

Abstract
Arrestins rapidly bind phosphorylated activated forms of their cognate G protein-coupled receptors (GPCRs), thereby preventing G protein coupling and often switching the signaling to other pathways. Amphipathic α-helix I (residues 100–111) has been implicated in receptor binding, but the mechanism of its action is not yet determined. Here we show that several mutations in the helix itself and adjacent hydrophobic residues in the body of the N-domain reduce arrestin1 binding to phosphorylated light-activated rhodopsin (PRh*). On the background of phosphorylation-independent mutants that bind with high affinity to both P-Rh* and light-activated unphosphorylated rhodopsin (Rh*), these mutations reduce the stability of the arrestin complex with P-Rh*, but not with Rh*. Using site-directed spin labeling we found that the local structure around α helix I changes upon binding to rhodopsin. However, the intra-molecular distances between α-helix I and adjacent α-strand I, or the rest of the N-domain, measured using double electron-electron resonance, do not change, ruling out relocation of the helix due to receptor binding. Collectively, these data demonstrate that α-helix I plays an indirect role in receptor binding, likely keeping α-strand I, carrying several phosphate-binding residues, in a position favorable for its interaction with receptor-attached phosphates.
Arrestin proteins bind active phosphorylated G protein-coupled receptors (GPCRs), terminating G protein-mediated signaling (1). In vivo the binding occurs within ~80 milliseconds in photoreceptors (2) and within a minute in other cells (3). Thus, the on-rate is rapid and roughly proportional to the arrestin concentration in the cytoplasm, which is ~0.1–0.2 mM for arrestin11 in rod photoreceptor outer segments (4–6) and 30–200 nM for non-visual arrestin2 and arrestin3 in the brain (7,8). In contrast, the dissociation of the arrestin-receptor complex appears to be a relatively slow process. In photoreceptors, arrestin1 apparently stays in complex with light-activated phosphorhodopsin (P-Rh*) for a long time, at least until P-Rh* decays into phosphoopsin (9,10), ensuring high fidelity of the quenching mechanism. Receptor-bound non-visual arrestins serve as adapters linking GPCRs to the internalization machinery (11,12) and G protein-independent signaling pathways (13,14). These functions depend on preferential interaction of trafficking and signaling proteins with bound, rather than free, arrestin (1,14). Receptor binding-induced conformational changes in arrestin (15–17) apparently underlie enhanced affinity of this form for several interaction partners (1,14,18). Thus, the conformation of arrestin associated with the receptor and the overall shape of the complex determines its functional capabilities.
In the basal conformation of the three arrestin subtypes with determined crystal structures, α-helix I interacts with the C-tail, β-strand I and adjacent N-domain elements (19–22) (Fig. 1A). Hydrophobic residues mediating this interaction are conserved from C. elegans to mammals (23). Receptor-attached phosphates bind several positively charged residues in β-strand I (21,24,25), destabilizing this interaction and inducing the release of the arrestin C-tail (16). Based on the assumption that this interaction is completely destabilized by receptor binding, and on the amphipathic nature of α-helix I resembling helices that many proteins use as membrane anchors, it was proposed that α-helix I may “swing out” to the side of the arrestin molecule where all of the other receptor-binding elements are localized (16,24–30) and then directly bind to the membrane (19). Subsequent findings in cell-based assays that α-helix I in arrestin2 is not required for arrestin mobilization to the receptor, but appears to be involved in its post-endocytic trafficking (31), did not unambiguously resolve the issue, because receptor binding of wild type and mutant arrestins was not quantitatively compared.
Fig. 1. Alanine substitutions in α-helix I affect arrestin1 binding to different functional forms of rhodopsin.
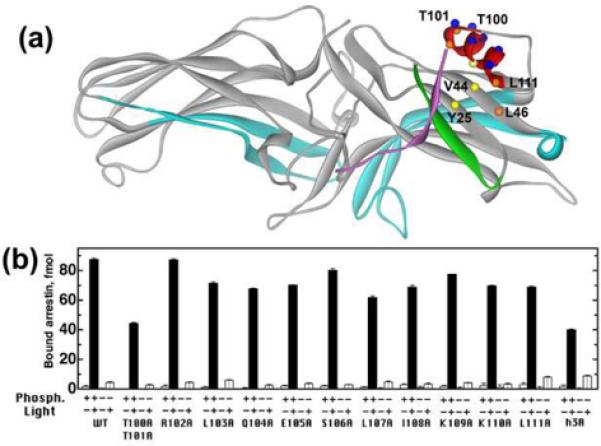
A. Molecular model of arrestin1 (1CF1, D-chain), color-coded, as follows: β-strand I, green; C-tail, magenta; receptor-binding elements implicated in receptor specificity, light blue. Residues mutated in this study are shown as spheres, color-coded, as follows: buried, yellow; partially exposed, orange; exposed, blue. B. Indicated functional forms of rhodopsin in native disc membranes (0.3 μg/assay) were incubated with 100 fmol of radiolabeled arrestin in 50 μl for 5 min at 37°C, and then bound arrestin was separated from free and quantified, as described in Methods. Means +/− SD of three experiments performed in duplicate are shown.
Using a combination of site-directed mutagenesis, direct measurements of receptor binding and the stability of the complex, site-directed spin labeling (SDSL), and intramolecular distance measurements in arrestin1, we demonstrate here that α-helix I does not substantially move relative to the N-domain upon receptor binding. Our data are consistent with an indirect role of helix I in arrestin interaction with phosphoreceptor, where it supports the position of β-strand I carrying multiple phosphate-binding residues in all arrestin proteins.
