

Abstract
The pathophysiology of schizophrenia may involve reduced NMDA receptor function and experimental models of NMDA receptor hypofunction have proven useful for characterizing neurobiological abnormalities potentially relevant to schizophrenia. The present study assessed behavioral responses and induction of Fos after administration of kainic acid to wild type mice (NR1+/+) and mice with genetically reduced NMDA receptor expression (NR1neo/neo). At a dose of 20 mg/kg kainic acid induced lethal seizures in 100% of the NR1neo/neo mice tested but produced no lethal seizures in the wild type mice. The NR1neo/neo mice also exhibited enhanced behavioral responses to kainic acid at a dose of 15 mg/kg but no lethal seizures were produced by this dose. A greater induction of Fos was observed in neocortical and limbic cortical regions of the NR1neo/neo compared to NR1+/+ mice after administration of 15 mg/kg kainic acid. In contrast, there were no differences between the genotypes in kainic acid induced of Fos in the amygdala, hippocampus, lateral septum, and nucleus accumbens. In order to determine if altered behavioral phenotypes of the NR1neo/neo mice could be related to increased sensitivity of kainate receptors to endogenous glutamate, effects of the highly selective kainate antagonist LY382884 were examined. The kainate antagonist reduced the exaggerated acoustic startle responses, deficits in prepulse inhibition of acoustic startle, and motor hyperactivity in the NR1neo/neo mice. These findings suggest that selective kainate receptor antagonists could be novel therapeutic candidates for schizophrenia.
Keywords: Schizophrenia, glutamate, kainic acid, NMDA receptor, hippocampus, cerebral cortex, kainic acid antagonist
1. Introduction
The NMDA hypofunction hypothesis of schizophrenia is supported by observations that NMDA antagonists produce a spectrum of behavioral effects in humans that mimic positive, negative and cognitive symptoms of schizophrenia (Javitt and Zukin, 1991; Krystal et al., 1994; Lahti et al., 1995; Olney and Farber, 1995; Lahti et al., 2001). In order to model endogenous NMDA receptor hypofunction, a mouse line was developed that expresses low levels of the NR1 subunit of the NMDA receptor (Mohn et al., 1999). Reduced expression of the NR1 gene was effected by incorporation of a bacterial neomysin resistance gene into intron 20 of the NR1 gene. The mice are referred to as NR1 hypomorphic or NR1 knock-down, since expression of the gene is reduced but not eliminated. The homozygous mutant animals are denoted as NR1neo/neo.
The NR1neo/neo mice exhibit behavioral phenotypes that support their utility to model certain behavioral characteristics relevant to schizophrenia. These phenotypes include reduced locomotor habituation in a novel environment (Mohn et al., 1999; Duncan et al., 2002), deficits in prepulse inhibition of acoustic startle (PPI) (Duncan et al., 2004; Fradley et al., 2005; Duncan et al., 2006a) and increased sensitivity to amphetamine-induced disruption of PPI (Moy et al., 2006). In addition, the NR1neo/neo mice show marked deficits in tests of social affiliation and social aggression (Mohn et al., 1999; Duncan et al., 2004).
NMDA receptor activation is an important regulatory signal during normal brain development (Schlaggar et al., 1993; Fox et al., 1996; Iwasato et al., 2000; Lee et al., 2005a; Lee et al., 2005b). Since reduced NMDA receptor function in the NR1neo/neo mice occurs over the life span of the animals, it is possible that the endogenous and developmental NMDA hypofunction could produce alterations in other glutamate receptor systems. We demonstrated previously that there was no apparent difference in the density of AMPA or kainate subtypes of glutamate receptors in the NR1neo/neo mice (Duncan et al., 2002). However, there is no information available about how the reduced NMDA receptor function in the NR1neo/neo mice might affect the functional sensitivity of non-NMDA subtypes of glutamate receptors.
The are indications that kainic acid receptors may be altered in schizophrenia patients (Meador-Woodruff and Healy, 2000; Scarr et al., 2005; Beneyto et al., 2007; Watis et al., 2008). The present study provides evidence for increased sensitivity of kainic acid receptors in the NR1neo/neo mice. Furthermore, a highly selective kainic acid receptor antagonist attenuated behavioral alterations in the mutant mice. These data suggest an increased sensitivity of kainate receptors to endogenous glutamate in the mutant mice may contribute to behavioral abnormalities of the animals.
2. Results
2.1 Behavioral Effects of Kainic Acid in wild type and NR1neo/neo mice
An initial study was conducted to test behavioral responses in wild type and mutant mice to 20 mg/kg kainic acid injected i.p. (Table 1). In the 4 wild type mice tested a range of behavioral effects were found, with each mouse exhibiting a different behavioral response as follows: 1) no obvious effect, 2) un-coordinated rear limb movements while walking and a brief period of rapid jumping 18 min after injection, 3) one brief period of forelimb clonus 28 min after injection, and 4) forelimb clonus followed by a motor seizure with lack of postural control lasting approximately 30 sec, 40 min after injection. After the single seizure, the mouse exhibited some locomotor activity but was mainly still during the following hour. Because of the short survival time (< 25 min) after kainic acid administration in the NR1neo/neo mice it was not possible to assess Fos for the 20 mg/kg dose.
Table 1.
Behavioral effects of 20 mg/kg kainic acid in NR1+/+ and NR1neo/neo mice
| Behavioral Response to 20 mg/kg Kainic Acid | NR1 +/+ | NR1 neo/neo |
|---|---|---|
| No obvious effect | 1/4 | 0/4 |
| Forelimb clonus not followed by seizure with lack of postural control | 1/4 | 0/4 |
| Forelimb clonus followed by at least one seizure with lack of postural control | 1/4 | 4/4 |
| Un-coordinated rear limb movements, brief period of rapid jumping | 1/4 | 0/4 |
| Lethal Seizure | 0/4 | 4/4 |
Mice were injected i.p. with kainic acid and observed for a 1 hr period.
Both wild type and NR1neo/neo groups consisted of 2 males and 2 females.
