

Abstract
Current clinical and preclinical anticancer formulations are limited by their use of toxic excipients and stability issues upon combining different drug formulations. We have found that poly(ethylene glycol)-block-poly(d,l lactic acid) (PEG-b-PLA) micelles can deliver multiple poorly water-soluble drugs at clinically relevant doses. Paclitaxel (PTX), etoposide (ETO), docetaxel (DCTX) and 17-AAG were solubilized individually in PEG-b-PLA micelles. Combinations of PTX/17-AAG, ETO/17-AAG, DCTX/17-AAG and PTX/ETO/17-AAG were also solubilized in PEG-b-PLA micelles. PEG-b-PLA micelles were characterized in terms of drug loading, size, stability and drug release. All anticancer agents in all combinations were all solubilized at the level of mg/mL and were stable for 24 hours in the 2 and 3 drug combination PEG-b-PLA micelles. The stability of the 2 and 3 drug combination PEG-b-PLA micelles was due to the presence of 17-AAG. In vitro, t1/2 values for 2 and 3 drug combination PEG-b-PLA micelles spanned 1-5 hrs. PEG-b-PLA micelles offer a promising alternative for combination drug therapy without formulation related side effects.
1. Introduction
Polymeric micelles are nanoscopic core/shell structures usually formed through the self-assembly of amphiphilic block copolymers (ABCs) (1). ABC micelles have a hydrophobic core surrounded by a hydrophilic outer shell. The inner core can be used as a storage site for poorly water-soluble drugs and can act as a nano-depot for these agents. This drug loaded inner core is protected by a biocompatible hydrophilic outer shell. Furthermore, heterogeneous functionalities can be introduced in each domain to facilitate drug loading and targeting. Over the past few years, ABC micelles have been used as drug carriers for poorly water-soluble drugs that result in improved pharmacokinetics (PK) for these drugs. These properties along with a proven safety record in humans has lead to a research spurring it as an alternate to commercial anti-cancer formulations (1-4). Examples of poorly water-soluble drugs in recent studies involving ABC micelles include amphotericin B, cyclosporin A, 4-hydroxyphenylretinamide, irinotecan, paclitaxel (PTX), propofol, and rapamycin (5-11).
Poorly water-soluble drugs like anticancer agents in preclinical development, e.g. 17-allylamino-17-demethoxygeldanamycin (17-AAG), and many in clinical practice, e.g. PTX, docetaxel (DCTX), and etoposide (ETO), require safe vehicles for drug solubilization and IV infusion. However, vehicles for IV drug infusion are often toxic, e.g. Cremophor EL (CrEL), and hamper progress in combination drug therapy involving poorly water-soluble anti-cancer agents. Combination therapy with current excipients runs the risk of precipitation and additive or synergistic toxicity caused by two or more vehicles for drug solubilization, e.g. CrEL for PTX and DMSO/lipid for 17-AAG. Hypersensitivity reactions occur in approximately 40% of patients that receive CrEL and PTX despite pre-medication with corticosteroids and histamine antagonists. CrEL-induced hypersensitivity reactions cause discontinuation of drug therapy, and are life-threatening in 1-3% of patients even with pre-medication with corticosteroids and histamine antagonists (12). Other serious toxicities associated with CrEL use include nephrotoxicity and neurotoxicity (13-15). In preclinical research, intraperitoneal injection of Taxol® and 17-AAG in rapid succession results in swift deaths in mice, mandating a 20 min separation of injections for combination drug therapy (16). It is noted that the most common grade 2 toxicity associated with a 1-or 1.5-hr IV infusion of 17-AAG/DMSO/lipid was bad odor and nausea in a Phase II clinical trial for melanoma (17). In a phase I clinical trial involving 17-AAG and trastuzumab, grade 3 hypersensitivity reactions in two patients associated with a 2-hr IV infusion of 17-AAG/CrEL/propylene glycol/ethanol resulted in discontinuation in the clinical trial despite pre-medication, noting a partial response in one of the patients (18).
Common IV formulations for anticancer agents include the use of CrEL and ethanol for PTX, ethanol and polyoxyethylene sorbitan monooleate (Tween 80) for DCTX (19) and Tween 80 for ETO (20). As noted, 17-AAG is a poorly water soluble and required DMSO/lipid or CrEL for administration in phase I/II clinical trials (21). Thus, there is a clear need to find alternate formulations and vehicles to deliver these agents without contributing to the side effects experienced by patients on chemotherapy.
PEG-b-PLA, an ABC, assembles readily in water into micelles and has been shown to raise the solubility of PTX from approximately 1 μg/mL to 10 mg/mL (9). PEG-b-PLA is less toxic than CrEL. However, a recent phase II clinical trial in metastatic breast cancer patients showed that PTX dosed as part of PEG-b-PLA micelles, without pre-medication with corticosteroids and histamine antagonists, does induce hypersensitivity reactions, albeit less severely than CrEL. PEG-b-PLA micelles increase the maximum tolerated dose of PTX in humans in comparison to CrEL, enhancing its anti-tumor efficacy (22). There is also evidence that PEG-b-PLA micelles impart linear PK for PTX, strongly contrasting with CrEL that induces a non-linear PK profile for PTX, i.e. lowering its clearance with dose escalation (12). Using these PEG-b-PLA micelles to raise the solubility of various anti-cancer agents is a potential delivery option, facilitating ease of entry into clinical trials in the cancer arena.
However, due to the heterogeneity of cancer cells as well as acquired drug resistance, single agent therapy is limited and combination chemotherapy has become a standard regimen to treat cancer patients. To be specific, drug combinations are beneficial in the view of retarding occurrence of resistant cell lines and wide coverage against multiple cell lines, resulting in maximum cell killing effect within acceptable toxicity (23). Synergistic drug combinations produce an even greater response rate or survival time than is possible with each drug used alone at its optimum dose. For example, 17-AAG, a prototype Hsp90 inhibitor had synergistic effects with a broad range of anticancer agents in different tumor cell lines (21, 24). 17-AAG causes a remarkable combinatorial depletion of multiple oncogenic proteins, e.g. Akt, ErbB-2, and Hif-1α, causing a blockage of cancer-causing and survival pathways, and there is keen interest in the combination of chemotherapy and 17-AAG (25).
In the case of 17-AAG and PTX it has been shown that 17-AAG sensitizes cancer cells to apoptosis induced by PTX, a mitotic inhibitor with anti-neoplastic activity, when the drugs are given together or when 17-AAG treatment was followed by exposure to PTX (26, 27). The combination of these two drugs was synergistic in different cancer cell lines and in mice (16, 25, 26, 28). Additionally in mice bearing H358 human non-small cell lung cancer xenografts PTX cytotoxicity was enhanced by 5-22 fold when combined with 17-AAG (29). The combination of PTX and 17-AAG was also evaluated in humans and showed enhanced efficacy and better tolerability profile as compared to PTX alone (30). In another case 17-AAG has also enhanced the activity of ETO, a topoisomerase II inhibitor, in vitro and in vivo (31). The combination of 17-AAG and ETO showed synergism in leukemia cells (32). Another study demonstrated the combination of 17-AAG and ETO decreased the IC50 of ETO by 10 fold in four different pediatric acute lymphoblastic leukemia cell lines (33).