EXPERIMENTAL PROCEDURES
Materials
[γ-32P]ATP, [14C]leucine, and [3H]leucine were purchased from DuPont NEN. All restriction enzymes were purchased from New England Biolabs. Sepharose 2B and all other chemicals were from sources previously described (10,32). Rabbit reticulocyte lysate was purchased from Ambion, and SP6 RNA polymerase was prepared as previously described (33). 11-cis-retinal was generously supplied by Dr. R. K. Crouch and the National Eye Institute.
Site-directed mutagenesis
Bovine arrestin1 cDNA (34) was a gift from Dr. T. Shinohara. The plasmid pARR-VSP was constructed and modified as described earlier (29,35). This pGEM2-based plasmid encodes bovine wild type arrestin1 with an "idealized" 5'-untranslated region (33) under control of a SP6 promoter. A modified pARR-VSP construct (pARR-SC) (36) was used for mutagenesis. All mutations were introduced by PCR using an appropriate mutagenizing oligonucleotide as a forward primer and an oligonucleotide downstream from the far restriction site to be used for subcloning as a reverse primer. Resulting fragments of various lengths and an appropriate primer upstream of the near restriction site were then used as reverse and forward primers, respectively, for the second round of PCR. Sites Bam HI (codons 47–49) and SalI (codons 145–146) were used for mutations in α-helix I, whereas Eco RI (in the 5'UTR) and Bam HI were used for mutations at positions 25, 44 and 46. The sequences of all constructs were confirmed by dideoxynucleotide sequencing.
In vitro transcription, translation, and evaluation of mutants' stability
Plasmids were linearized with HindIII before in vitro transcription to produce mRNAs encoding full-length arrestin proteins. In vitro transcription and translation were performed as described (32,37). All arrestin proteins were labeled by incorporation of [3H]leucine and [14C]leucine with the specific activity of the mix 1.5–3 Ci/mmol, resulting in the specific activity of arrestin proteins within the range of 66–85 Ci/mmol (145–187 dpm/fmol). The translation of every mutant used in this study produced a single labeled protein band with the expected mobility on SDS-PAGE. The relative stability of all mutants used in this study (evaluated as described in (35)) exceeds 80% of wild type arrestin.
Rhodopsin preparations
Urea-treated rod outer segment membranes were prepared, phosphorylated with rhodopsin kinase and regenerated with 11-cis-retinal as described (10). The stoichiometry of phosphorylation for the rhodopsin preparations used in these studies was 3.8 mol phosphate/mol rhodopsin.
Direct binding assay
Arrestin1 binding to rhodopsin was performed as described (25,37). Briefly, in vitro translated tritiated arrestins (100 fmol) were incubated in 50 mM Tris-HCl, pH 7.5, 0.5 mM MgCl2, 1.5 mM dithiothreitol, 50 mM potassium acetate with 7.5 pmol (0.3 μg) of the various functional forms of rhodopsin in a final volume of 50 μl for 5 min at 37°C either in the dark or under room light. The samples were immediately cooled on ice and loaded under dim red light onto 2 ml Sepharose 2B columns equilibrated with 10 mM Tris-HCl, pH 7.5, 100 mM NaCl. Bound arrestin eluted with the rod outer segments in the void volume (between 0.5 – 1.1 ml). Nonspecific binding determined in the presence of 0.3 μg liposomes was subtracted.
Stability of the arrestin-P-Rh* complex was determined as described (10,38). Briefly, a 10-fold concentrated sample of rhodopsin and arrestin was incubated for 5 min at 37°C under room light, cooled on ice and diluted 10-fold with ice-cold 50 mM Tris-HCl, pH 7.5, 0.5 mM MgCl2, 1.5 mM dithiothreitol, 50 mM potassium acetate. The samples were kept on ice and at indicated times aliquots equivalent to the standard binding assay sample were loaded onto Sepharose 2B columns and processed as above.
Site-directed spin labeling EPR spectroscopy
Site-directed mutagenesis and arrestin expression and purification were performed as described (32). Cysteines were introduced in positions of interest in the background of fully functional cysteine-less arrestin1: ASA-CL (C63A, C128S, C143A) or VSV-CL (C63V, C128S, C143V) (16,39–41). Arrestin1 cysteine mutants in 50mM MOPS, 100mM NaCl, pH7.2 buffer were labeled with a ten-fold molar excess of 2,2,5,5-tetramethylpyrroline-3-yl-methanethiosulfonate spin label (MTSL; Toronto Research Chemicals) overnight at 4°C followed by removal of excess label by extensive dialysis as described (16,39). The product of the reaction is the nitroxide side chain designated R1. CW EPR spectroscopy was carried out at X-band on a Bruker EleXsys 500 fitted with a super high Q cavity. Samples (10 μL) were contained in a glass capillary and spectra were recorded at room temperature over 100 G at a microwave power of 10 mW, modulation amplitude of 1 G, and typically were signal averaged 25–36 times.
Accessibility measurements
Accessibility of the R1 nitroxide to collision with the paramagnetic reagents O2 and NiEDDA was determined using the power saturation method (42). Samples were typically 5 μL and contained in a gas-permeable TPX capillary. Data were collected on an X-band Varian E102 Century series spectrometer using a loop-gap resonator (Medical Advances) at room temperature under the flow of nitrogen in the presence and absence of 20 mM nickel ethylenediaminediacetate (NiEDDA) or air and analyzed using LabView software written by Dr. Christian Altenbach (UCLA) according to procedures previously described (42). The analysis provides the ratio of the rate constants for collision (exchange) of the nitroxide with the two reagents, i.e. R = kex (O2)/kex(NiEDDA).