Strikingly different effects were found for the 20 mg/kg dose of kainic acid in the NR1neo/neo mice. All 4 of NR1neo/neo mice tested exhibited forelimb clonus within 10 min after injection and lethal motor seizures occurred in each of the mice within 25 min after injection of the compound. This was an unexpected finding since in previous studies with other strains of mice we have never seen lethal seizures induced by this dose of kainic acid (unpublished observations).
In the next experiment, effects of kainic acid at a dose of 15 mg/kg were tested in the wild type and NR1neo/neo mice. For this dose, there was again a wide range of responses among individual mice (Table 2). For the 9 wild type mice tested, 4 exhibited no obvious signs of seizure activity, 2 exhibited one brief period of forelimb clonus, and 3 exhibited jumping and/or forelimb clonus followed by 1–2 seizures (duration 5–10 sec) with loss of postural control. For the NR1neo/neo mice, all 6 of the animals tested had at least 1 seizure with loss of postural control. The number of seizures ranged from 1–9 for the different mice and, in most cases, the duration of the seizures was 5–20 seconds. However, one of the NR1neo/neo exhibited almost continuous seizures for approximately 10 min and was therefore euthanized. Fos induction as described below was not assessed for this mouse.
Table 2.
Behavioral effects of 15 mg/kg kainic acid in NR1+/+ and NR1neo/neo mice
| Behavioral Response to 15 mg/kg Kainic Acid | NR1 +/+ | NR1 neo/neo |
|---|---|---|
| No Obvious Response | 4/9 | 0/6 |
| Forelimb Clonus/Rigid Tail | 2/9 | 0/6 |
| Seizure with Loss of Postural Control | 3/9 | 6/6 |
Mice were injected i.p. with kainic acid and observed for a 1 hr period.
The wild type group consisted of 5 males and 4 females. The NR1neo/neo group consisted of 2 males and 4 females.
2.2 Effects of Kainic Acid on Fos in wild type and NR1 hypomorphic mice
In wild type mice, kainic acid at a dose of 15 mg/kg induced a robust induction of Fos in select brain regions, including the hippocampal formation, amygdala, cingulate cortex, and nucleus accumbens. Results of the quantitative analyses are summarized in Figure 1. In the amygdala, staining for Fos was observed in all regions, including basolateral, lateral, central and medial, and cortical nuclei (Figure 2). In the hippocampus, pyramidal neurons in CA1-4 fields expressed the protein, as did neurons scattered in stratum radiatum and stratum oriens (Fig 3). Intense Fos staining was also observed in the granule cells of the dentate gyrus. Very few neurons were stained in neocortical regions of wild type mice after administration of kainic acid, although the cingulate cortex did show numerous labeled cells (Figures 2 and 3). Despite considerable individual variation in the behavioral responses to the drug, the variability in Fos induction was relatively low and no obvious relationship between behavioral responses and Fos induction was apparent in the wild type mice. The data in Figure 1 include all mice given 15 mg/kg kainic acid, including those that exhibited no obvious signs of seizure activity.
Figure 1.
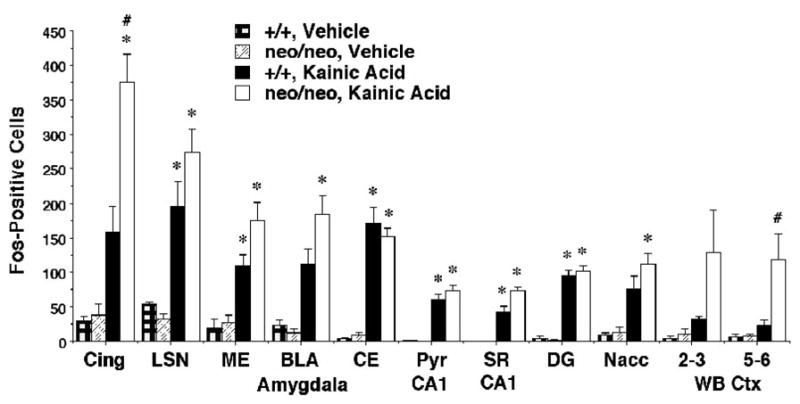
Fos induction in the cingulate cortex, lateral septum, nuclei of the amygdala, and other regions in NR1+/+ and NR1neo/neo mice following treatment with vehicle or kainic acid. Mice were injected with kainic acid (15 mg/kg i.p.) and perfused 2 hr later. Data are from all mice tested, including wild type mice that did not exhibit behavioral signs of seizures. Abbreviations: Cing, cingulate cortex; LSN, lateral septal nucleus; ME, medial nucleus; BLA, basolateral nucleus; CE, central nucleus; CA1 Pyr, hippocampal CA1 stratum pyramidale; CA1 SR, hippocampal CA1 stratum radiatum; DG, dentate gyrus of the hippocampal formation; Nacc, nucleus accumbens; WB 2–3, whisker barrel region of somatosensory cortex layers 2 and 3; WB 5–6, whisker barrel region of somatosensory cortex layers 5 and 6. *p<0.05, comparison with respective vehicle group; #p<0.05, comparison with adjacent +/+, kainic acid group.
Figure 2.
