.2741/2398. [DOI] [PubMed] [Google Scholar]
- 23.Kouzine F, Sandrod S, Elisha-Feil Z, Levens D. Nat Struct Mol Biol. 2008;15:146. doi: 10.1038/nsmb.1372. [DOI] [PubMed] [Google Scholar]
- 24.Sun D, Hurley LH. J Med Chem. 2009;52:2863. doi: 10.1021/jm900055s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halder K, Chowdhury S. Nucleic Acids Res. 2005;33:4466. doi: 10.1093/nar/gki750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simonsson T, Pribylova M, Vorlickova M. Biochem Biophys Res Commun. 2000;278:158. doi: 10.1006/bbrc.2000.3783. [DOI] [PubMed] [Google Scholar]
- 27.Williamson JR, Raghuraman MK, Cech TR. Cell. 1989;59:871. doi: 10.1016/0092-8674(89)90610-7. [DOI] [PubMed] [Google Scholar]
- 28.Gueron M, Leroy JL. Curr Opin Struct Biol. 2000;10:326. doi: 10.1016/s0959-440x(00)00091-9. [DOI] [PubMed] [Google Scholar]
- 29.Leroy JL, Gehring K, Kettani A, Gueron M. Biochemistry. 1993;32:6019. doi: 10.1021/bi00074a013. [DOI] [PubMed] [Google Scholar]
- 30.Chen L, Cai L, Zhang X, Rich A. Biochemistry. 1994;33:13540. doi: 10.1021/bi00250a005. [DOI] [PubMed] [Google Scholar]
- 31.Huppert JL, Balasubramanian S. Nucleic Acids Res. 2005;33:2908. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huppert JL, Balasubramanian S. Nucleic Acids Res. 2007;35:406. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huppert JL, Bugaut A, Kumar S, Balasubramanian S. Nucleic Acids Res. 2008;36:6260. doi: 10.1093/nar/gkn511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huppert JL. Biochimie. 2008;90:1140. doi: 10.1016/j.biochi.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 35.Yadav VK, Abraham JK, Mani P, Kulshrestha R, Chowdhury S. Nucleic Acids Res. 2008;36:D381. doi: 10.1093/nar/gkm781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mani P, Yadav VK, Das SK, Chowdhury S. PLoS ONE. 2009;4:e4399. doi: 10.1371/journal.pone.0004399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verma A, Halder K, Halder R, Yadav VK, Rawal P, Thakur RK, Mohd F, Sharma A, Chowdhury S. J Med Chem. 2008;51:5641. doi: 10.1021/jm800448a. [DOI] [PubMed] [Google Scholar]
- 38.Phan AT, Modi YS, Patel DJ. J Am Chem Soc. 2004;126:8710. doi: 10.1021/ja048805k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seenisamy J, Rezler EM, Powell TJ, Tye D, Gokhale V, Joshi CS, Siddiqui-Jain A, Hurley LH. J Am Chem Soc. 2004;126:8702. doi: 10.1021/ja040022b. [DOI] [PubMed] [Google Scholar]
- 40.Ambrus A, Chen D, Dai J, Jones RA, Yang D. Biochemistry. 2005;44:2048. doi: 10.1021/bi048242p. [DOI] [PubMed] [Google Scholar]
- 41.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Proc Natl Acad Sci USA. 2002;99:11593. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang D, Hurley LH. Nucleosides, Nucleotides, Nucleic Acids. 2006;25:951. doi: 10.1080/15257770600809913. [DOI] [PubMed] [Google Scholar]
- 43.Hurley LH, Von Hoff DD, Siddiqui-Jain A, Yang D. Semin Oncol. 2006;33:498. doi: 10.1053/j.seminoncol.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 44.Ou TM, Lu YJ, Zhang C, Huang ZS, Wang JH, Tan JH, Chen Y, Ma DL, Wong KY, Tang JC, Chan AS, Gu LQ. J Med Chem. 2007;50:1465. doi: 10.1021/jm0610088. [DOI] [PubMed] [Google Scholar]
- 45.Kang HJ, Park HJ. Biochemistry. 2009;48:7392. doi: 10.1021/bi9006836. [DOI] [PubMed] [Google Scholar]
- 46.Dexheimer TS, Carey SS, Zuohe S, Gokhale VM, Hu X, Murata LB, Maes EM, Weichsel A, Sun D, Meuillet EJ, Montfort WR, Hurley LH. Mol Cancer Ther. 2009;8:1363. doi: 10.1158/1535-7163.MCT-08-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.González V, Guo K, Hurley LH, Sun D. J Biol Chem. 2009;284:23622. doi: 10.1074/jbc.M109.018028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun D, Guo K, Rusche JJ, Hurley LH. Nucleic Acids Res. 2005;33:6070. doi: 10.1093/nar/gki917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo K, Gokhale V, Hurley LH, Sun D. Nucleic Acids Res. 2008;36:4598. doi: 10.1093/nar/gkn380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rankin S, Reszka AP, Huppert J, Zloh M, Parkinson GN, Todd AK, Ladame S, Balasubramanian S, Neidle S. J Am Chem Soc. 2005;127:10584. doi: 10.1021/ja050823u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernando H, Reszka AP, Huppert J, Ladame S, Rankin S, Venkitaraman AR, Neidle S, Balasubramanian S. Biochemistry. 2006;45:7854. doi: 10.1021/bi0601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phan AT, Kuryavyi V, Burge S, Neidle S, Patel DJ. J Am Chem Soc. 2007;129:4386. doi: 10.1021/ja068739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shirude PS, Okumus B, Ying L, Ha T, Balasubramanian S. J Am Chem Soc. 2007;129:7484. doi: 10.1021/ja070497d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qin Y, Rezler EM, Gokhale V, Sun D, Hurley LH. Nucleic Acids Res. 2007;25:7698. doi: 10.1093/nar/gkm538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo K, Pourpak A, Beetz-Rogers K, Gokhale V, Sun D, Hurley LH. J Am Chem Soc. 2007;129:10220. doi: 10.1021/ja072185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cogoi S, Xodo LE. Nucleic Acids Res. 2006;34:2536. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Armond R, Wood S, Sun D, Hurley LH, Ebbinghaus SW. Biochemistry. 2005;44:16341. doi: 10.1021/bi051618u. [DOI] [PubMed] [Google Scholar]
- 58.Palumbo SL, Ebbinghaus SW, Hurley LH. J Am Chem Soc. 2009;131:10878. doi: 10.1021/ja902281d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palumbo S, Memmott RM, Uribe DJ, Krotova-Khan Y, Hurley LH, Ebbinghaus SW. Nucleic Acids Res. 2008;36:1755. doi: 10.1093/nar/gkm1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu Y, Sugiyama H. Nucleic Acids Res. 2006;34:949. doi: 10.1093/nar/gkj485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mathur V, Verma A, Maiti S, Chowdhury S. Biochem Biophys Res Commun. 2004;320:1220. doi: 10.1016/j.bbrc.2004.06.074. [DOI] [PubMed] [Google Scholar]
- 62.Cashman DJ, Buscaglia R, Freyer MW, Dettler J, Hurley LH, Lewis EA. J Mol Model. 2008;14:93. doi: 10.1007/s00894-007-0254-z. [DOI] [PubMed] [Google Scholar]
- 63.Simonsson T, Pribylova M, Vorlickova M. Biochem Biophys Res Commun. 2000;278:158. doi: 10.1006/bbrc.2000.3783. [DOI] [PubMed] [Google Scholar]
- 64.Pataskar SS, Dash D, Brahmachari SK. J Biomol Struct Dyn. 2001;19:307. doi: 10.1080/07391102.2001.10506741. [DOI] [PubMed] [Google Scholar]
- 65.Godde F, Toulme JJ. Biochemistry. 1998;37:13765. doi: 10.1021/bi9811967. [DOI] [PubMed] [Google Scholar]
- 66.Curd FHS, Landquist JK, Rose FL. J Chem Soc. 1948:1759. [PubMed] [Google Scholar]
- 67.Hoffer M. Chem Ber. 1960;93:2777. [Google Scholar]
- 68.Rolland V, Kotera M, Lhomme J. Syn Commun. 1997;27:3505. [Google Scholar]
- 69.Ross S, Burrows C. Nucleic Acids Res. 1996;24:5062. doi: 10.1093/nar/24.24.5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maxam AM, Gilbert W. Methods Enzymol. 1980;65:499. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 71.Manzini G, Yathindra N, Xodo LE. Nucleic Acids Res. 1994;22:4634. doi: 10.1093/nar/22.22.4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cantor CR, Warshaw MM, Shapiro H. Biopolymers. 1970;9:1059. doi: 10.1002/bip.1970.360090909. [DOI] [PubMed] [Google Scholar]
- 73.Qin Y, Hurley LH. Biochimie. 2008;90:1149. doi: 10.1016/j.biochi.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hazel P, Huppert J, Balasubramanian S, Neidle S. J Am Chem Soc. 2004;126:16405. doi: 10.1021/ja045154j. [DOI] [PubMed] [Google Scholar]
- 75.Risitano A, Fox KR. Nucleic Acids Res. 2004;32:2598. doi: 10.1093/nar/gkh598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bugaut A, Balasubramanian S. Biochemistry. 2008;47:689. doi: 10.1021/bi701873c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bevilacqua PC, Blose JM. Annu Rev Phys Chem. 2008;59:79. doi: 10.1146/annurev.physchem.59.032607.093743. [DOI] [PubMed] [Google Scholar]
- 78.Legault P, Pardi A. J Am Chem Soc. 1994;116:8390. [Google Scholar]
- 79.Cai Z, Tinoco I., Jr Biochemistry. 1996;35:6026. doi: 10.1021/bi952985g. [DOI] [PubMed] [Google Scholar]
- 80.Groebe DR, Uhlenbeck OC. Nucleic Acids Res. 1988;16:11725. doi: 10.1093/nar/16.24.11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haasnot CAG, Hilbers CW, van der Marel GA, van Boom JH. J Biosci. 1985;8:767. [Google Scholar]
- 82.Uhlenbeck O. Nature. 1990;346:613. doi: 10.1038/346613a0. [DOI] [PubMed] [Google Scholar]
- 83.Zwieb C. J Biol Chem. 1992;267:15650. [PubMed] [Google Scholar]