Distance measurements and data analysis
DEER measurements were performed using a Bruker EleXsys 580 spectrometer equipped with a 2 mm split-ring resonator at 80K. Before insertion into the resonator, samples (200μM) containing 20% glycerol as a cryoprotectant were flash-frozen in sealed quartz capillaries (1.5 mm I.D. × 1.8 mm O.D.) using liquid nitrogen. Rod outer segment membranes (200μg) (prepared as described (40)), phosphorylated or nonphosphorylated, were pelleted at 16,000×g for 30min. The pellet was resuspended with spin labeled arrestins in the dark to final concentrations of 450μM and 200μM, respectively. For PRh* and Rh* binding, samples were exposed to 500 nm light for 5 min at room temperature before freezing. The four pulse DEER pulse sequence (π/2)ν1 – τ1 – (π)ν1 – t – (π)ν2 – τ1+τ2− t – (π)ν1 – τ2 – echo was used (43,44). The ELDOR pulse (ν2; 16 ns) was positioned at the centerfield maximum of the echo detected nitroxide spectrum, whereas the π/2 and π observe pulses (ν1; 8 and 16 ns) were positioned at the low field line of the spectrum (ν1-ν2 = 70 MHz). A phase cycle +x/−x was applied to the first π/2 observe pulse to remove baseline offsets. Echo decay data were analyzed using the DeerAnalysis2006 package (available at http://www.epr.ethz.ch/software) (45). For an excellent review of the DEER method directed at a general audience, the reader is referred to that by Jeschke (43). For arrestin in solution and bound to rhodopsin in membranes, the background echo decay was corrected using a homogeneous three-dimensional or two-dimensional spin distribution, respectively.
RESULTS
The effects of mutations on arrestin1 binding to rhodopsin
To assess the role of α-helix I in the arrestin-rhodopsin interaction, we performed alanine scanning mutagenesis of this region and tested the binding of the mutants to P-Rh* and to the three non-preferred functional forms: phosphorylated inactive (dark) (P-Rh), light activated unphosphorylated (Rh*), and dark unphosphorylated (Rh) rhodopsin. Each point mutation in the helix except R102A reduces arrestin1 binding to P-Rh* by 10–20% (Fig.1B). Alanine substitutions of exposed residues (Arg102, Ser106 and Lys109) produce smaller effects than mutations of the buried or partially buried hydrophobic residues (Leu103, Leu107, Ile108, Leu111), although the removal of the two exposed threonines (Thr100, 101) is as detrimental to the binding as the triple mutation eliminating leucines 103, 107, and 111 aligned on one side of the helix (referred to below as the h3A mutation)(Fig.1B). Some of these changes parallel expected reductions in protein stability due to the mutations. It is has been shown that Ala substitutions of bulky hydrophobic residues at buried sites produce internal cavities known to destabilize proteins (46). This likely explains the effects of Ala substitutions at 103, 107 and 111 (or partially buried site 108), while mutation of the solvent exposed Thr 101 disrupts an important H-bond with Asp 42. However, the effects of mutations of exposed residues that are not involved in intra-molecular interactions could reflect their role in receptor binding.
Interestingly, alanine substitutions of leucines 103, 107, and 111 moderately enhance arrestin1 binding to Rh*, consistent with a role for helix 1 in maintaining arrestin in its basal state through interactions with the N-domain and the C-tail, which are disrupted during arrestin activation (20,24). The destabilization due to the Ala mutations would increase flexibility and facilitate local structural changes that favor Rh* binding. Unlike other known “activating” mutations (29,35,38), neither alanine substitutions of individual leucines 103, 107, and 111 nor the h3A mutation significantly increase arrestin1 binding to dark P-Rh (Fig.1B).
In the basal state of arrestin1 helix I is localized on the side of the N-domain (Fig. 1A), anchored by its interactions with the arrestin C-tail, β-strand I, and adjacent nonpolar residues on the body of the N-domain, namely Val44, Leu46, and Tyr25. These residues face the hydrophobic surface of helix I (Leu103, Leu107 and Leu111) and form the hydrophobic core of the fold in this region. To test whether Val44, Leu46 or Tyr25 play a role in receptor interaction, we changed them to alanines individually and in combinations and then tested the binding of these mutants. Individual V44A and L46A mutations slightly reduce arrestin1 binding to P-Rh* (by about 10%), whereas the V44A/L46A double mutation reduces it by almost 30% (Fig.2A). None of the mutations affect arrestin1 selectivity, i.e., their binding to non-preferred forms of rhodopsin does not change. In contrast, Y25A does not appreciably affect P-Rh* binding, but enhances the binding to dark P-Rh and Rh*, consistent with its role in the stabilization of the three-element interaction inferred from the crystal structure (20). The triple mutation Y25A/V44A/L46A decreases arrestin1 binding to P-Rh* to the same extent as the V44A/L46A double mutation and simultaneously enhances its binding to dark P-Rh and Rh* to the same extent as the Y25A mutation alone (Fig.2A). Thus, the effects of Y25A and V44A/L46A (the reduction in selectivity and inhibition of P-Rh* binding, respectively) appear to be independent of one another.
Fig. 2. Alanine substitutions of the residues anchoring α-helix I to the N-domain differentially affect arrestin binding.

The binding of indicated mutants in the context of WT (A) and constitutively active arrestin1 (B, C) to different functional forms of rhodopsin was determined, as in Fig.1. Means +/− SD of three experiments performed in duplicate are shown. Abbreviations: 3A, F375A/V376A/F377A triple mutation; h3A, L103A/L107A/L111A triple mutation; 2A, V44A/L46A double mutation.