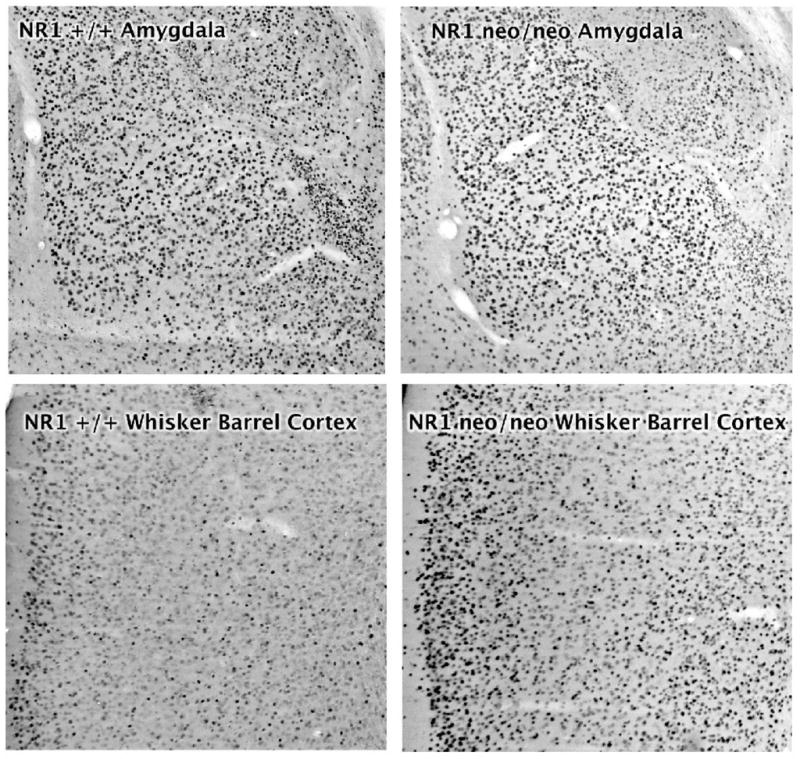
Fos induction in the amygdala (top panels) and whisker barrel somatosensory cortex (bottom panels) in NR1+/+ and NR1neo/neo mice. Mice were injected with kainic acid (15 mg/kg) i.p and perfused 2 hour later.
Figure 3.
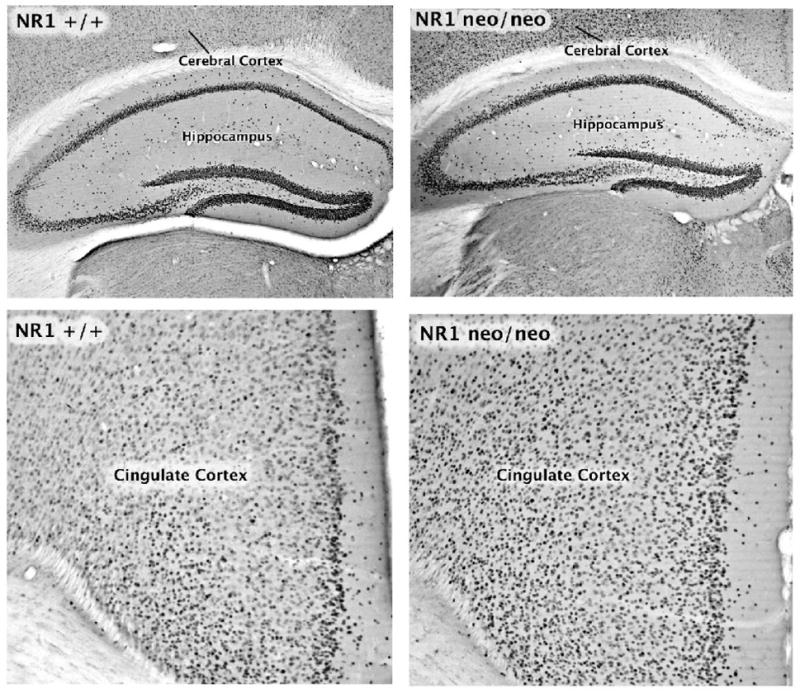
Fos induction in the hippocampus (top panels) and cingulate cortex (bottom panels) in NR1+/+ and NR1neo/neo mice. Mice were injected with kainic acid (15 mg/kg) i.p and perfused 2 hour later.
For the NR1neo/neo mice given 15 mg/kg kainic acid, staining for Fos showed a similar distribution as observed for the wild type mice in the hippocampus, amygdala, lateral septum, and nucleus accumbens (Figure 1). In the cingulate cortex the number of darkly stained cells was significantly greater in the mutant compared to the wild type animals (Figures 1 and 3). In neocortical regions such as the somatosensory cortex, the number of Fos-positive cells after kainic acid was dramatically greater in the NR1neo/neo compared to the wild type mice (Figures 1 and 2).
A repeated measures ANOVA conducted on cell counts confirmed highly significant effects of kainic acid on Fos expression [main effect of treatment, F(1,16)=53.41, p<0.0001; and treatment × brain region interaction, F(10,160)=6.27, p<0.0001], as well as a significant genotype × brain region interaction [F(10,160)=2.03, p=0.034]. Greater sensitivity to the effects of kainic acid were observed in the NR1neo/neo group in two brain regions: the cingulate cortex [main effect of genotype, F(1,16)=6.303, p=0.0232; genotype × treatment interaction, F(1,16)=5.36, p=0.034] and the whisker barrel region of somatosensory cortex, layers 5 and 6 [main effect of genotype, F(1,16)=4.57, p=0.048]. In layers 2–4 of the somatosensory cortex, the genotype × treatment interaction closely approached significance [F(1,16)=4.37, p=0.053].
2.3 Effects of LY382884 on acoustic startle and PPI in wild type and NR1neo/neo mice
As previously demonstrated (Duncan et al., 2004), the NR1neo/neo mice exhibited increased startle response and decreased PPI in comparison to wild type mice (Figure 4). Within-genotype analyses indicated that the highly selective kainate receptor antagonist LY382884 significantly reduced the startle response in the NR1neo/neo mice [main effect of treatment, F(1,24)=5.44, p=0.029; treatment × decibel level interaction, F(6,144)=3.02, p=0.0083], but not in the wild type mice. Examination of Figure 6 shows that the effect of the drug was to essentially normalize the startle response of the mutant mice to the level of the wild type controls. LY382884 increased PPI in both the NR1+/+ mice F(1,23)=4.34, p=0.049] and the NR1neo/eno mutants [F(1,24)=5.33, p=0.029].
Figure 4.