However, despite the advantages of combination chemotherapy, one of the main problems associated with clinical use is the complicated regimens that must be administered to patients. As most anti-cancer drugs are poorly water-soluble and utilize toxic excipients to enhance their solubility, combining two or three drugs can be challenging in clinical practice, owing to compatibility and stability issues (23). Thus, using PEG-b-PLA micelles to rationally design and deliver chemotherapeutic regimens instead of single anti-cancer agents might be a better approach to overcome these formulation related and clinical challenges. In our previous work, we successfully solubilized 17-AAG in PEG-b-PLA micelles (34). The PK profile of 17-AAG in these micelles was similar to CrEL in rats, without the attendent toxicity observed with the CrEL formulation (no deaths versus 35% mortality for CrEL) (34). Based on this platform our goal is to create PEG-b-PLA micellar systems that can simultaneouly delivery multiple anticancer agents, like PTXl, DCTX, or ETO by co-solublizing them with 17-AAG to generate safer, more stable formulations for potentially synergistic combination chemotherapy (Fig. 1).
Fig. 1.
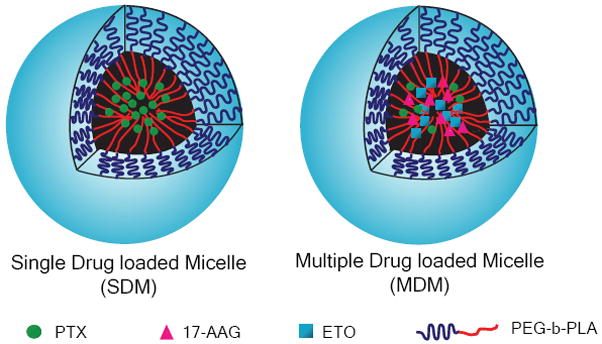
Schematic representation of single and multiple drug loaded PEG-b-PLA micelles
2.Materials & Methods
2.1. Materials
PEG-b-PLA (Mn PEG and PLA are 4.2 K and 1.9 K respectively, PDI = 1.05) was purchased from Advanced Polymer Materials Inc (Montreal, CAN). Paclitaxel was obtained from LKT laboratories Inc (St. Paul, MN). Docetaxel and 17-AAG was purchased from LC Laboratories (Woburn, MA). Etoposide, DMSO-d6 and D2O were purchased from Sigma-Aldrich Inc (St. Louis, MO). All other materials were obtained from Fisher Scientific Inc (Fairlawn, NJ). All reagents were HPLC grade.
2.2.Methods
2.2.1.Preparation and characterization of drug-loaded PEG-b-PLA micelles
Single drug micelles (SDM) were prepared by adding 2.0 mg of PTX, DCTX, ETO or 17-AAG and 15 mg of PEG-b-PLA in a 5.0 mL round bottom flask. The drug-polymer mixture was dissolved in 0.50 mL acetonitrile (ACN). The ACN was removed at 60 °C under reduced pressure on a rotary evaporator resulting in the formation of a homogenous film. The drug-polymer film was rehydrated with 0.50 mL of DD H2O at 60 °C with gentle agitation resulting in a clear solution of drug loaded PEG-b-PLA micelles. The micellar solution was filtered using a 0.45 μm filter, and the micelles were characterized in terms of size and loading by Dynamic Light Scattering (DLS)and HPLC respectively.
PEG-b-PLA was also used to prepare multiple drug micelles (MDM) with different drugs combinations of PTX/17-AAG, DCTX/17-AAG, ETO/17-AAG and PTX/ETO/17-AAG. MDMs were prepared similarly to the SDMs by mixing 2.0 mg of each drug with 15 mg of the polymer. The procedure for the preparation and characterization of these micelles was identical to the SDMs.
Drug(s) to polymer w/w percent was calculated for SDM and MDM. PEG-b-PLA micelles were freeze dried, weighed and the amount of drug(s) in the freeze dried sample was quantified by HPLC. The drug w/w percent was calculated as the mass of the drug(s)to the mass of polymer in the freeze dried sample multiplied by 100.
2.2.2. Quantification of drug loading in SDM and MDM by reverse phase-HPLC
The content of drug loaded in PEG-b-PLA micelles was quantified by reverse phase HPLC. The HPLC system used for quantifying was a Shimadzu prominence HPLC system (Shimadzu, JP), consisting of a LC-20AT pump, SIL-20AC HT autosampler, CTO-20AC column oven and a SPD-M20A diode array detector. A sample of 10 μL was injected into a Zorbax SB-C8 Rapid Resolution cartridge (4.6×75mm, 3.5 micron, Agilent). The column temperature was maintained at 40 °C throughout the run. Two HPLC methods were developed to quantify the amount of the drug(s) loaded in PEG-b-PLA micelles. The first method was developed to quantify PTX,DCTX and 17-AAG in SDM or MDM. The mobile phase was an isocratic mixture of 45% ACN and 54% aqueous phase containing 0.1% phosphoric acid and 1% methanol in DD H2O. The run time was 10 min, the flow rate was 1.0 mL/min and the detection was at 227 nm for PTX and DCTX while 17 AAG was detected at 333 nm. The retention time of 17-AAG, PTX, and DCTX were 5.6, 6.8 and 8.1 min, respectively, while the limit of detection (LOD) of the three drugs by this method were 1.00, 0.52, and 0.43 μg/mL, respectively. The second HPLC method was developed to quantify PTX, ETO and 17-AAG in SDM or MDM. The mobile phase was a gradient mixture of ACN and aqueous phase containing 0.1% phosphoric acid and 1% methanol in DD H2O. The run time was 15 min, the flow rate was 1.0 mL/min and the detection was at 227 nm for PTX and ETO while 17-AAG was detected at 333 nm. The retention time of ETO, PTX and 17-AAG were 3.3, 8.0 and 8.5 min respectively while LOD of the three drugs were 0.55, 0.47 and 0.43 μg/mL, respectively. With both HPLC methods sample were injected twice and reproducible and rapid separation of the drugs was achieved.
2.2.3.Dynamic Light Scattering (DLS) measurements of SDM and MDM
The size of the micelles was determined by DLS using a ZETASIZER Nano-ZS (Malvern Instruments Inc., UK) equipped with He-Ne laser (4mW, 633 nm) light source and 90° angle scattered light collection configuration. The drug loaded micellar solutions were diluted 20 times with DD H2O and the samples was equilibrated for 2 min at 25 °C before the measurements. Final PEG-b-PLA concentration was approximately 1.5 mg/mL. The hydrodynamic diameter of PEG-b-PLA micelles was calculated based on the Stokes-Einstein equation. Correlation function was curve-fitted by cumulant method to calculate mean size and polydispersion index(PDI). All measurements were repeated three times, and volume-weighted particle sizes were presented as the average diameter with standard deviation.