Several structurally distinct “constitutively active” mutants of arrestin1 that bind with high affinity not only to P-Rh*, but also to Rh* and dark P-Rh, have been described (17,24,29,30,35,36,38,47). To test whether alanine substitutions in the helix and the body of the N-domain affect the binding to the non-preferred forms of rhodopsin, we combined them with a potent activating triple mutation F375A/V376A/F377A (referred to below as 3A). We found that the V44A/L46A double mutation in combination with 3A reduces arrestin1 binding to dark P-Rh, P-Rh*, and Rh* to a similar extent (Fig.2B,C), suggesting that Val44 and Leu46 play a role in high-affinity binding of the 3A mutant to all three functional forms of rhodopsin. The Y25A mutation both in combination with 3A and V44A/L46A/3A marginally reduces arrestin1 binding to dark P-Rh and Rh*, but not to P-Rh* (Fig.2B), suggesting that Tyr25 (located on the same side of the N-domain as Val44 and Leu46) may also play a minor role in the interaction. Since both h3A and the V44A/L46A double mutation significantly reduce P-Rh* binding, we tested combinations of the two (Fig.2C). We found that detrimental effects of these two mutations are nearly additive in the context of WT arrestin1 and the “constitutively active” 3A mutant. In the latter case the binding to dark P-Rh and Rh* appears to be affected as much as the binding to P-Rh* (Fig.2C).
To gain further insight into the functional role of the three leucines in the helix and Val44 and Leu46 we tested whether alanine substitutions of these residues affect the dissociation rate of the arrestin-receptor complex. To this end, we performed the binding assay at ten times higher than standard concentrations of both arrestin and P-Rh*. The samples were then cooled on ice, diluted 10-fold with ice-cold binding buffer, and the amount of bound arrestin1 was determined at various time points in aliquots equivalent to a standard binding assay (Fig.3A). The half-life of the P-Rh* complex with WT arrestin1 was found to be 130 ± 19 min, whereas the V44A/L46A double mutation reduces it to 77 ± 14 min (p<0.001). The same mutation in combination with h3A reduces the half-life of the complex to a similar extent (from 171 ± 17 min for h3A to 120 ± 11 min for h3A/V44A/L46A, p<0.01). These experiments clearly suggest that Val44 and Leu46 play a role in the stabilization of the complex. In contrast to V44A/L46A, the h3A mutation decreases arrestin1 selectivity (i.e. enhances its binding to the non-preferred forms of rhodopsin, dark P-Rh* and Rh*). We have previously found that mutations with this effect increase the apparent half-lives in dissociation experiments, primarily due to poor discrimination between P-Rh* and phosphoopsin into which P-Rh* decays (38). However, even the most promiscuous arrestin1 mutants demonstrate very low affinity for the product of Rh* decay, unphosphorylated opsin (38). Therefore to gauge the role of the h3A mutation itself we performed similar experiments with Rh*. To obtain more meaningful data, in these experiments we used h3A, V44A/L46A, and the combination of these two mutations not on the background of WT arrestin1 (with relatively low affinity for Rh*) but on the background of the 3A mutation, which yields high Rh* binding. The 3A mutation not only “equalizes” all mutants with regard to Rh* binding, but it also confers similar reduced selectivity, thereby eliminating the “stabilizing” effect of the h3A mutation associated with the loss of selectivity. We found that the V44A/L46A double mutation does not significantly decrease the stability of the arrestin1-P-Rh* complex, whereas the h3A mutation reduces its half-life almost by half (Fig.3B). Importantly, neither mutation appreciably affects the half-life of the arrestin1-Rh* complex (Fig.3C), indicating that intact α-helix I stabilizes the complex only in the presence of rhodopsin-attached phosphates.
Fig. 3. Elimination of hydrophobic residues on the a-helix I/N-domain interface selectively reduces the stability of arrestin complex with P-Rh*.

Binding of the indicated mutants to P-Rh* (A, B) or Rh* (C) was measured after the complex was allowed to dissociate for the indicated time at 4°C. Means +/− SD of three experiments performed in duplicate are shown. Half-life of the complex (t1/2) was determined using a single-exponential decay equation in GraphPad Prizm. T1/2 for curves shown in panel A for wild type (WT), h3A, h3A-2A, and 2A arrestin was 130±19, 171±17, 120±11**, and 77±14*** min, respectively (**, p<0.01, as compared to h3A; ***, p<0.001, as compared to WT). T1/2 for curves shown in panel B for 3A, h3A, 2A–3A, and 2A-3A-h3A arrestins was 9.1±1.4, 4.9±1.1**, 8.7±1.4, and 4.5±1.1** h, respectively (**, p<0.01, as compared to 3A and 2A–3A). T1/2 for curves shown in panel C for 3A, h3A, 2A–3A, and 2A-3A-h3A arrestins was 73±6, 77±9, 68±7, and 72±7 min, respectively (there were no statistically significant differences). The dissociation of mutants with reduced selectivity (B, C) was measured in the presence of 1 mM NH2OH, which chemically reacts with all-trans-retinal in light-activated rhodopsin, thereby preventing its re-binding to opsin. Abbreviations: 3A, F375A/V376A/F377A triple mutation; h3A, L103A/L107A/L111A triple mutation; 2A, V44A/L46A double mutation.
Structural changes involving α-helix I upon binding to P-Rh*
Binding and dissociation data suggest that α-helix I is directly or indirectly involved in rhodopsin binding. However, the helix is localized on the “side” of the molecule, whereas all earlier identified receptor-binding elements were mapped to the concave surfaces of the two arrestin domains (16,25,27,28) (Fig.1A). One model reconciling these findings is that the helix (attached to the body of the N-domain via long multi-residue “connectors”) swings out of its basal position and reaches the receptor-facing side of the molecule, perhaps interacting with the receptor directly or with the membrane surface.