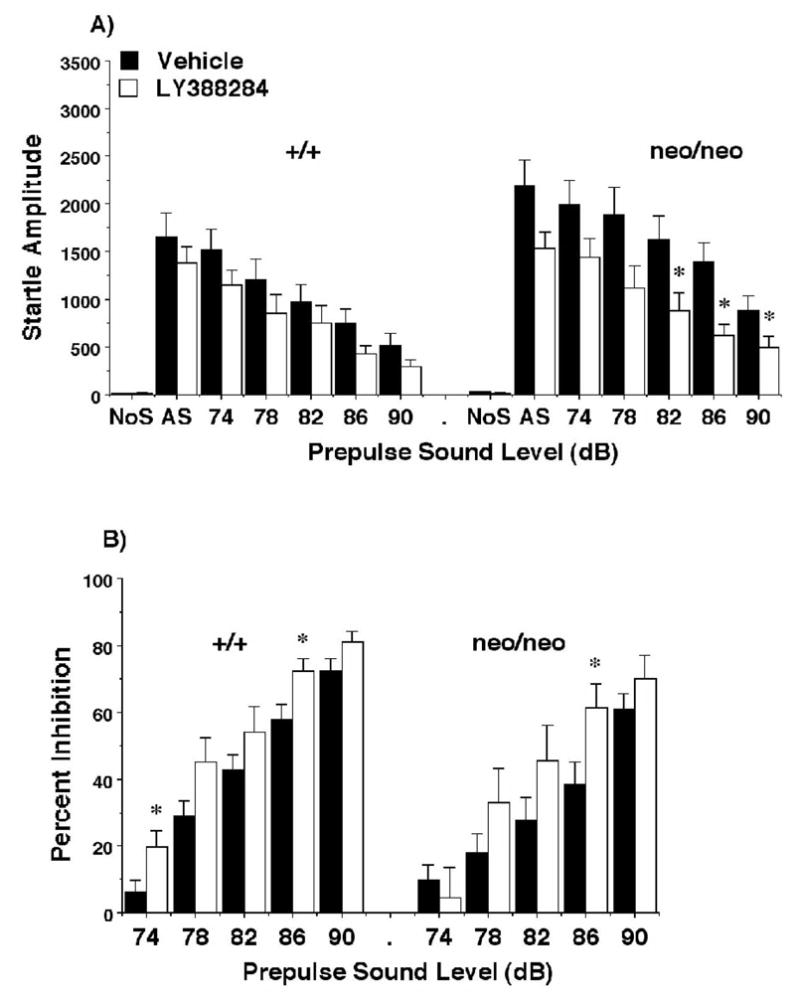
Amplitude (A) and prepulse inhibition (B) of acoustic startle responses following LY382884 (100 mg/kg) in NR1+/+ and NR1neo/neo mice. Data shown are means (+ SEM) for each group. Trials included no stimulus (No S) trials and acoustic startle stimulus (AS; 120 dB) alone trials. Within-genotype ANOVAs indicated significant effects of LY382884 on startle amplitude in the NRneo/neo (p=.029) and on PPI for the NR1+/+ (p= .049) and NR1neo/neo mice (p=0.029). * p < 0.05, comparison with vehicle score at same stimulus level.
Figure 6.

Rearing activity during a 1-hr session in an open field. Data shown are means (± SEM) for each group. Within-genotype repeated measures ANOVA indicated that LY388284 significantly reduced activity in the mutant mice (p < .006) but not in the wild type mice * p < 0.05, comparison with vehicle score at same time point.
2.4 Effects of LY382884 on open field activity in wild type and NR1neo/neo mice
As previously reported (Mohn et al., 1999) the NR1neo/neo mice were hyperactive in the open field in comparison to the wild type mice (Figures 5 and 6). Separate within-genotype analyses for the measure of horizontal activity indicated that LY382884 significantly reduced activity in the mutant mice [treatment × time interaction, F(11,264)=1.93, p=0.036], without any significant effects in the NR1+/+ group. For the rearing measure, the overall repeated measures ANOVA revealed a significant genotype × treatment interaction [F(1,47)=5.86, p=0.0194]. LY382884 reversed increased levels of rearing in the NR1neo/neo mice [main effect of treatment, F(1,24)=9.07, p=0.006], without any impact on activity in the controls.
Figure 5.

Horizontal activity during a 1-hr session in an open field following LY382884. Data shown are means (± SEM) for each group. Within-genotypes repeated measures ANOVAs indicated that LY388284 significantly (p=0.036) reduced activity in the mutant mice but not in the wild type mice. * p < 0.05, comparison with vehicle score at same time point.
3. Discussion
NMDA receptors are well documented to play a crucial role in brain development and function. The present study explored the possibility that chronic and developmental NMDA receptor hypofunction in NR1neo/neo mice would alter responses to systemic administration of kainic acid. For both doses of kainic acid tested (15 and 20 mg/kg), the NR1neo/neo mice showed markedly greater behavioral responses compared to the wild type mice. This was most striking for the 20 mg/kg dose, which produced lethal seizures in 100% of the mutant animals but in none of the wild type mice. As noted above, the effects at 20 mg/kg kainic acid in the mutants were highly unexpected since previous studies with other strains found no lethal seizures after this dose.
The short survival time of the NR1neo/neo mice after injection of 20 mg/kg kainic acid precluded assessment of Fos for this dose. For the 15 mg/kg dose, in accord with the greater behavioral responses of the NR1neo/neo mice, Fos induction was substantially greater compared to wild type mice in limbic cortical and neocortical regions. However, Fos induction was similar in the mutant and wild type mice in the amygdala, hippocampus, lateral septum, and nucleus accumbens in response to kainic acid. Further work is required to determine mechanisms responsible for the differential sensitivity of brain regions to Fos induction by kainic acid in the NR1neo/neo mice.
In previous autoradiographic studies of 3H-kainate binding, we found no apparent differences in receptor density or binding pattern between the NR1neo/neo and wild type mice (Duncan et al., 2002). Thus, the functional supersensitivity to kainic acid in the NR1neo/neo mice is not reflected in a change in 3H-kainate binding. These results illustrate the difficulties in the interpreting functional changes from receptor binding data.
The increased seizure sensitivity of the NR1 hypomorphic mice to kainic acid might not be predicted since NMDA antagonists block behavioral measures of kainic acid-induced seizures (for example see Clifford et al., 1990). These observations underscore the distinctions between the NR1neo/neo genetic model of NMDA hypofunction and the effects of acute administration of NMDA antagonists.
The increased Fos induction that was observed in limbic cortical and neocortical regions in response to kainic acid in the NR1neo/neo mice could be due to selectively increased sensitivity of kainate receptors in those regions. Alternatively, enhanced excitatory drive of cortical neurons from subcortical structures, due to increased kainate sensitivity in regions such as the hippocampus and amygdala, could also account for our findings. Although Fos induction was slightly greater in the NR1neo/neo mice for some nuclei of the amygdala, the greatly exaggerated response in cortical regions in comparison indicates an enhanced spread of activity into the cerebral cortex in the mutant mice.
Another possible explanation for the much greater kainic acid-induced Fos in cerebral cortical regions the NR1neo/neo could be that different sub-classes of kainate receptors are differentially affected (indirectly) by the mutation. Native kainate receptors, like other ionotropic glutamate receptors, are composed of different sub-units which are assembled to create functionally distinct receptors that have different neuroanatomical distributions (for reviews see (Jane et al., 2009)). There are 5 different subunits that can be assembled to form native kainate receptors. In the most recent IUPHAR nomenclature they are designated as Gluk1-5 (formally Gluk1-3 were designated GluK5-7 and Gluk4-5 were KA1 and KA2). Gluk3 and Gluk5 are expressed at a considerably higher level in the cerebral cortex than the other subunits. Whether functional sensitivity of kainate receptors with different subunit composition are differentially affected in the NR1neo/neo mice will require further study.
In contrast to the similar or enhanced Fos responses to kainic acid in the NR1neo/neo mice, induction of Fos in the mutant mice after challenge with sub-seizure doses of a selective NMDA agonist (tertrazol 5yl-gycine) was greatly reduced in cerebral cortical regions, hippocampus, and amygdala (Inada et al., 2007; Duncan et al., 2008). However, we were surprised to find that there was no decreased sensitivity to seizure-producing effects of tetrazol 5yl-glycine in the mutant mice (Duncan et al., 2008). Despite the marked reduction in tetrazol 5yl glycine-induced Fos in regions noted above, the mutant and wild type mice showed robust and equivalent induction of Fos in the ventral medial and arcuate nuclei of the hypothalamus, lateral septum, and nucleus of the solitary tract. These findings suggest that the reduction in NR1 subunit expression in the NR1neo/neo mice may not be neuroanatomically uniform and that structures not usually associated with seizures may mediate NMDA agonist-induced seizures.
The divergent effects of an NMDA agonist and kainic acid on cortical Fos induction in the two genotypes suggest that the NR1 hypomorphic mutation produces a selective increased response to kainic acid. Further work is required to determine whether the increased responses to systemic kainic acid administration in the mutants is due to increased electrophysiological sensitivity of kainate receptors or to modification of circuits involving the receptors.
It is unclear how the findings of increased sensitivity to kainic acid in the cerebral cortex might relate to pathophysiology of schizophrenia. However, if schizophrenia is associated with chronic and developmental NMDA receptor hypofunction, it is conceivable that functional supersensitivity of kainate receptors could emerge as a consequence, as observed in the mouse model.
There is evidence for alterations in kainate receptors from postmortem studies of brains of schizophrenia patients (Meador-Woodruff and Healy, 2000; Scarr et al., 2005; Beneyto et al., 2007; Watis et al., 2008). However, there is considerable inconsistency among different published reports. For example, 3H-kainate binding was increased in the orbital frontal cortex (Deakin et al., 1989) but reduced in the dorsolateral prefrontal cortex (Scarr et al., 2005), and unchanged in the anterior cingulate cortex (Zavitsanou et al., 2002). As noted above, it is not always possible to predict changes in function from static measures of receptor binding or mRNA expression. It is therefore impossible to know from the postmortem binding and expression studies how the reported changes might relate to kainate receptor sensitivity in schizophrenia.
A consistent finding in postmortem studies in schizophrenia is a reduced number of GABAergic neurons (Beasley and Reynolds, 1997; Beasley et al., 2002; Zhang and Reynolds, 2002; Sakai et al., 2008). In an elegant double-labeled in situ hybridization study, the number of GABAergic neurons expressing the Gluk1 (GluR5) subunit of the kainate receptor were reduced by approximately 40% in the anterior cingulate cortex of schizophrenia patients (Woo et al., 2007). Furthermore, the numerical density of neurons exhibiting immunoreactivity for kainic acid receptors was reduced in the orbital frontal cortex of schizophrenia patients (Garey et al., 2006). Since excessive activation of kainate receptors is known to produce neurotoxic effects, one could speculate that supersensitivity of kainate receptors could be involved in the reported reduced number of neurons expressing kainate receptors.
As noted in the introduction, increased acoustic startle responses and reduced PPI are robust and consistent behavioral phenotypes the NR1neo/neo mice. We have demonstrated that both typical and atypical antipsychotics increase PPI in the mutant mice but only the atypical drugs reduced the exaggerated startle response in the animals (Duncan et al., 2006a; Duncan et al., 2006b). The present study examined effects of LY382884, a highly selective kainate receptor antagonist, to determine if the abnormalities of the mutant mice in the PPI paradigm could be related to increased kainate receptor sensitivity to endogenous glutamate. LY382884 is a relatively low potency drug with poor penetrability to the central nervous system but is highly selective for the kainate receptors containing the Gluk1 (GluR5) subunit, with no appreciable affinity for or activity at AMPA receptors (O’Neill et al., 1998; Smolders et al., 2002). The drug is active in models of anxiety (Alt et al., 2007) and epilepsy (Smolders et al., 2002; Barton et al., 2003). In the NR1neo/neo mice LY382884 reduced the exaggerated startle response, PPI deficits, and locomotor hyperactivity produced by the mutation. These results suggest that some of the behavioral abnormalities in the mutant mice may relate to increased activity of endogenous glutamate at kainate receptors. Furthermore, the behavioral profile of the kainate antagonist in the NR1neo/neo mice is similar to that of atypical antipsychotic drugs described above. The results provide the first preclinical indication that selective kainate receptors antagonists could have antipsychotic activity.
Although a high dose of LY382884 (100 mg/kg) was given, the drug had no significant effect on startle response amplitudes or motor activity in the wild type mice. This dose was maximally effective in the 6 Hz seizure model while exhibiting no behavioral toxicity in the rotarod test (Barton et al., 2003). It will be of interest to test additional selective kainate antagonists in future studies in the NR1 hypomorphic mice and in other animal models relevant to schizophrenia. Such work could provide further support for the idea that selective kainate antagonists represent novel candidate therapeutic agents for schizophrenia.