2.2.4. Turbidity measurements of SDM and MDM
Turbidity measurements were used to evaluate the physical stability of drug loaded PEG-b-PLA micelles. A CARY 50 BIO UV-Visible spectrophotometer equipped with dip probe was used to measure turbidity. Micellar solutions were diluted six times, for a final concentrion of PEG-b-PLA of 5 mg/mL, with DD H2O and filtered through 0.45 μm filter. The absorbance of each sample was recorded at 650 nm and collected over 24 hr at ambient temperature. Each measurement was performed in triplicate.
2.2.5. 1H NMR spectroscopy of SDM and MDM
1H NMR spectroscopy was used to confirm the incorporation of drugs into PEG-b-PLA micelles. Individual drugs or multiple drugs in PEG-b-PLA polymer films were prepared as described in section 2.2.1. The formed film was solubilized in 0.70 mL of DMSO-d6 or in 0.70 mL D2O warmed to 60 °C, and the 1H NMR spectrum recorded for each sample.
1H NMR measurements were performed on UNITY INOVA NMR spectrometers (Varian, USA) model operating at 400 MHz normal proton frequencies. Sample temperature was regulated for all measurements and was set at 25 °C. The spectrometer was equipped with FTS Systems preconditioning device (composed of refrigerating unit, internal temperature controller and inclusion transfer line). To control pre-cooling or pre-heating of the compressed and dried air used as temperature control medium: final temperature regulation of the sample was achieved within the NMR probe. Acquisition parameters were adjusted on a case-by-case basis to provide adequate signal-to-noise ratio and spectral resolution, the latter typically at 0.5 ppB/point for 1D High-resolution proton. More specifically, protons were excited by a single π/2 pulse followed by detection of the proton signal.
2.2.6.In vitro release profiles of drug(s) from SDM and MDM
The release profile of PTX, DCTX, ETO and 17-AAG from PEG-b-PLA micelles was evaluated by a dialysis method. SDMs or MDMs were prepared and characterized as mentioned in section 2.2.1. Post micelle preparation each sample was diluted with DD H2O, to yield about 0.10 mg/mL of each drug. A volume of 2.5 mL of the prepared sample was loaded into a 3 ml Slide-A-Lyzer® (Thermo Scientific Inc.) dialysis cassette with a MWCO of 20,000 g/mol. Four cassettes were used in each experiment. The cassettes were placed in 2.0 L of buffer which was changed every 3 hr to ensure sink conditions for drug(s) and polymer. A sample of 100 μL was drawn from each cassette at various sampling time intervals and then replaced with 100 μL of fresh buffer. The sampling time intervals were 0, 0.5, 2, 3, 6, 9, 12 and 24 hr. The amount of drug(s)in each sample was quantified by HPLC as per section 2.2.2.
2.2.7. Data Analysis
Statistical analysis was performed using one-way ANOVA at 5 % significance level combined with Tukey’s Multiple Comparison Test or t-test at 5 % significance level. Curve-fitting analysis using one phase exponential association was used to calculate the half-life (t1/2) of drug in in vitro drug release experiments. Both analyses were performed using GraphPad Prism version 5.00 for Windows, GraphPad Software, San Diego California USA, www.graphpad.com
3. Results
3.1 Preparation and characterization of drug-loaded PEG-b-PLA micelles
SDMs were prepared for PTX, DCTX, ETO or 17-AAG with PEG-b-PLA. The solubility of all drugs in the micelles was starkly significantly higher as compared to their intrinsic solubility in water Table 1 and Fig.2. PTX solubility increased from 0.0003 mg/mL (35, 36) to 3.50 mg/mL, DCTX solubility increased from 0.0055 mg/mL (35)to 4.27 mg/mL. ETO solubility also increased from 0.0580 mg/mL (37) to 3.17 mg/mL. 17-AAG solubility increased from 0.1000 gm/mL (38)to 4.21 mg/mL.
Table 1.
Physical characterization of SDM and MDM. (n = 3 ± SD).
| Drug(s) in SDM or MDM | Solubility (mg/ml) | Drug to polymer w/w (%) | Total drug(s) to polymer w/w (%) | Micelle diameter (nm ± SD) |
|---|---|---|---|---|
| PTX | 3.54 ± 0.32 * | 10.3 ± 0.9 | 10.3 ± 0.9 | 38.8 ± 0.6 |
| DCTX | 4.27 ± 0.44 * | 11.5 ± 0.5 | 11.5 ± 0.5 | 37.3 ± 1.7 |
| ETO | 3.31 ± 0.15 * | 9.6 ± 0.6 | 9.6 ± 0.6 | 32.6 ± 1.0 |
| 17-AAG | 3.90 ± 0.28 | 11.3 ± 0.3 | 11.3 ± 0.3 | 39.3 ± 2.9 |
| PTX | 3.92 ± 0.17 | 13.4 ± 0.9 | 25.9 ± 1.6 | 38.9 ± 1.1 |
| 17-AAG | 3.88 ± 0.29 | 12.4 ± 0.8 | ||
| DCTX | 4.62 ± 0.44 | 12.6 ± 1.6 | 25.8 ± 2.2 | 39.0 ± 0.8 |
| 17-AAG | 4.01 ± 0.08 | 13.3 ± 0.6 | ||
| ETO | 3.49 ± 0.24 | 12.6 ± 0.9 | 25.0 ± 2.2 | 35.3 ± 1.2 |
| 17-AAG | 4.21 ± 0.38 | 13.3 ± 0.6 | ||
| PTX | 3.50 ± 0.20 | 11.5 ± 0.5 | 35.6 ± 1.5 | 36.5 ± 0.5 |
| ETO | 3.17 ± 0.04 | 12.0 ± 0.5 | ||
| 17-AAG | 3.61 ± 0.33 | 12.1 ± 0.5 | ||
denotes statistical significance at p<0.05 compared to the compound’s intrinsic solubility
Fig. 2.