To explore these possibilities SDSL was used to monitor changes in the environment of both the polar and nonpolar surfaces of the amphipathic α-helix I upon binding to P-Rh*. To this end we replaced Leu103, Glu105, Ile108 and Leu111 one at a time with a nitroxide side chain (R1, Fig.4, inset). The locations of these residues in α-helix I are shown in Fig. 4A. The EPR spectra of the spin labeled arrestin1 mutants in solution (black traces) and bound to P-Rh* (red traces) are shown in Fig.4B; spectra for Leu103R1 and Leu111R1 were previously published (16) and are reproduced here for reference. Consistent with its buried location, the spectrum of Leu103R1 in solution reflects a relatively immobilized state of the nitroxide. Glu105R1 and Ile108R1 have similar EPR spectra arising from a combination of a relatively mobile and less mobile component, indicative of local tertiary interactions with neighboring residues and consistent with their locations on the outside of α-helix I (Fig. 4). The spectrum of Leu111R1 is remarkable; the narrow center linewidth indicates fast motion while the well-resolved hyperfine extrema suggest a high ordering. Indeed, the spectrum can be reasonably well simulated with a fast z-axis anisotropic motion of high order (red trace in Fig. 1S) but a minor second component may be present. Although the native leucine residue is at a relatively buried location (side chain solvent accessibility ≈ 24%), it is at the end of the helix and an R1 side chain modeled at this site (with the same Cα-Cβ dihedral angle as the native residue) projects into the solvent but in a constrained environment (Fig. 1S), consistent with the motion implied by the spectrum. Upon arrestin binding to P-Rh*, the motion of R1 at each site becomes more restricted (red traces, Fig. 4B), indicating that either there is a structural rearrangement of this local area upon receptor binding or some population comes into direct contact with P-Rh*, perhaps with the highly flexible C-terminus of rhodopsin that is believed to displace the C-tail of arrestin and could therefore lie in proximity to α-helix I.
Fig. 4. The effects of arrestin1 binding to P-Rh* on the mobility of spin label in α-helix I.

A. Arrestin1 structure (1CF1, D-chain) with elements color-coded as follows: α-helix I, red; β-strand I, green; C-tail, magenta; receptor-binding elements, light blue. Inset: The structure of the R1 spin label side chain. B. For each spin-labeled arrestin, normalized EPR spectra of R1 at indicated positions in the absence (black traces) or presence of P-Rh* (red traces) are compared.
In addition to R1 mobility, the relative accessibility of the nitroxide to collision with the paramagnetic species O2 (non-polar) and NiEDDA (polar) serves as a sensitive measure of the local environment. The relative accessibility, R = kex(O2)/kex(NiEDDA), where kex is the rate constant for collisional encounters, is readily determined using power saturation (see Methods). For R1 completely exposed to solvent, R ≈ 6 – 8 due to the larger diffusion coefficient of O2 relative to NiEDDA. As R1 becomes increasingly buried in a protein core, R increases due to the differential exclusion of the larger NiEDDA from the crevices between side chains (48); values of R as large as ≈ 60 have been observed at completely buried sites (42). Similarly large values of R are observed for R1 exposed to the fluid interior of the bilayer due to the higher solubility of the non-polar O2 in the bilayer relative to NiEDDA (49). Relative accessibilities were determined for Leu103R1 and Leu111R1 in α-helix I for arrestin1 in solution and bound to P-Rh*. In solution, the R value of 7.4 for Leu111R1 confirms that the nitroxide is completely solvent exposed as surmised from the nitroxide mobility and modeling. Leu103R1 is at a buried site in the structure, consistent with the larger R of 25.5. Upon binding to P-Rh*, Leu103R1 becomes more solvent exposed (R = 11.3) while Leu111R1 changes very little (R = 6.3). These results complement the mobility data and collectively they suggest relatively small structural rearrangements involving α-helix I. Had the hydrophobic face of α-helix I become exposed to the fluid non-polar interior of a membrane upon binding to P-Rh*, the R value for Leu103R1 would have increased rather than decreased.
To directly test whether α-helix I actually moves out of its basal position upon binding to P-Rh*, we constructed doubly spin labeled mutants to measure distances between α-helix I and β-strand I (16/108, 12/108) and between α-helix I and the center of the receptor-binding cavity of the N-domain (111/173). In addition, distance measurements were made between α-helix I and the arrestin1 C-tail (103/376) (Fig. 5A,B). In this latter case, distance changes are anticipated based on our earlier results that showed a large scale motion of the C-tail upon binding to P-Rh* (16). Distance measurements were made using DEER spectroscopy (see Methods). Figure 5C shows DEER data for the double mutants investigated. For each double mutant the corrected echo amplitude as a function of time (the dipolar evolution, left column), the dipolar spectrum (the Fourier transform of the dipolar evolution, center column), and the interspin distance distribution derived from analysis of the dipolar evolution and dipolar spectrum (right column) are shown.
Fig. 5. α-helix I does not appreciably move upon arrestin1 binding to P-Rh*.