4. Experimental Procedures
4.1 Animals
NR1neo/neo mice were produced initially on a mixed genetic background by incorporating a neomysin resistance gene into an intron 20 of the NR1 locus as described in detail (Mohn et al., 1999). This insertion mutation greatly reduced expression of the NR1 gene in all brain regions thus far examined. Homozygotes carrying the NR1 hypomorphic mutation do not breed effectively.
Therefore, the mutant homozygotes must be generated by breeding heterozygotic mice. When the NR1 hypomorphic mutation was induced in pure C57BL/6J or 129SvEv mice the mutants did not gain weight at the same rate as wild type mice and the frequency of mutants born was less than expected. To overcome these problems, the strategy of breeding F1 hybrids was developed to generate the NR1neo/neo mice (Duncan et al., 2004).
For the current study animals from “stock” colonies of F1 hybrid 129SvEv NR1+/neo and C57BL/6J NR1+/neo mice (identified by PCR) were used to set up matings to generate experimental animals, i.e., F1 hybrid NR1neo/neo and NR1+/+ littermates. In all cases a 129SvEv+/neo female was bred to a C57BL/6J+/neo male. All of the F1 NR1neo/neo and NR1+/+ offspring of these litters are genetically identical (see below) at all loci except at the NR1 gene and the NR1neo/neo mice exhibit marked reductions in NMDA receptor binding in all regions examined (Duncan et al., 2004). Since the 129SvEv mice were the breeding females, the mitochondria will of 129SvEv origin in the F1 hybrids. Since C57BL/6J mice were the breeding males, the Y chromosome of the F1 hybrids will be of C57BL/6J origin. For the X chromosome and all other chromosomes, the maternal chromosome will be of 129SvEv origin and the paternal will be of C57BL/6J origin. Therefore the experimental and control animals are genetically identical with the caveat that the F1 hybrid NRneo/neo mice are homozygous for genes very tightly linked to the NR1 gene, and the controls will have one copy of 129SvEv and one copy of C57BL/6J genes for these tightly linked genes. The C57BL/6J heterozygote mice used to generate the F1 hybrids are at N20 and therefore this linked region is predicted to be very small. Both male and female mice were used in the studies. Mice were 60–75 days at the time of testing.
4.2 Drugs
Kainic acid (Ocean Produce International, Shelburne, Nova Scotia, Canada) was dissolved in saline and injected i.p. LY382884 (3S,4aR,6S,8aR-6-((4-carboxyphenyl)methyl)-1,2,3,4,4a,5,6,7,8,8a-decahydroisoquinoline-3-carboxylic acid) was a gift from Eli Lilly and Company, Indianapolis, IN, suspended in 10% cyclodextrin and injected i.p.
4.3 Fos immunohistochemistry after kainic acid
Mice were observed for signs of seizure activity for a period of 60 min after the kainic acid injection. Two hours after administration of kainic acid mice were perfused for immunocytochemical assessment of Fos protein. Procedures were performed according to the previously published protocols (Duncan et al., 1993; Miyamoto et al., 2004). Mice were deeply anesthetized with Nembutal (80 mg/kg i.p.) two hours after saline or kainic acid injection. Mice were then perfused through the left cardiac ventricle with ice-cold 100mM sodium phosphate-buffered saline (PBS, pH=7.4) for 1 min, at a rate of 5 ml/min, followed by 4% paraformaldehyde for 3 min at the same rate of perfusion. The brains were removed and fixed with 4% paraformaldehyde overnight. Coronal sections (50 μm) of the forebrain were cut with a vibratome and placed in PBS. Sections were treated with 5% normal goat serum [Vector Laboratories, Burlingame, CA] and 0.1% Triton X-100 in PBS for 30 min. and then were incubated for 72 hours at 4 degrees C with a rabbit Fos antibody (gift from Dr. Peter Petruz). After incubation with the Fos antibody, sections were incubated for 1h with biotinylated anti-rabbit IgG [Vector Laboratories, Burlingame, CA]. Sections were then incubated with avidin-biotin complex [Vectastain Elite ABC kit; Vector Laboratories] for 1h. Sections were then placed in a solution containing 0.05% 3,3′-diamino-benzidene tetra-hydrochloride, 0.005% cobalt chloride, 0.008% nickel ammonium sulfate, and 0.02% hydrogen peroxide.
4.4 Quantification of Fos expression
Cells exhibiting nuclear staining for Fos in selected brain regions were counted at a magnification of 200x by an experimenter blind to the treatment group. The location of the areas used within each brain region for assessing Fos cells were guided by the atlas of (Franklin and Paxinos, 1997). Darkly stained cells in strictly defined areas of the various brain regions were counted with the aid of an eyepiece grid. Depending on the size of the regions of interest different sized areas were counted. The brain regions assessed and grid areas measured (in microns) were: lateral septal nucleus (250 × 500), nucleus accumbens (250 × 250), cingulate cortex (500 × 500), whisker barrel region of somatosensory cortex layers 2–4 (400 × 500), whisker barrel region of somatosensory cortex layers 5 and 6 (400 × 400), medial nucleus of amygdala (250 × 500), central nucleus of amygdala (25 × 500), basolateral nucleus of amygdala (250 × 500), granule cell layer of dentate gyrus (50 × 250), and CA1 pyramidal cell layer (50 × 250).
4.5 Effects of LY382884 on acoustic startle and prepulse inhibition
The acoustic startle measure was based on the reflexive whole-body flinch, or startle response, following exposure to a sudden noise. Animals were tested with a San Diego Instruments SR-Lab system using the procedure described by Paylor and Crawley (1997). Briefly, mice (4–7 months of age) were placed in a small Plexiglas cylinder within a larger, sound-attenuating chamber (San Diego Instruments). The cylinder was seated upon a piezoelectric transducer, which allowed vibrations to be quantified and displayed on a computer. The chamber included a house light, fan, and a loudspeaker for the acoustic stimuli (bursts of white noise). Background sound levels (70 dB) and calibration of the acoustic stimuli were confirmed with a digital sound level meter (San Diego Instruments).