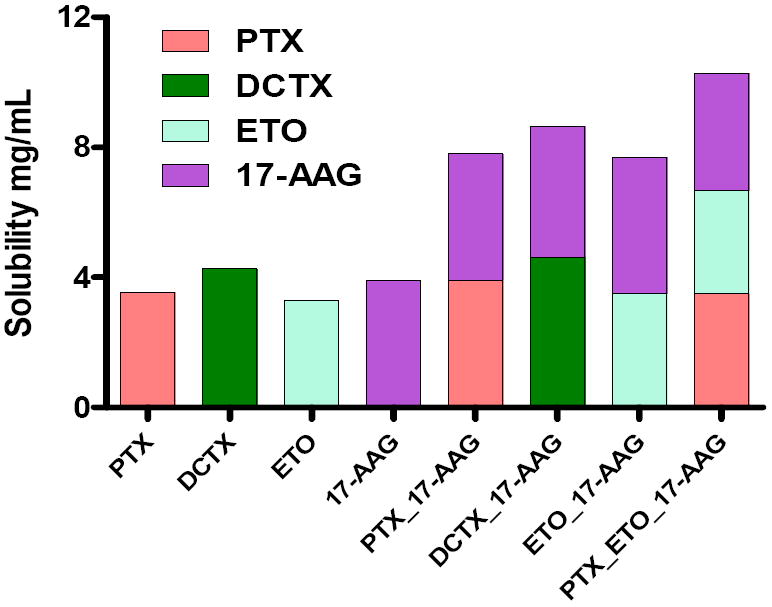
Aqueous solubility of drug(s) in SDM and MDM (n = 3)
PEG-b-PLA was used to prepare MDM with different drugs combinations of PTX/ 17-AAG, DCTX/17-AAG, ETO/17-AAG and PTX/ETO/17-AGG. The magnitude of solubility enhancement for all drugs in the MDM micelles was similar to the SDMs. The presence of multiple drugs within PEG-b-PLA micelles did not adversely affect the apparent solubility enhancement of the individual drugs in a statistically significant manner Table 1 and Fig.2.
The drug(s) to polymer w/w percents of SDM and MDM are listed in Table 1. For SDM, PTX, DCTX, ETO and 17 –AAG w/wpercentswere11.3, 11.5, 9.6 and 11.3% respectively. The w/w percents for all drugs in the MDM were statistically the same as the SDM. For all SDMs and MDMs approximately100% of the initial amount of the drug(s) and polymer was recovered as loaded PEG-b-PLA micelles. The capacity of these PEG-b-PLA micelles to incorporate drugs increased as the number drugs being loaded increased. For SDM the loading capacity % w/w was approximately 10%, with two drug MDM the loading capacity of the micelle increased to approximately 25 % w/w and with three drug MDM the loading capacity was approximately 35 % w/w.
3.2 DLS measurements of SDM and MDM
The size of unloaded PEG-b-PLA micelle, SDMs and MDMs measured by DLS are listed in Table 1. All micelles exhibited a unimodal distribution with a size range of 30 -40 nm. PDI of all SDMs and MDMs were below 0.2, indicating narrow particle size distribution.
3.3 Turbidity measurements of SDM and MDM
The physical stability of SDM and MDM was evaluated by turbidity measurements. The increase in turbidity, as measured by changes in absorbance at 650 nm, over time correlates with drug precipitation following release from PEG-b-PLA micelles. The turbidity measurements were further supported by HPLC and DLS measurements. SDM with 17-AAG is physically stable over 24 hrs without a change in particle size or drug loss Table 2. SDM with PTX (Fig. 3), DCTX & ETO were stable for approximately 6 hrs. HPLC data shows that at 24 hrs approximately 84%, 73 % and 68 %of PTX, DCTX and ETO precipitated, respectively. SDM solutions exhibited a cloudy, white appearance post 24 hrs indicative of drug precipitation. DLS data also showed aggregate formation (data not shown).
Table 2.
Initial solubility and solubility at 24 hours of drug(s) in SDM and MDM as assessed by HPLC (n = 3 ± SD).
| Drug(s) in SDM or MDM | Initial Solubility (mg/ml) | Solubility @ 24 hr (mg/ml) | % w/w drug(s) retained @ 24 hr |
|---|---|---|---|
| PTX | 3.54 ± 0.32 * | 0.57 ± 0.07 * | 16.2 ± 1.0 |
| DCTX | 4.27 ± 0.44 * | 1.14 ± 0.03 * | 26.8 ± 3.3 |
| ETO | 3.31 ± 0.15 * | 1.07 ± 0.16 * | 32.3 ± 3.4 |
| 17-AAG | 3.90 ± 0.28 | 3.84 ± 0.18 | 98.6 ± 2.4 |
| PTX | 3.92 ± 0.17 | 3.86 ± 0.15 | 98.5 ± 0.3 |
| 17-AAG | 3.88 ± 0.29 | 3.77 ± 0.28 | 96.9 ± 0.2 |
| DCTX | 4.62 ± 0.44 | 4.45 ± 0.13 | 96.3 ±1.8 |
| 17-AAG | 4.01 ± 0.08 | 3.83 ± 0.17 | 95.5 ± 2.7 |
| ETO | 3.49 ± 0.24 | 3.28 ± 0.19 | 94.1 ± 1.5 |
| 17-AAG | 4.21 ± 0.38 | 3.95 ± 0.39 | 93.9 ± 1.1 |
| PTX | 3.50 ± 0.20 | 3.46 ± 0.18 | 98.7 ± 1.3 |
| ETO | 3.17 ± 0.04 | 3.11 ± 0.06 | 98.2 ± 1.1 |
| 17-AAG | 3.61 ± 0.33 | 3.52 ± 0.33 | 97.5 ± 1.4 |
denotes statistical significance at p<0.05
Fig. 3.
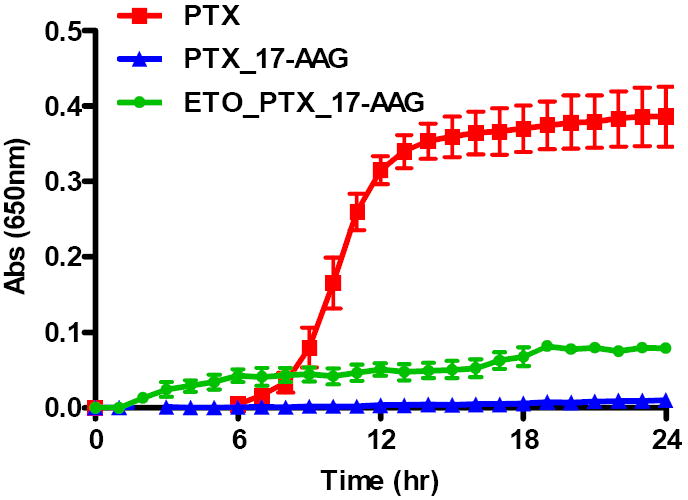
Turbidity measurements of SDM with PTX, PTX/17-AAG MDM & PTX/ETO/17-AAG MDM (n = 3 ± SD)
MDM with PTX and 17-AAG were more stable against precipitation than SDM with PTX alone over a 24 hr period (Fig. 3), noting drug retention of approximately 97 % Table 2. DCTX/17-AAG and ETO/17-AAG have similar results for stability and drug retention within the micelle of approximately 96% and 94 % respectively Table 2. At 24 hrs these MDMs solutions were visually clear and DLS data confirmed the particle size, remained between 30 -40 nm (data not shown).
In the 3-drug combination, MDM of PTX/ETO/17-AAG was stable for 24 hrs (Fig 3). Drug loading data indicated that all drugs were retained above 97 % of their initial loading value at 24 hrs. MDM solutions were visually clear and particle size did not change significantly at 24 hrs (data not shown).