A, B. Arrestin1 structure (1CF1, D-chain) with elements color-coded as in Fig. 4. R1 side chain is modeled at the indicated positions. C. The inter-nitroxide distances between pairs of spin labels within the arrestin1 molecule in solution (black traces) and in complex with P-Rh* (green traces) were measured by DEER spectroscopy as described in Methods. The DEER experiment measures the magnetic dipolar interaction between nitroxides as a modulation of an electron spin echo amplitude; the frequency of modulation is directly proportional to r−3, where r is the inter-spin distance. The primary data is the echo amplitude as a function of time. The dipolar evolution function (left column) is obtained after subtraction of an exponentially decaying background due to spins with randomly distributed inter-spin distances (e.g., interactions between adjacent molecules). In each case the best fits are shown as red traces. Fourier transformation of the dipolar evolution function gives the dipolar spectrum (central column); the splitting between the extrema is proportional to r−3. The data for free (black lines) and P-Rh*-bound arrestin1 (green lines) are overlayed. The gray bars highlight the region of the spectrum corresponding to an electron spin echo envelope modulation (ESEEM) signal, reflecting electron/proton interactions and not related to the interspin distance. This is quite pronounced in the 16R1/381R1 spectrum. Simultaneous fitting of the dipolar evolution function and dipolar spectrum yields the experimental inter-spin distance distribution (in angstroms, right column). The distributions were obtained using Tikhonov regularization.
The location of α-helix I relative to nearby β-strand I is revealed by the distances between Ile12R1/Ile108R1 and Ile16R1/Ile108R1. Strong dipolar interaction is apparent in each case, as judged from the rapid decay of the dipolar evolution (Fig. 5C). The interspin distance distributions are peaked at ≈18Å and 26Å, respectively; for Ile16R1/Ile108R1 there is second population at ≈22Å. These distances agree well with values determined from models of R1 in the crystal structure of arrestin 1 (20Å and 26Å, respectively; Fig. 5A). The second population in Ile16R1/Ile108R1 could arise from rotamer populations of R1. On binding to P-Rh* there is essentially no change in distance for Ile12R1/Ile108R1, but a broadening of the distribution and a small shift in population to shorter distances for Ile16R1/Ile108R1 which may be related to the flexibility of the sequence in which 16R1 resides (16). Thus, rhodopsin binding does not appear to significantly change the position of α-helix I relative to β-strand I. While α-helix I does not move relative to β-strand I, this finding does not exclude the possibility of both elements moving together. To test whether this is the case, the distance between Leu111R1 in α-helix I and Leu173R1 in the N domain was determined by DEER (Fig. 5C). For arrestin1 in solution the interspin distance distribution was remarkably narrow and centered at 28Å, close to the value estimated from modeling (27Å, Fig. 5A). The narrow width can be attributed to the high order of Leu111R1 noted above and the fact that Leu173R1 is highly immobilized (16). The binding to P-Rh* does not change the distance distribution. Collectively, these data demonstrate that α-helix I does not significantly shift out of its basal position upon P-Rh* binding.
In free arrestin1, a strong magnetic dipolar interaction between Leu103R1 in α-helix I and Val376R1 in the C-tail, and between Ile16R1 in β-strand I and Ala381 in the C-tail is evident, as indicated by the extremely sharp drop-off of the dipolar evolution and the broad dipolar spectrum (Fig. 5C). The distance distributions are centered around ≈19Å, consistent with the inter-nitroxide distance for R1 residues modeled in the crystal structure (20) (19Å and 20Å respectively, Fig. 5). Remarkably, binding to P-Rh* leads to a dramatic increase in the widths of the distributions with contributions that extend all the way from 20 to 50Å. It should be noted that the collection time for the DEER data (2–2.5 μs) in these measurements is insufficient to accurately determine the width of distances beyond about 45Å, but the major point is that there are increases in the populations of spins at distances well beyond the 20Å seen in the free arrestin1. Since data presented above establish that neither Ile16R1 nor Leu103R1 move upon P-Rh* binding, it is the C-tail that is moving. These data confirm and extend our earlier CW EPR data on the inter-spin distance of Ile16R1/Ala381R1 that showed an increase in the distance, beyond the detection limit of ≈20Å, upon binding to P-Rh * (16).
DISCUSSION
Arrestins are a relatively small protein family (only four subtypes in mammals), but appear to be almost as ubiquitous as G protein-coupled receptors, being present in virtually every eukaryotic cell (23). Because of the high biological importance of arrestin-receptor interactions, receptor-binding elements of arrestin proteins were mapped by many groups using a wide variety of methods (16,25–30,38,50–52). All of these elements are localized on the same side of the molecule: the concave surfaces of the two arrestin domains (Fig.1A) (1,18). There is one exception: α-helix I and/or the elements connecting it to the body of the N-domain were implicated in arrestin1 binding to rhodopsin (53) and arrestin2 and arrestin3 mediated receptor trafficking (31). The amphipathic nature of this helix in all arrestin proteins (19–22) suggested the idea that it may “anchor” arrestin to the membrane, thereby stabilizing the arrestin complex with its cognate receptor (19). An alternative explanation is that the helix affects receptor binding indirectly, through its interactions with other arrestin elements.
Here we quantitatively evaluated the effects of mutations in the α-helix I on arrestin1 binding to different functional forms of rhodopsin, on the half-life of the arrestin1-rhodopsin complex, and investigated corresponding structural changes using SDSL and distance measurements. Systematic alanine scanning mutagenesis of α-helix I revealed that mutations in multiple positions significantly reduce wild type arrestin1 binding to P-Rh* and the binding of constitutively active mutants to P-Rh*, dark P-Rh, and Rh* (Figs. 1, 2). Alanine substitutions of several bulky hydrophobic residues that “anchor” the helix to the body of the N-domain are also detrimental to receptor binding (Fig.2). Mutations of this type often create cavities or packing defects, producing large changes in the thermodynamic stability of proteins (46). Effects of these mutations on protein-protein interactions may be indirect, mediated by the global destabilization of the affected partner. However, we found that alanine substitutions reduce the stability of the arrestin complex with P-Rh*, but not with Rh* (Fig. 3), suggesting that α-helix I is primarily involved in the binding to the phosphorylated receptor.