The selective kainate receptor antagonist LY382884 was injected i.p. at a dose of 100 mg/kg, 25 min before placing mice in the startle chambers. This dose was chosen based on discussion with Eli Lilly and published studies regarding the specificity and potency of the compound in in vivo animal models of seizures and anxiety (Smolders et al., 2002; Barton et al., 2003; Alt et al., 2007). The number of mice tested was 25 NR1+/+ (11 given vehicle and 14 given drug) and 26 NR1neo/neo (14 given vehicle and 12 given drug).
Each test session consisted of 42 trials, presented following a five-minute habituation period. Seven different types of trials were presented: no-stimulus (NoS) trials, trials with the acoustic startle stimulus (40 ms; 120 dB) alone, and trials in which a prepulse stimulus (20 ms; either 74, 78, 82, 86, or 90 dB) had onset 100 ms before the onset of the startle stimulus. The different trial types were presented in blocks of 7, in randomized order within each block, with an average intertrial interval of 15 seconds (range: 10 to 20 seconds). Measures were taken of the startle amplitude for each trial, defined as the peak response during a 65-msec sampling window that began with the onset of the startle stimulus. An overall analysis was performed for each subject’s data for levels of prepulse inhibition at each prepulse sound level (calculated as 100 − [(response amplitude for prepulse stimulus and startle stimulus together/response amplitude for startle stimulus alone) × 100]).
4.6 Open field test
Immediately following the acoustic startle test, mice were given a 1-hr test in a photocell-equipped automated open field (40 cm × 40 cm × 30 cm; Versamax system, Accuscan Instruments). Activity chambers were contained inside sound-attenuating boxes, equipped with houselights and fans. Measures were taken of horizontal activity and number of rearing movements at 5-min intervals during the test.
4.7 Statistics
Data for fos expression were first analyzed using a repeated measures Analysis of Variance (ANOVA), with the factors genotype (+/+ or neo/neo), treatment (vehicle or kainic acid), and brain region (the repeated measure). Separate 2-way ANOVAs were then conducted for each brain region, with the factors genotype and treatment. Behavioral data were first analyzed using an overall repeated measures ANOVA, with the factors genotype (+/+ or neo/neo), treatment (vehicle or drug), and either decibel level (the repeated measure for the acoustic startle test) or time (the repeated measure for the open field test). Separate repeated measures ANOVAs were then conducted for each genotype, with the factor treatment. Fishers Protected Least Significant Difference (PLSD) tests were conducted between group means only when a significant F value was found in the repeated measures ANOVA. For all comparisons, significance was set at p<0.05.
Acknowledgments
Supported by MH063398, MH080069, and HD03110 from the NIH. The technical assistance of Joseph Farrington, Geoffrey Shafer, and Randy Nonneman are appreciated greatly.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alt A, Weiss B, Ornstein PL, Gleason SD, Bleakman D, Stratford RE, Witkin JM. Anxiolytic-like effects through a GLUKS kainate receptor mechanism. Neuropharmacology. 2007;52:1482–1487. doi: 10.1016/j.neuropharm.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Barton ME, Peters SC, Shannon HE. Comparison of the effect of glutamate receptor modulators in the 6 Hz and maximal electroshock seizure models. Epilepsy Research. 2003;56:17–26. doi: 10.1016/j.eplepsyres.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Reynolds GP. Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophrenia Research. 1997;24:349–355. doi: 10.1016/s0920-9964(96)00122-3. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Zhang ZJ, Patten I, Reynolds GP. Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biological Psychiatry. 2002;52:708–715. doi: 10.1016/s0006-3223(02)01360-4. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology. 2007;32:1888–1902. doi: 10.1038/sj.npp.1301312. [DOI] [PubMed] [Google Scholar]
- Clifford DB, Olney JW, Benz AM, Fuller TA, Zorumski CF. Ketamine, Phencyclidine, and Mk-801 Protect against Kainic Acid-Induced Seizure-Related Brain-Damage. Epilepsia. 1990;31:382–390. doi: 10.1111/j.1528-1157.1990.tb05492.x. [DOI] [PubMed] [Google Scholar]
- Deakin JFW, Slater P, Simpson MDC, Gilchrist AC, Skan WJ, Royston MC, Reynolds GP, Cross AJ. Frontal Cortical and Left Temporal Glutamatergic Dysfunction in Schizophrenia. Journal of Neurochemistry. 1989;52:1781–1786. doi: 10.1111/j.1471-4159.1989.tb07257.x. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Inada K, Farrington JS, Koller BH. Seizure responses and induction of Fos by the NMDA Agonist (tetrazol-5-yl)glycine in a genetic model of NMDA receptor hypofunction. Brain Research. 2008;1221:41–48. doi: 10.1016/j.brainres.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GE, Johnson KB, Breese GRT. Topographic patterns of brain activity in response to swim stress: Assessment by 2-deoxyglucose uptake and expression of Fos-like immunoreactivity. J Neuroscience. 1993;13:3932–3943. doi: 10.1523/JNEUROSCI.13-09-03932.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GE, Miyamoto S, Gu HB, Lieberman JA, Koller BH, Snouwaert JN. Alterations in regional brain metabolism in genetic and pharmacological models of reduced NMDA receptor function. Brain Research. 2002;951:166–176. doi: 10.1016/s0006-8993(02)03156-6. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Moy SS, Lieberman JA, Koller BH. Effects of haloperidol, clozapine, and quetiapine on sensorimotor gating in a genetic model of reduced NMDA receptor function. Psychopharmacology. 2006a;184:190–200. doi: 10.1007/s00213-005-0214-1. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Moy SS, Lieberman JA, Koller BH. Typical and atypical antipsychotic drug effects on locomotor hyperactivity and deficits in sensorimotor gating in a genetic model of NMDA receptor hypofunction. Pharmacology Biochemistry and Behavior. 