3.4 1H NMR spectroscopy of SDM and MDM
1H NMR measurements were used to confirm the incorporation of drugs into PEG-b-PLA micelles. 1H NMR spectra in DMSO-d6 for individual drugs and drug combinations showed all the prominent resonance peaks representative of the drug(s) and those of PLA and PEG blocks. In contrast only the PEG resonance peaks were detected in D2O while PLA and drug(s) resonance peaks were absents due to the restricted mobility of the PLA and drug(s) molecules within the core of the micelle which is indicative of drug(s) incorporation into PEG-b-PLA micelles (39).
PEG-b-PLA was identified by proton resonances at 3.4-3.6 ppm from ethylene oxide of the PEG group and the proton resonances at 5.0-5.1 for the lactic acid group (39, 40). 17-AAG was identified by proton resonance at 0.7 ppm for the methyl groups. PTX was identified by proton resonances at 1.0 ppm for its methyl groups at C16 and C17. Lastly, ETO was identified by proton resonances between 6 – 7 ppm for the aromatic protons of the benzene rings. A representative group of NMR spectra for PTX/ETO/17-AAG and polymer in DMSO-d6 and MDM with PTX/ETO/17-AAG in D2O have been provided (Fig. 4).
Fig. 4.
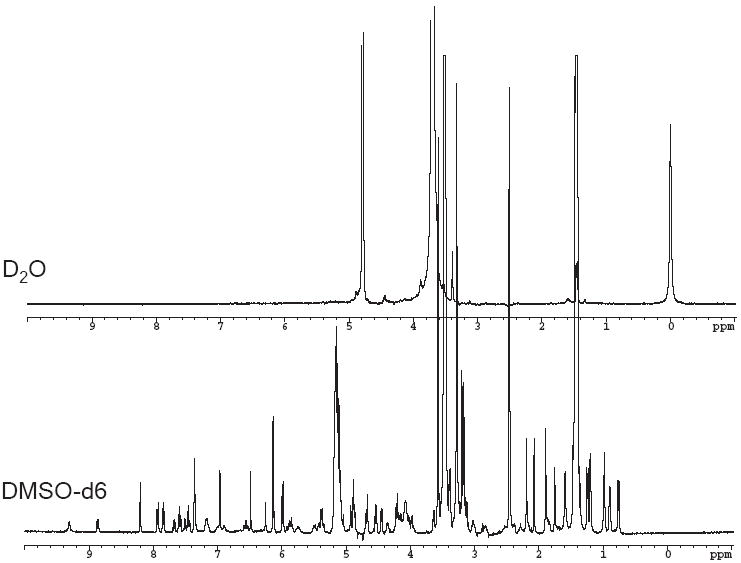
1H NMR spectra of PTX/ETO/17-AAG in (A) D2O (B) DMSO-d6
3.5 In vitro release profiles of drug(s) from SDM and MDM
Drug release data from SDM containing 17-AAG, ETO showed that over 90% of the drug was release from PEG-b-PLA micelle in 6 hrs (Fig. 5A). Over 90% of DCTX was released from SDM in 9 hrs (Fig. 5A). Rapid precipitation of PTX from the SDM during the dialysis experiment precluded garnering of any meaningful data. MDM containing PTX and 17-AAG showed over 72 % PTX release in 24 hrs while more than 90% of 17-AAG was released in 9 hrs (Fig. 5B). MDM containing ETO and 17-AAG showed over 90 % drug release in 6 and 9 hrs, respectively (Fig. 5C). MDM containing DCTX and 17-AAG showed over 90 % release in 12 and 9 hrs, respectively (Fig. 5D). MDM containing the three drug combination of PTX/ETO/17-AAG showed approximately 80% release of PTX in 24 hrs and over 90 % release for ETO and 17-AAG in 6 and 9 hrs, respectively (Fig. 5E).
Fig. 5.
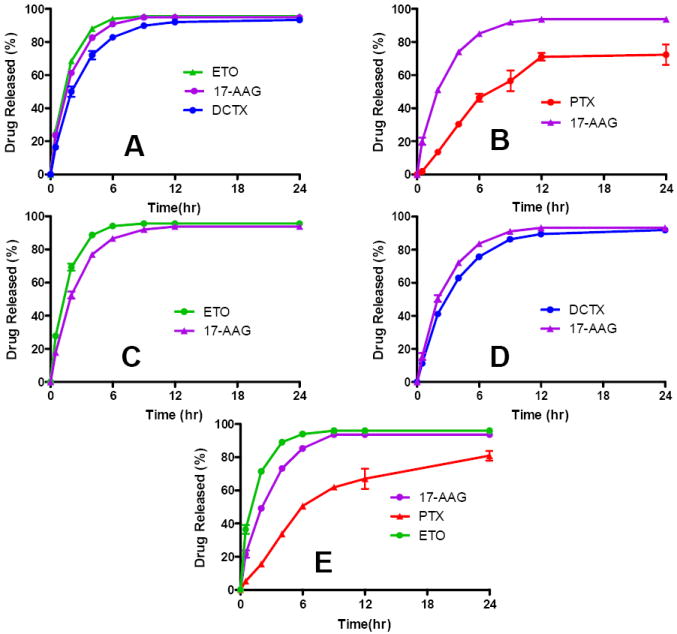
In vitro drug release kinetics of (A) SDM containing ETO, DCTX or 17-AAG, (B) MDM with PTX/17-AAG, (C) MDM with ETO/17-AAG, (D) MDM with DCTX/17-AAG and (E) MDM with PTX/ETO/17-AAG (n = 4 ± SD)
The in vitro drug release data was curve fitted using one phase exponential association (GraphPad Prism). The first order rate constant derived from the curve-fitting was used to calculate the t1/2 of the drug release from PEG-b-PLA micelles. The data for the SDM and MDM are presented in Table 3 along with the goodness of fit and log oil-in-water partition coefficient values for all drugs. The data in Table 3 lists the first-order rate constants and the t1/2 values for all SDM and MDM except for SDM with PTX. This again was due to the rapid precipitation of PTX from the SDM during drug release test, which precluded the possibility of garnering any useful data.
Table 3.