Site-directed spin labeling EPR spectroscopy demonstrated that the mobility of the spin label in several positions in the helix changes upon P-Rh* binding (Fig.4), as does solvent accessibility, demonstrating changes in local structure. In the basal arrestin conformation, α-helix I interacts with β-strand I, the body of the N-domain, and the arrestin C-tail (19–22). Functional (24,35,37,38,47) and structural data (16) indicate that this interaction is disrupted upon arrestin binding to the receptor. Therefore, we tested whether upon receptor binding α-helix I swings out of its position and moves to the side of arrestin containing the rest of the known receptor-binding elements (Fig. 1A). To this end, we employed DEER spectroscopy to measure inter-spin distances between nitroxide side chains in doubly labeled free and receptor-bound arrestin1. No appreciable changes in distances were observed between different positions in α-helix I and β-strand I or between α-helix I and the body of the N-domain (Fig. 5). These data clearly indicate that in receptor-bound arrestin α-helix I remains very close to its basal position relative to two different parts of the N-domain, ruling out the possibility of a large movement. On the other hand, the distance between α-helix I and the arrestin C-tail is increased to the same extent as the distance between β-strand I and the C-tail (Fig. 5). These changes apparently reflect solely the movement of the C-tail due to interaction with P-Rh* (16).
Despite the lack of movement of α-helix I, both the mutagenesis and SDSL data indicate an involvement of the helix in receptor binding. Simultaneous contacts of the receptor with α-helix I and the rest of the receptor-binding elements in the arrestin molecule cannot be rationalized in the context of the traditional model of the complex where the concave sides of the two arrestin domains are envisioned facing the plane of the membrane (1,14,18). A rhodopsin dimer (54) with α-helix I binding one receptor and the other elements interacting with a second receptor cannot solve the problem, either: we have recently established that both in vitro with only two purified proteins present and in rod photoreceptors of living mice arrestin1 “saturates” rhodopsin at 1:1 ratio (5). Since only arrestin1 monomer can bind rhodopsin (40) because “sister” subunits shield large portions of receptor-binding surface in dimers and tetramers (41), this ratio cannot be accounted for by an interaction of arrestin1 dimer with rhodopsin dimer. Therefore, these data establish that each rhodopsin molecule binds its own arrestin (5). In this context, the simplest model compatible with our results is that α-helix I supports the optimal positioning of three identified phosphate-binding residues on β-strand I, lysines 14 and 15 (24) and arginine 18 (21), for the interaction with receptor-attached phosphates. This scenario readily explains our finding that the same mutations in α-helix I that destabilize the arrestin1 complex with P-Rh* have no appreciable effect on the arrestin1-Rh* complex (Fig. 3). Remarkable structural conservation in the arrestin family (19–22) and the similarity of the mechanisms that regulate receptor binding (21,35,36,55–57) make it very likely that this model is applicable to the interaction of other arrestins with their cognate receptors. Recent studies show that retromer subunits Vps26A and Vps26B have an “arrestin fold” (58,59), although the interactions of these subunits with known partners are not regulated by phosphorylation. Despite remarkable structural similarity, arrestin elements that play key role in phosphate binding, such as the cluster of positive charges in the β-strand I, the “polar core” between the two domains, and the “detachable” C-tail, are missing in Vps26 subunits (58,59). Interestingly, Vps26 subunits also do not have an equivalent of the arrestin α-helix I, suggesting that it plays a specific role in arrestin interactions with phosphorylated binding partners.
Receptor interacts with an extensive arrestin surface, engaging numerous residues (16,25,27,28,52). In view of this, the magnitude of the impact of helix mutations on arrestin1 binding (Figs. 1,2) and the stability of its complex with P-Rh* (Fig.3) is remarkable. If these effects are indirect, our results suggest that the exact positioning of the β-strand I plays an unexpectedly important role in high-affinity receptor binding. An alternative explanation could be a model where arrestin molecules come to the cytoplasmic tip of the receptor at a sharp angle, allowing the concave surfaces of both arrestin domains and its α-helix I to make simultaneous contacts with the receptor. However, to be fully compatible with the data this explanation requires an additional assumption, that arrestin complexes with P-Rh* and Rh* are structurally different. This model implies that the interaction of rhodopsin-attached phosphates with the three positive charges in β-strand I orient arrestin in such a way as to maximize rhodopsin contacts with the helix. When the phosphates are absent (i.e., in Rh*), arrestin orientation in the complex is different, reducing the contribution of α-helix I to the interaction. Although this model is more speculative, several recent reports indicate that arrestin can form more than one type of complex with the same receptor. In particular, arrestin1 complexes with “normal” P-Rh* carrying 3–4 phosphates and heavily phosphorylated rhodopsin are different (10). Several studies showed that the functional capabilities of the complex of non-visual arrestins with their cognate receptors is regulated by differential receptor phosphorylation (60–62). Recent findings that many non-receptor partners, such as protein kinases c-Raf-1, MEK1, ERK2, ASK1, MKK4, JNK3, and ubiquitin ligase Mdm2, engage both arrestin domains (63,64), which are believed to move relative to each other upon receptor binding (17), provides a plausible structural explanation of different functional capabilities of arrestin-receptor complexes with distinct shapes. Although the formation of an arrestin1-rhodopsin complex of any shape is likely sufficient to preclude further G protein activation by simple steric exclusion (65), recent unexpected findings that visual arrestins 1 and 4 interact with multiple non-receptor signaling proteins (64,66–68), similar to their non-visual cousins (13), suggest that the particular shape of the arrestin1-rhodopsin complex may affect its additional functional capabilities with important biological consequences in photoreceptor cells.