2006b;85:481–491. doi: 10.1016/j.pbb.2006.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GE, Moy SS, Perez A, Eddy DM, Zinzow WM, Lieberman JA, Snouwaert JN, Koller BH. Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav Brain Res. 2004;153:507–519. doi: 10.1016/j.bbr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Fox K, Schlaggar BL, Glazewski S, Oleary DDM. Glutamate receptor blockade at cortical synapses disrupts development of thalamocortical and columnar organization in somatosensory cortex. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:5584–5589. doi: 10.1073/pnas.93.11.5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradley RL, O’Meara GF, Newman RJ, Andrieux A, Job D, Reynolds DS. STOP knockout and NMDA NR1 hypomorphic mice exhibit deficits in sensorimotor gating. Behavioural Brain Research. 2005;163:257–264. doi: 10.1016/j.bbr.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1997. [Google Scholar]
- Garey LJ, Von Bussmann KA, Hirsch SR. Decreased numerical density of kainate receptor-positive neurons in the orbitofrontal cortex of chronic schizophrenics. Experimental Brain Research. 2006;173:234–242. doi: 10.1007/s00221-006-0396-8. [DOI] [PubMed] [Google Scholar]
- Inada K, Farrington JS, Moy SS, Koller BH, Duncan GE. Assessment of NMDA receptor activation in vivo by Fos induction after challenge with the direct NMDA agonist (tetrazol-5-yl)glycine: effects of clozapine and haloperidol. Journal of Neural Transmission. 2007;114:899–908. doi: 10.1007/s00702-007-0628-5. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–731. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jane DE, Lodge D, Collingridge GL. Kainate receptors: Pharmacology, function and therapeutic potential. Neuropharmacology. 2009;56:90–113. doi: 10.1016/j.neuropharm.2008.08.023. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. American Journal of Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Archives of General Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Weiler MA, Tamara MB, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. 2001;25:455–467. doi: 10.1016/S0893-133X(01)00243-3. [DOI] [PubMed] [Google Scholar]
- Lee LJ, Iwasato T, Itohara S, Erzurumlu RS. Exuberant thalamocortical axon arborization in cortex-specific NMDAR1 knockout mice. Journal of Comparative Neurology. 2005a;485:280–292. doi: 10.1002/cne.20481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LJ, Lo FS, Erzurumlu RS. NMDA receptor-dependent regulation of axonal and dendritic branching. Journal of Neuroscience. 2005b;25:2304–2311. doi: 10.1523/JNEUROSCI.4902-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Healy DJ. Glutamate receptor expression in schizophrenic brain. Brain Research Reviews. 2000;31:288–294. doi: 10.1016/s0165-0173(99)00044-2. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Snouwaert JN, Koller BH, Moy SS, Lieberman JA, Duncan GE. Amphetamine-induced Fos is reduced in limbic cortical regions but not in the caudate or accumbens in a genetic model of NMDA receptor hypofunction. Neuropsychopharmacology. 2004;29:2180–2188. doi: 10.1038/sj.npp.1300548. [DOI] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Moy SS, Perez A, Koller BH, Duncan GE. Amphetamine-induced disruption of prepulse inhibition in mice with reduced NMDA receptor function. Brain Research. 2006;1089:186–194. doi: 10.1016/j.brainres.2006.03.073. [DOI] [PubMed] [Google Scholar]
- O’Neill MJ, Bond A, Ornstein PL, Ward MA, Hicks CA, Hoo K, Bleakman D, Lodge D. Decahydroisoquinolines: novel competitive AMPA/kainate antagonists with neuroprotective effects in global cerebral ischaemia. Neuropharmacology. 1998;37:1211–1222. doi: 10.1016/s0028-3908(98)00134-8. [DOI] [PubMed] [Google Scholar]
- Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Archives of General Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- Sakai T, Oshima A, Nozaki Y, Ida I, Haga C, Akiyama H, Nakazato Y, Mikuni M. Changes in density of calcium-binding-protein-immunoreactive GABAergic neurons in prefrontal cortex in schizophrenia and bipolar disorder. Neuropathology. 2008;28:143–150. doi: 10.1111/j.1440-1789.2007.00867.x. [DOI] [PubMed] [Google Scholar]
- Scarr E, Beneyto M, Meador-Woodruff JH, Dean B. Cortical glutamatergic markers in schizophrenia. Neuropsychopharmacology. 2005;30:1521–1531. doi: 10.1038/sj.npp.1300758. [DOI] [PubMed] [Google Scholar]
- Schlaggar BL, Fox K, Oleary DDM. Postsynaptic Control of Plasticity in Developing Somatosensory Cortex. Nature. 1993;364:623–626. doi: 10.1038/364623a0. [DOI] [PubMed] [Google Scholar]
- Smolders I, Bortolotto ZA, Clarke VRJ, Warre R, Khan GM, O’Neill MJ, Ornstein PL, Bleakman D, Ogden A, Weiss B, Stables JP, Ho KH, Ebinger G, Collingridge GL, Lodge D, Michotte Y. Antagonists of GLU(K5)-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nature Neuroscience. 2002;5:796–804. doi: 10.1038/nn880. [DOI] [PubMed] [Google Scholar]
- Watis L, Chen SH, Chua HC, Chong SA, Sim K. Glutamatergic abnormalities of the thalamus in schizophrenia: a systematic review. Journal of Neural Transmission. 2008;115:493–511. doi: 10.1007/s00702-007-0859-5. [DOI] [PubMed] [Google Scholar]
- Woo TUW, Shrestha K, Ainstrong C, Minns MM, Walsh JP, Benes FM. Differential alterations of kainate receptor subunits in inhibitory interneurons in the anterior cingulate cortex in schizophrenia and bipolar disorder. Schizophrenia Research. 2007;96:46–61. doi: 10.1016/j.schres.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavitsanou K, Ward PB, Huang XF. Selective alterations in ionotropic glutamate receptors in the anterior cingulate cortex in schizophrenia. Neuropsychopharmacology. 2002;27:826–833. doi: 10.1016/S0893-133X(02)00347-0. [DOI] [PubMed] [Google Scholar]
- Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophrenia Research. 2002;55:1–10. doi: 10.1016/s0920-9964(01)00188-8. [DOI] [PubMed] [Google Scholar]