Curve fitting of in vitro drug(s) release from SDM and MDM (n = 4, mean ± SD).
| Drug(s) in SDM or MDM | first-order rate constant (hr-1) | t1/2 (hr) | goodness of fit (r2) | log P |
|---|---|---|---|---|
| PTX | - | - | - | 3.0 |
| DCTX | 0.379 | 1.83 | 0.993 | 2.4 |
| 17-AAG | 0.525 | 1.32 | 0.999 | 1.3 |
| ETO | 0.636 | 1.09 | 0.999 | 1.0 |
| PTX | 0.138 | 5.01 | 0.938 | 3.0 |
| 17-AAG | 0.398 | 1.74 | 0.996 | 1.3 |
| DCTX | 0.288 | 2.41 | 0.996 | 2.4 |
| 17-AAG | 0.375 | 1.85 | 0.996 | 1.3 |
| ETO | 0.657 | 1.06 | 0.997 | 1.0 |
| 17-AAG | 0.414 | 1.67 | 0.997 | 1.3 |
| PTX | 0.136 | 5.10 | 0.973 | 3.0 |
| ETO | 0.785 | 0.88 | 0.992 | 1.0 |
| 17-AAG | 0.386 | 1.80 | 0.995 | 1.3 |
4. Discussion
PEG-b-PLA micelles were able to successfully solubilize all chemotherapeutic agents alone or in combination with other drugs at clinically relevant levels. Given the toxicity associated with common formulation vehicles used, like CrEL, ethanol, DMSO and Tween 80 to name a few, this formulation provides a safer and less toxic alternative. Additionally, the presence of multiple drugs within the same micelles did not adversely effect the solubility enhancement achieved by individual drugs. This is due to the high loading capacity of PEG-b-PLA micelles as seen by the increasing % w/w contributions of the drug in forming these micelles. This allows for combination chemotherapy within one carrier system, which has not been previously attempted due to solubility and stability issues. Initial solubility studies have indicated that PEG-b-PLA micelles have the capacity to solubilize multiple drugs allowing for concomitant delivery of potentially synergistic chemotherapeutic combinations without the attendant clinical issues currently seen while using multiple drug regimens.
The stability of these formulations was also evaluated using optical density measurements coupled with HPLC and DLS. Assessing the stability of PEG-b-PLA micelles is crucial to determine if the formulation is stable long enough for handling and IV administration. As the data shows the SDM are stable for at least 6 hrs while the 17-AAG micelles were stable for 24 hrs. The two drug MDM combinations showed greater stability. The presence of 17-AAG in these micelles helps stabilize the formulation and confers greater stability of the chemotherapeutic agents at the same level of solubilization as seen with the SDM. All two drug MDM combinations retained over 94 %of their drug loading at 24 hrs. The three drug combination MDM showed the highest degree of stability at 24 hrs with over 97 % of drug loaded being retained. For all PEG-b-PLA micelles that were stable at 24 hrs there was no significant shift in their size as determined by DLS. The stability data is extremely promising and indicates that these MDM formulations are able to deliver clinically relevant doses of there chemotherapeutic agents in a clinically relevant time frame.
We were able to confirm the formation of micelles and the localization of the drug(s) within the micellar core using 1H NMR spectroscopy. The data in Fig. 4B clearly shows the proton resonances for all drugs and PEG-b-PLA in DMSO-d6. In contrast Fig. 4A had suppressed proton resonances for the PLA block and drugs with only the PEG block peaks clearly visible. The presence of the drug(s) within the micelle core restricts the mobility of the drug(s) molecules and results in a loss of drug proton resonances. Characterization of the in vitro release kinetics of chemotherapy of individual drugs and in combination for PEG-b-PLA micelles is important to assess the stability and release pattern of the drug(s) from the micelle under sink conditions. In previous work, the in vitro release of 17-AAG from PEG-b-PLA micelles was fairly rapid (on the scale of a few hrs), consistent with a low impact of PEG-b-PLA micelles on the PK of 17-AAG in rats (34). In this work, in vitro release of DCTX, ETO, or 17-AAG from PEG-b-PLA micelles spans several hrs with t1/2 ranging from 1.09 hr for ETO,1.32 hr for 17-AAG, and 1.83 hr for DCTX (Fig. 5), corresponding well with the oil/water partition coefficients of anticancer agents Table 3. PTX precipitates during release from PEG-b-PLA micelles, preventing an estimation of t1/2. For 2-drug combinations, release of 17-AAG is faster than PTX and DCTX and slower than ETO, which has the lowest log P (Fig. 5) Table 3. For the 3-drug combination, t1/2 values for ETO, 17-AAG, and PTX are 0.88, 1.80, and 5.10 hrs, respectively, for PEG-b-PLA micelles (Fig. 5E), corresponding well with the oil/water partition coefficients of these anticancer agents.
The interesting phenomena seen with all the two drug and these combinations is the ability of 17-AAG to stabilize PEG-b-PLA micelles. This phenomenon is starkly evidenced by the stability of PTX in the two drug and three drug combinations, while itself being prone precipitation in the SDM formulation. The novel finding of ability of 17-AAG to maintain the stability of PTX and other hydrophobic drugs in PEG-b-PLA micelles for at least 24 hours bears further investigation. This insight might lead to designing better combination chemotherapeutic carriers in the future.
Another facet of the in vitro release kinetics suggests that variation between the release of anti-cancer agents from the micelles is diffusion-controlled and not due to the break down of PEG-b-PLA micelles, where drug release kinetics are expected to be similar for the two and three drug combinations. However, t1/2 values for PEG-b-PLA micelles suggest that drug release will be fairly rapid and comparable in vivo, owing to poor stability of PEG-b-PLA micelles in the presence of serum proteins, specifically α- and β-globulins (41). In earlier work, PEG-b-PLA micelles filled with a pair of fluorescent probes, DiIC18 and DiOC18, lose fluorescence resonance energy transfer after IV injection within 15 min, indicating rapid release of DiIC18 and DiOC18 in vivo. As it turns out, α- and β-globulins induce a rapid loss of fluorescence energy transfer of DiIC18 and DiOC18 for PEG-b-PLA micelles in vitro as well, probably due to disruption of PEG-b-PLA micelles. These results indicate that PEG-b-PLA micelles will release pairs of chemotherapy and 17-AAG within 1 hr in vivo due to a loss of integrity, caused by α- and β-globulins, although effects of dilution and other components in blood must also be considered.
The promising nature of these results will be tested in the near future in cancer cell lines to determine cytotoxic concentrations for these combinations. This will allow us to further determine if any additive and/or synergistic effects are seen with these combinations. Further validation in murine tumor models will help test the hypothesis that concurrent combination drug release from PEG-b-PLA micelles may provide linear PKs for PTX and 17-AAG and potentially synergistic anti-tumor efficacy given higher maximum tolerated doses in comparison to DMSO/lipid and CrEL (16). Studies with other two and three drug combinations in PEG-b-PLA micelles will also be conducted in murine tumor models.