Supplementary Material
A. Experimental (black trace) and simulated (red trace) spectra of R1 in position 111. B. Space-filling model of arrestin1 (1CF1, D-chain) with 111R1 shown as a stick model to illustrate its projection into the solvent. The C-tail and β-strand I are shown in magenta and dark green, respectively.
{kind=link}
Acknowledgements
The authors are grateful to Drs. T. Shinohara and Rosalie K. Crouch for bovine rod arrestin cDNA and 11-cis-retinal, respectively, and to M.-G. Velez for the construction of α-helix mutants. This study was supported by NIH grants EY11500 (VVG), EY05216 and the Jules Stein Professorship Endowment (WLH); GM70642 (CSK); SMH was supported by Training Grant GM07628.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
We use systematic names of arrestin proteins: arrestin1 (historically called S-antigen, 48 kDa protein, and visual or rod arrestin), arrestin2 (β-arrestin or β-arrestin1), arrestin3 (β-arrestin2), and arrestin4 (cone or X-arrestin).
References
- 1.Gurevich VV, Gurevich EV. Pharm Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krispel CM, Chen D, Melling N, Chen YJ, Martemyanov KA, Quillinan N, Arshavsky VY, Wensel TG, Chen CK, Burns ME. Neuron. 2006;51:409–416. doi: 10.1016/j.neuron.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 3.Barak LS, Ferguson SS, Zhang J, Caron MG. J Biol Chem. 1997;272:27497–27500. doi: 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- 4.Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanson SM, Gurevich EV, Vishnivetskiy SA, Ahmed MR, Song X, Gurevich VV. Proc Nat Acad Sci USA. 2007;104:3125–3128. doi: 10.1073/pnas.0610886104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gurevich VV, Hanson SM, Gurevich EV, Vishnivetskiy SA. How Rod Arrestin Achieved Perfection: Regulation of its Availability and Binding Selectivity. In: Fliesler SJ, Kisselev O, editors. Signal transduction in the retina. CRC Press; Boca Raton, FL: 2007. [Google Scholar]
- 7.Gurevich EV, Benovic JL, Gurevich VV. Neuroscience. 2002;109:421–436. doi: 10.1016/s0306-4522(01)00511-5. [DOI] [PubMed] [Google Scholar]
- 8.Gurevich EV, Benovic JL, Gurevich VV. J Neurochem. 2004;91:1404–1416. doi: 10.1111/j.1471-4159.2004.02830.x. [DOI] [PubMed] [Google Scholar]
- 9.Palczewski K, McDowell JH, Jakes S, Ingebritsen TS, Hargrave PA. J Biol Chem. 1989;264:15770–15773. [PubMed] [Google Scholar]
- 10.Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, Gurevich VV. J Biol Chem. 2007;282:32075–32083. doi: 10.1074/jbc.M706057200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodman OB, Jr., Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Nature. 1996;383(6599):447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 12.Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson s. S. G., Caron MG, Barak LS. Proc Nat Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lefkowitz RJ, Shenoy SK. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 14.Gurevich VV, Gurevich EV. Structure. 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 15.Schleicher A, Kuhn H, Hofmann KP. Biochemistry. 1989;28:1770–1775. doi: 10.1021/bi00430a052. [DOI] [PubMed] [Google Scholar]
- 16.Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Proc Natl Acad Sci U S A. 2006;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vishnivetskiy SA, Hirsch JA, Velez M-G, Gurevich YV, Gurevich VV. J. Biol. Chem. 2002;277(46):43961–43968. doi: 10.1074/jbc.M206951200. [DOI] [PubMed] [Google Scholar]
- 18.Gurevich VV, Gurevich EV. Trends Pharmacol Sci. 2004;25:59–112. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Structure. 2001;9(9):869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 20.Hirsch JA, Schubert C, Gurevich VV, Sigler PB. Cell. 1999;97(2):257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 21.Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 22.Milano SK, Pace HC, Kim YM, Brenner C, Benovic JL. Biochemistry. 2002;41(10):3321–3328. doi: 10.1021/bi015905j. [DOI] [PubMed] [Google Scholar]
- 23.Gurevich EV, Gurevich VV. Genome Biol. 2006;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vishnivetskiy SA, Schubert C, Climaco GC, Gurevich YV, Velez M-G, Gurevich VV. J. Biol. Chem. 2000;275(52):41049–41057. doi: 10.1074/jbc.M007159200. [DOI] [PubMed] [Google Scholar]
- 25.Hanson SM, Gurevich VV. J Biol Chem. 2006;281:3458–3462. doi: 10.1074/jbc.M512148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM, Benovic JL. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- 27.Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. J. Biol. Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- 28.Pulvermuller A, Schroder K, Fischer T, Hofmann KP. J Biol Chem. 2000;275:37679–37685. doi: 10.1074/jbc.M006776200. [DOI] [PubMed] [Google Scholar]
- 29.Gurevich VV, Benovic JL. J. Biol. Chem. 1995;270(11):6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- 30.Gurevich VV, Benovic JL. Mol Pharmacol. 1997;51:161–169. doi: 10.1124/mol.51.1.161. [DOI] [PubMed] [Google Scholar]
- 31.Dinh DT, Qian H, Seeber R, Lim E, Pfleger K, Eidne KA, