

Abstract
In this paper we hypothesize a novel mechanism by which brain injury occurs after intracerebral hemorrhage. We propose the following mechanism: 1) heme derived from extravasated erythrocytes is degraded into bilirubin and bilirubin oxidation products (BOXes). 2) Bilirubin and BOXes activate microglia and astrocytes, which are cells with immune functions in the brain. This activation leads to release of cytokines and activation of leukocyte adhesion molecules on the luminal surface of cerebral endothelial cells. 3) Leukocytes then traffic into the brain. 4) Leukocytes, activated glial cells and cytokines contribute to injury processes. We present preliminary data in support of our hypothesis that BOXes activate glia.
INTRODUCTION
Intracerebral hemorrhage (ICH) is a bleeding into the brain parenchyma and accounts for approximately 15% of all stokes. ICH is associated with high mortality and morbidity and has a one-year survival rate of only 30% (1,2). It is recognized that the mass effect of an expanding hematoma and subsequent edema account for only a portion of brain injury following ICH (2). Indeed, there is evidence suggesting that both inflammation and heme products have a role in brain injury after ICH (3,4) although their relative contributions remain unclear at this time.
Inflammation has been well-documented after ICH, with a number of peripheral leukocyte cell types trafficking into the central nervous system (CNS) parenchyma. These cells include macrophages, neutrophils and CD4 T cells (5,6). Additionally, the proinflammatory cytokines interleukin (IL)-1β and tumor necrosis factor-α (TNF-α) are expressed at increased levels in the brain after ICH; these cytokines are probably derived in part from activated microglia (4). Finally, immune system pre-activation with polyinosinic–polycytidilic acid (polyI:C) increases neuronal cell death in experimental ICH in mice (7). Conversely, macrophage/microglial inhibitory factor (MIF) reduces brain injury volume after ICH as assessed by breakdown of the blood brain barrier and edema (8). We posit that the sum of the above events provides a body of evidence that supports the putative role of inflammation in the pathogenesis of ICH-induced brain injury.
It is also evident that the heme catabolism pathway contributes to brain injury (3). Mice deficient in hemeoxygenase (HO)-1, the inducible, rate-limiting enzyme in heme catabolism, experience reduced injury, neutrophil infiltration and microglial activation (9). It is thought that some of the injury resulting from heme breakdown is due to the release of free ferrous iron. Ferrous iron can participate in the Fenton reaction to convert peroxide into the hydroxyl radical, causing damage to a number of biomolecules found in the CNS (10). While this pathway probably occurs, we hypothesize an additional role of heme-derived products in the pathogenesis of brain injury after ICH.
The contributions of inflammation and heme catabolism to brain injury after ICH have been largely investigated separately. Below we present our hypothesis that heme products and immume system interact to exacerbate brain injury after ICH. In short, we hypothesize that 1) heme is converted into unconjugated bilirubin (UBR) and bilirubin oxidation products (BOXes), which 2) activate microglia and astrocytes, leading to liberation of proinflammatory cytokines and chemokines and 3) the recruitment of peripheral leukocytes into the CNS. 4) The result is an immune mediated complication occurring post–ICH.
HYPOTHESIS
Our hypothesis is that blood products lead to activation of glial cells, release of cytokines and recruitment of peripheral leukocytes, which contribute to injury processes. This hypothesis synthesizes multiple components working together, is summarized in Figure 1 and described in detail below.
Figure 1.

This figure schematizes our hypothesis: Erythrocytes lyse and release their hemoglobin after ICH. Heme is liberated from hemoglobin and is converted into UBR and BOXes. UBR and BOXes activate microglia and astrocytes, leading to the release of cytokines and chemokines and subsequent activation of the endothelium and trafficking of leukocytes into the brain parenchyma. Activated leukocytes, glia and cytokines then contribute to brain injury.
1. Heme is converted into bilirubin and BOXes
Heme catabolism is well-described post ICH. Heme is metabolized to biliverdin by HO-1 or HO-2; HO-1 is induced by oxidative stress and is upregulated after ICH, while HO-2 is constitutively active (11). Biliverdin is reduced to UBR by biliverdin reductase, which is ubiquitous in the brain (11) and not rate limiting. UBR is oxidized to BOXes by reactive oxygen species or cytochrome c oxidase, likely via one-electron oxidation mechanisms (12–14). Notably, both bilirubin and BOXes are found in the brain after experimental ICH (1,14) as well as clinically post subarachnoid hemorrhage (15).