5. Conclusions
Current chemotherapeutic agents in clinical and pre-clinical situations require dosing with harsh excipients that can cause severe formulation related side effects. Additionally stability concerns create issues with administering combination chemotherapy simultaneously. Our formulation using PEG-b-PLA offers a novel alternative to the current commercial formulations. We have shown that we can successfully combine up to three chemotherapeutic agents in one carrier system at clinically relevant concentrations and with 24 hour stability. Another promising find of this work is the ability of 17-AAG to maintain the stability of different hydrophobic drugs in the carrier system for 24 hours.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aliabadi HM, Shahin M, Brocks DR, Lavasanifar A. Disposition of Drugs in Block Copolymer Micelle Delivery Systems: From Discovery to Recovery. Clinical Pharmacokinetics. 2008;47:619–634. doi: 10.2165/00003088-200847100-00001. [DOI] [PubMed] [Google Scholar]
- 2.Kwon GS. Editorial for Theme Section on Polymeric Micelles for Drug Delivery. Pharmaceutical Research. 2008;25:2053–2055. doi: 10.1007/s11095-008-9644-x. [DOI] [PubMed] [Google Scholar]
- 3.Torchilin VP. Structure and design of polymeric surfactant-based drug delivery systems. Journal of Controlled Release. 2001;73:137–172. doi: 10.1016/s0168-3659(01)00299-1. [DOI] [PubMed] [Google Scholar]
- 4.Kataoka K, Kwon GS, Yokoyama M, Teruo O, Sakurai Y. Block copolymer micelles as vehicles for drug delivery. Journal of Controlled Release Special Issue Proceedings of the Second European Symposium on Controlled Drug Delivery. 1993;24:119–132. [Google Scholar]
- 5.Vakil R, Kwon GS. Poly(ethylene glycol)-b-Poly(E-caprolactone) and PEG-Phospholipid Form Stable Mixed Micelles in Aqueous Media. Langmuir. 2006;22:9723–9729. doi: 10.1021/la061408y. [DOI] [PubMed] [Google Scholar]
- 6.Aliabadi HM, Mahmud A, Sharifabadi AD, Lavasanifar A. Micelles of methoxy poly(ethylene oxide)-b-poly([var epsilon]-caprolactone) as vehicles for the solubilization and controlled delivery of cyclosporine A. Journal of Controlled Release. 2005;104:301–311. doi: 10.1016/j.jconrel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 7.Okuda T, Kawakami S, Yokoyama M, Yamamoto T, Yamashita F, Hashida M. Block copolymer design for stable encapsulation of N-(4-hydroxyphenyl)retinamide into polymeric micelles in mice. International Journal of Pharmaceutics. 2008;357:318–322. doi: 10.1016/j.ijpharm.2008.01.044. [DOI] [PubMed] [Google Scholar]
- 8.Matsumura Y. Poly (amino acid) micelle nanocarriers in preclinical and clinical studies. Advanced Drug Delivery Reviews “Clinical Developments in Drug Delivery Nanotechnology”. 2008;60:899–914. doi: 10.1016/j.addr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Kim SC, Kim DW, Shim YH, Bang JS, Oh HS, Kim SW, Seo MH. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. Journal of Controlled Release. 2001;72:191–202. doi: 10.1016/s0168-3659(01)00275-9. [DOI] [PubMed] [Google Scholar]
- 10.Ravenelle F, Vachon P, Rigby-Jones AE, Sneyd JR, Le Garrec D, Gori S, Lessard D, Smith DC. Anaesthetic effects of propofol polymeric micelle: a novel water soluble propofol formulation. British Journal of Anaesthesia. 2008;101:186–193. doi: 10.1093/bja/aen147. [DOI] [PubMed] [Google Scholar]
- 11.Forrest ML, Won C-Y, Malick AW, Kwon GS. In vitro release of the mTOR inhibitor rapamycin from poly(ethylene glycol)-b-poly([epsilon]-caprolactone) micelles. Journal of Controlled Release. 2006;110:370–377. doi: 10.1016/j.jconrel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Tije AJt, Verweij J, Loos WJ, Sparreboom A. Pharmacological Effects of Formulation Vehicles: Implications for Cancer Chemotherapy. Clinical Pharmacokinetics. 2003;42:665–685. doi: 10.2165/00003088-200342070-00005. [DOI] [PubMed] [Google Scholar]
- 13.Weiss R, Donehower R, Wiernik P, Ohnuma T, Gralla R, Trump D, Baker J, Jr, Van Echo D, Von Hoff D, Leyland-Jones B. Hypersensitivity reactions from taxol. J Clin Oncol. 1990;8:1263–1268. doi: 10.1200/JCO.1990.8.7.1263. [DOI] [PubMed] [Google Scholar]
- 14.Lorenz W, Reimann H-J, Schmal A, Dormann P, Schwarz B, Neugebauer E, Doenicke A. Histamine release in dogs by Cremophor EL ® and its derivatives: Oxethylated oleic acid is the most effective constituent. Inflammation Research. 1977;7:63–67. doi: 10.1007/BF01964882. [DOI] [PubMed] [Google Scholar]
- 15.Dye D, Watkins J. Suspected anaphylactic reaction to Cremophor EL. British Medical Journal. 1980;280:1353. doi: 10.1136/bmj.280.6228.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solit DB, Basso AD, Olshen AB, Scher HI, Rosen N. Inhibition of Heat Shock Protein 90 Function Down-Regulates Akt Kinase and Sensitizes Tumors to Taxol. Cancer Res. 2003;63:2139–2144. [PubMed] [Google Scholar]
- 17.Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, Teitcher J, Wolchok JD, Germino FJ, Krown SE, Coit D, Rosen N, Chapman PB. Phase II Trial of 17-Allylamino-17-Demethoxygeldanamycin in Patients with Metastatic Melanoma. Clin Cancer Res. 2008;14:8302–8307. doi: 10.1158/1078-0432.CCR-08-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Bagatell R, Ma W, Wheler J, Rosen N, Norton L, Cropp GF, Johnson RG, Hannah AL, Hudis CA. Combination of Trastuzumab and Tanespimycin (17-AAG, KOS-953) Is Safe and Active in Trastuzumab-Refractory HER-2 Overexpressing Breast Cancer: A Phase I Dose-Escalation Study. J Clin Oncol. 2007;25:5410–5417. doi: 10.1200/JCO.2007.11.7960. [DOI] [PubMed] [Google Scholar]
- 19.Hennenfent and KL, Govindan R. Novel formulations of taxanes: a review. Old wine in a new bottle? Ann Oncol. 2006;17:735–749. doi: 10.1093/annonc/mdj100. [DOI] [PubMed] [Google Scholar]
- 20.Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. European Journal of Cancer. 1998;34:1514–1521. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 21.Solit DB, Chiosis G. Development and application of Hsp90 inhibitors. Drug Discovery Today. 2008;13:38–43. doi: 10.1016/j.drudis.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Lee KS, Chung HC, Im SA, Park YH, Kim CS, Kim S-B, Rha SY, Lee MY, Ro J. Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Research and Treatment. 2008;108:241–250. doi: 10.1007/s10549-007-9591-y. [DOI] [PubMed] [Google Scholar]
- 23.Finley RS, Balmer C. Optimizing Chemotherapy Outcome. In: Finleyand RS, Balmer C, editors. Concepts in oncology therapeutics. American Society of Heallth-System Pharmacists; Bethesda, MD: 1998. pp. 87–89. [Google Scholar]
- 24.Zhou Y, Feng L, Mueller T, Liu F, Johnson RG., Jr 17-AAG (KOS-953), an inhibitor of Hsp90 function, significantly enhances the cytotoxicity of anticancer drugs: An effective approach for combination therapy. AACR Meeting Abstracts. 2004;2004:458-b. [Google Scholar]
- 25.Xiao L, Rasouli P, Ruden DM. Possible Effects of Early Treatments of Hsp90 Inhibitors on Preventing the Evolution of Drug Resistance to Other Anti-Cancer Drugs. Current Medicinal Chemistry. 2007;14:223–232. doi: 10.2174/092986707779313372. [DOI] [PubMed] [Google Scholar]
- 26.Munster PN, Basso A, Solit D, Norton L, Rosen N. Modulation of Hsp90 Function by Ansamycins Sensitizes Breast Cancer Cells to Chemotherapy-induced Apoptosis in an RB-and Schedule-dependent Manner. Clin Cancer Res. 2001;7:2228–2236. [PubMed] [Google Scholar]
- 27.Nguyen DM, Chen A, Mixon A, Schrump DS, Roth SJA. Sequence-dependent enhancement of paclitaxel toxicity in non-small cell lung cancer by 17-allylamino 17-demethoxygeldanamycin. J Thorac Cardiovasc Surg. 1999;118:908–915. doi: 10.1016/s0022-5223(99)70061-9. [DOI] [PubMed] [Google Scholar]
- 28.Sain N, Krishnan B, Ormerod MG, De Rienzo A, Liu WM, Kaye SB, Workman P, Jackman AL. Potentiation of paclitaxel activity by the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin in human ovarian carcinoma cell lines with high levels of activated AKT. Mol Cancer Ther. 2006;5:1197–1208. doi: 10.1158/1535-7163.MCT-05-0445. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen DM, Lorang D, Chen GA, Stewart JH, Tabibi E, Schrump DS. Enhancement of paclitaxel-mediated cytotoxicity in lung cancer cells by 17-allylamino geldanamycin: in vitro and in vivo analysis. The Annals of Thoracic Surgery. 2001;72:371–379. doi: 10.1016/s0003-4975(01)02787-4. [DOI] [PubMed] [Google Scholar]
- 30.Ramalingam SS, Egorin MJ, Ramanathan RK, Remick SC, Sikorski RP, Lagattuta TF, Chatta GS, Friedland DM, Stoller RG, Potter DM, Ivy SP, Belani CP. A Phase I Study of 17-Allylamino-17- Demethoxygeldanamycin Combined with Paclitaxel in Patients with Advanced Solid Malignancies. Clin Cancer Res. 2008;14:3456–3461. doi: 10.1158/1078-0432.CCR-07-5088. [DOI] [PubMed] [Google Scholar]
- 31.Barker CR, Hamlett J, Pennington SR, Burrows F, Lundgren K, Lough R, Watson AJM, Jenkins John R. The topoisomerase II-Hsp90 complex: A new chemotherapeutic target? International Journal of Cancer. 2006;118:2685–2693. doi: 10.1002/ijc.21717. [DOI] [PubMed] [Google Scholar]
- 32.Yao Q, Weigel B, Kersey J. Synergism between Etoposide and 17-AAG in Leukemia Cells: Critical Roles for Hsp90, FLT3, Topoisomerase II, Chk1, and Rad51. Clin Cancer Res. 2007;13:1591–1600. doi: 10.1158/1078-0432.CCR-06-1750. [DOI] [PubMed] [Google Scholar]
- 33.Hawkins LM, Jayanthan AA, Naredran A. Effects of 17-Allylamino-17-Demethoxygeldanamycin (17-AAG) on Pediatric Acute Lymphoblastic Leukemia (ALL) with Respect to Bcr-Abl Status and Imatinib Mesylate Sensitivity. Pediatric Research. 2005;57:430–437. doi: 10.1203/01.PDR.0000153871.45184.19. [DOI] [PubMed] [Google Scholar]
- 34.Xiong MP, Yanez JA, Kwon GS, Davies NM, Laird Forrest M. A cremophor-free formulation for tanespimycin (17-AAG) using PEO-b-PDLLA micelles: Characterization and pharmacokinetics in rats. Journal of Pharmaceutical Sciences. 2008 doi: 10.1002/jps.21509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ali SM, Hoemann MZ, Aube Jeffrey, Georg GI, Mitscher LA, Jayasinghe LR. Butitaxel Analogues: Synthesis and Structure-Activity Relationships. Journal of Medicinal Chemistry. 1997;40:236–241. doi: 10.1021/jm960505t. [DOI] [PubMed] [Google Scholar]
- 36.Lee SC, Huh KM, Lee J, Cho YW, Galinsky RE, Park K. Hydrotropic Polymeric Micelles for Enhanced Paclitaxel Solubility: In Vitro and In Vivo Characterization. Biomacromolecules. 2007;8:202–208. doi: 10.1021/bm060307b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas S, Brightman F, Gill H, Lee S, Boris Pufong Simulation modelling of human intestinal absorption using Caco-2 permeability and kinetic solubility data for early drug discovery. Journal of Pharmaceutical Sciences. 2008;97:4557–4574. doi: 10.1002/jps.21305. [DOI] [PubMed] [Google Scholar]
- 38.Tian Z-Q, Liu Y, Zhang D, Wang Z, Dong SD, Carreras CW, Zhou Y, Rastelli G, Santi DV, Myles DC. Synthesis and biological activities of novel 17-aminogeldanamycin derivatives. Bioorganic & Medicinal Chemistry. 2004;12:5317–5329. doi: 10.1016/j.bmc.2004.07.053. [DOI] [PubMed] [Google Scholar]
- 39.Blanco E, Bey EA, Dong Y, Weinberg BD, Sutton DM, Boothman DA, Gao J. [beta]-Lapachone-containing PEG-PLA polymer micelles as novel nanotherapeutics against NQO1-overexpressing tumor cells. Journal of Controlled Release “Proceedings of the Thirteenth International Symposium on Recent Advances in Drug Delivery Systems”. 2007;122:365–374. doi: 10.1016/j.jconrel.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heald CR, Stolnik S, Kujawinski KS, De Matteis C, Garnett MC, Illum L, Davis SS, Purkiss SC, Barlow RJ, Gellert PR. Poly(lactic acid)-Poly(ethylene oxide) (PLA-PEG) Nanoparticles: NMR Studies of the Central Solidlike PLA Core and the Liquid PEG Corona. Langmuir. 2002;18:3669–3675. [Google Scholar]
- 41.Chen H, Kim S, He W, Wang H, Low PS, Park K, Cheng J-X. Fast Release of Lipophilic Agents from Circulating PEG-PDLLA Micelles Revealed by in Vivo Forster Resonance Energy Transfer Imaging. Langmuir. 2008;24:5213–5217. doi: 10.1021/la703570m. [DOI] [PubMed] [Google Scholar]