2. Bilirubin and BOXes activate resident immune cells of the brain, microglia and astrocytes, leading to production of cytokines and chemokines
In vitro, UBR leads to production of TNF-α and IL-1β by microglia and astrocytes as well as subsequent cell death in both cell types and glutamate release by astrocytes (16–18). The signaling mechanisms that mediate the effects of UBR on astrocytes may be due to nuclear factor (NF)-κB activation by MAP kinases (19). Necrotic cell death and glutamate release may increase inflammation further and lead to excitotoxic injury, respectively. We hypothesize that BOXes have similar effects on cytokine release by microglia and astrocytes due to the structural similarity among these molecules. In addition, we have preliminarily observed increased astrocyte glial fibrillary acidic protein (GFAP) immunoreactivity when supplementing blood with BOXes in our mouse ICH model (Figure 2).
Figure 2.
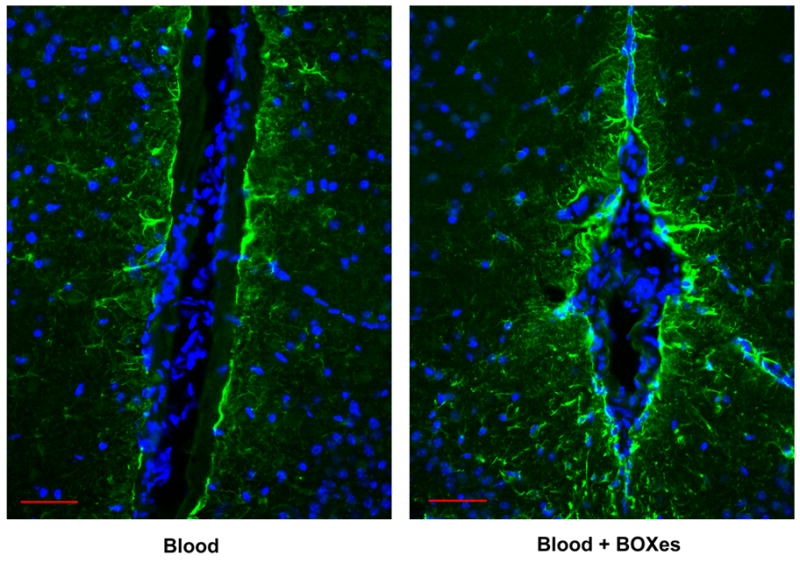
BOXes increase GFAP immunoreactivity. Mice were infused with 10 μL of autologous whole blood or 10 μL of blood containing 1.3 nmol of BOXes. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC). Details of the surgical procedures can be found in reference 6. Astrocytes were stained with an anti-GFAP antibody (green) and nuclei were stained with DAPI (blue). Scale bar equals 50 μm.
3. Peripheral leukocytes are recruited to the brain
TNF-α and IL-1β are involved in activation of leukocytes as well as activation of the endothelium, promoting leukocyte extravasation and trafficking into tissues (20). In addition, TNF-α and IL-1β induce astrocyte production of RANTES (CCL5) and monocyte chemoattractant protein-1 (MCP-1) (21). Microglia are stimulated by TNF-α and IL-1β and produce the chemokines MCP-1 and IL-8 (22). RANTES is chemotactic for monocytes, neutrophils and T cells, IL-8 is chemotactic for T cells and neutrophils and MCP-1 is chemotactic for monocytes and activated T cells (23,24). Therefore, many of the key molecules involved in leukocyte chemotaxis and trafficking may be present after ICH. Bilirubin and BOXes may directly stimulate microglia and astrocytes to release these chemokines or their release may be indirectly mediated by TNF-α and IL-1β. Another possibility is that cytokines and chemokines originating from the brain are released into the circulation, further contributing to leukocyte recruitment. There are limited data describing levels of cytokines and chemokines in peripheral blood after ICH. However, one study has found elevated TNF-α and IL-6 in the plasma of ICH patients (4). In a mouse model of ischemic stroke elevated IL-6 and MCP-1 were found in the plasma after stroke (25). Therefore, we hypothesize that cytokines and chemokines originating from microglia and astrocytes activate the endothelium and are chemotactic for peripheral leukocytes, both of which increase inflammation after ICH. Because the blood brain barrier is disrupted post ICH, leukocyte extravasation and cytokine/chemokine release into the blood would be facilitated (26).
4. Brain injury occurs as a result of peripheral leukocytes, glial cells and cytokines
Activated microglia, macrophages and neutrophils produce superoxide via NADPH oxidase (27,28). Superoxide itself may contribute to oxidative injury, but it is also the precursor to hydrogen peroxide, the hydroxyl radical and peroxynitrite (27). Mice deficient in the gp91phox subunit of NADPH oxidase have reduced injury after ICH as measured by decreased edema, mortality and behavioral deficit (29). Cytokines released from astrocytes and peripheral leukocytes may be injurious by further activating and recruiting immune cells. Cytokines, particularly TNF-α, are directly cytotoxic as a result of an interaction between the TNF receptor and TRAIL, an early signaling component of the apoptotic cascade (30). Finally, the inflammatory process will contribute to increases in local blood flow through vasodilatation, leading to increased edema. Paradoxically the increased blood flow could also increase free oxygen levels with increased opportunity to produce more reactive oxygen species and more bilirubin and BOXes. A resulting positive feedback system might therefore result in even greater damage.
DISCUSSION
The mechanisms of brain injury following ICH are likely complex. In this paper we have proposed one mechanism whereby injury may occur. We hypothesize the heme products UBR and BOXes activate the resident immune cells of the brain, microglia and astrocytes, which in turn are involved in the recruitment of peripheral leukocytes leading to brain injury. The importance of heme products is suggested by studies in which HO-1 is deleted or inhibited, leading to decreased ICH-related pathology (9). Many of the protective effects of inhibiting HO-1 are thought to be mediated through decreased ferrous iron, which may reduce formation of the hydroxyl radical via the Fenton reaction. However, decreased UBR and BOXes may also be responsible for this protective effect.
Our proposed hypothesis is readily testable. It is known that UBR and BOXes are present in the brain after ICH (11,14), therefore it is reasonable to manipulate levels of UBR and BOXes and observe the effects on microglial and astrocyte activation, cytokine levels and cell death of both neurons and glia. It is known that mircoglia are involved in the pathogenesis of ICH-induced injury because inhibition by MIF reduces damage volume (8). This and other methods such as inhibition of NADPH oxidase or neutralizing antibodies to specific cytokines would help delineate which deleterious effects of UBR and BOXes are direct effects of these molecules and which are due to their interaction with microglia and astrocytes. This distinction is important because UBR is directly neurotoxic (31).
What has been missing in much of the post-ICH injury literature is why there is a potent but delayed inflammatory response post ICH that is independent of the ‘mass effect’ (2). When we link the chemical, cellular and cytokine events as an interacting cascade we see that additional cellular injury will be the result. With the previous reports of 12 to 46 hours required for UBR and BOXes production, this time frame is consistent with the current hypothesis (14). Because there is not significant hemolysis in rodent models of ICH until after day 1 (11), it is understandable why the increase in UBR and BOXes levels is delayed. This delay represents a possible time window for therapeutic intervention aimed at preventing some of the morbidity associated with ICH. Indeed, because our hypothesis presents a mechanism contributing to ICH-induced brain injury, a better understanding of this putative pathway may suggest therapeutic targets as well as more complete explanations for the beneficial effects of inhibiting HO-1.
Acknowledgments
This work was supported by NIH grants R01NS050569 and NIH U54EB007954 to JFC.
Footnotes
Conflicts of Interest: We have no conflicts of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Spontaneous intracerebral hemorrhage. N Engl J Med. 2001;344(19):1450–60. doi: 10.1056/NEJM200105103441907. [DOI] [PubMed] [Google Scholar]
- 2.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5(1):53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 3.Koeppen AH, Dickson AC, Smith J. Heme oxygenase in experimental intracerebral hemorrhage: the benefit of tin-mesoporphyrin. J Neuropathol Exp Neurol. 2004;63(6):587–97. doi: 10.1093/jnen/63.6.587. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27(5):894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- 5.Xue M, Del Bigio MR. Intracerebral injection of autologous whole blood in rats: time course of inflammation and cell death. Neurosci Lett. 2000;283(3):230–2. doi: 10.1016/s0304-3940(00)00971-x. [DOI] [PubMed] [Google Scholar]
- 6.Loftspring MC, McDole J, Lu A, Clark JF, Johnson AJ. Intracerebral hemorrhage leads to infiltration of several leukocyte populations with concomitant pathophysiological changes. J Cereb Blood Flow Metab. 2009;29(1):137–43. doi: 10.1038/jcbfm.2008.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xue M, Del Bigio MR. Immune pre-activation exacerbates hemorrhagic brain injury in immature mouse brain. J Neuroimmunol. 2005;165(1–2):75–82. doi: 10.1016/j.jneuroim.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Tsirka SE. Tuftsin fragment 1–3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke. 2005;36(3):613–8. doi: 10.1161/01.STR.0000155729.12931.8f. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Dore S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007;130(Pt 6):1643–52. doi: 10.1093/brain/awm095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark JF, Loftspring M, Wurster WL, Pyne-Geithman GJ. Chemical and biochemical oxidations in spinal fluid after subarachnoid hemorrhage. Front Biosci. 2008;13:1806–12. doi: 10.2741/2801. [DOI] [PubMed] [Google Scholar]
- 11.Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23(6):629–52. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- 12.Loftspring MC, Wurster WL, Pyne-Geithman GJ, Clark JF. An in vitro model of aneurysmal subarachnoid hemorrhage: oxidation of unconjugated bilirubin by cytochrome oxidase. J Neurochem. 2007;102(6):1990–5. doi: 10.1111/j.1471-4159.2007.04667.x. [DOI] [PubMed] [Google Scholar]
- 13.Kranc KR, Pyne GJ, Tao L, Claridge TD, Harris DA, Cadoux-Hudson TA, et al. Oxidative degradation of bilirubin produces vasoactive compounds. Eur J Biochem. 2000;267(24):7094–101. doi: 10.1046/j.1432-1327.2000.01812.x. [DOI] [PubMed] [Google Scholar]
- 14.Clark JF, Loftspring M, Wurster WL, Beiler S, Beiler C, Wagner KR, et al. Bilirubin oxidation products, oxidative stress, and intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:7–12. doi: 10.1007/978-3-211-09469-3_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pyne-Geithman GJ, Morgan CJ, Wagner K, Dulaney EM, Carrozzella J, Kanter DS, et al. Bilirubin production and oxidation in CSF of patients with cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25(8):1070–7. doi: 10.1038/sj.jcbfm.9600101. [DOI] [PubMed] [Google Scholar]
- 16.Juliet PA, Mao X, Del Bigio MR. Proinflammatory cytokine production by cultured neonatal rat microglia after exposure to blood products. Brain Res. 2008;1210:230–9. doi: 10.1016/j.brainres.2008.02.099. [DOI] [PubMed] [Google Scholar]
- 17.Gordo AC, Falcao AS, Fernandes A, Brito MA, Silva RF, Brites D. Unconjugated bilirubin activates and damages microglia. J Neurosci Res. 2006;84(1):194–201. doi: 10.1002/jnr.20857. [DOI] [PubMed] [Google Scholar]
- 18.Fernandes A, Silva RF, Falcao AS, Brito MA, Brites D. Cytokine production, glutamate release and cell death in rat cultured astrocytes treated with unconjugated bilirubin and LPS. J Neuroimmunol. 2004;153(1–2):64–75. doi: 10.1016/j.jneuroim.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Fernandes A, Falcao AS, Silva RF, Gordo AC, Gama MJ, Brito MA, et al. Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J Neurochem. 2006;96(6):1667–79. doi: 10.1111/j.1471-4159.2006.03680.x. [DOI] [PubMed] [Google Scholar]
- 20.Burger D, Dayer JM. Cytokines, acute-phase proteins, and hormones: IL-1 and TNF-alpha production in contact-mediated activation of monocytes by T lymphocytes. Ann N Y Acad Sci. 2002;966:464–73. doi: 10.1111/j.1749-6632.2002.tb04248.x. [DOI] [PubMed] [Google Scholar]
- 21.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36(2):180–90. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 22.Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81(3):302–13. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- 23.Luther SA, Cyster JG. Chemokines as regulators of T cell differentiation. Nat Immunol. 2001;2(2):102–7. doi: 10.1038/84205. [DOI] [PubMed] [Google Scholar]
- 24.Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, et al. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52(1):145–76. [PubMed] [Google Scholar]
- 25.Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006;26(5):654–65. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- 26.Wagner KR, Xi G, Hua Y, Kleinholz M, de Courten-Myers GM, Myers RE, et al. Lobar intracerebral hemorrhage model in pigs: rapid edema development in perihematomal white matter. Stroke. 1996;27(3):490–7. doi: 10.1161/01.str.27.3.490. [DOI] [PubMed] [Google Scholar]
- 27.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–9. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 28.Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8(9–10):1583–96. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- 29.Tang J, Liu J, Zhou C, Ostanin D, Grisham MB, Neil Granger D, et al. Role of NADPH oxidase in the brain injury of intracerebral hemorrhage. J Neurochem. 2005;94(5):1342–50. doi: 10.1111/j.1471-4159.2005.03292.x. [DOI] [PubMed] [Google Scholar]
- 30.Wang S. The promise of cancer therapeutics targeting the TNF-related apoptosis-inducing ligand and TRAIL receptor pathway. Oncogene. 2008;27(48):6207–15. doi: 10.1038/onc.2008.298. [DOI] [PubMed] [Google Scholar]
- 31.Daood MJ, McDonagh AF, Watchko JF. Calculated free bilirubin levels and neurotoxicity. J Perinatol. 2009;29 (Suppl 1):S14–9. doi: 10.1038/jp.2008.218. [DOI] [PubMed] [Google Scholar]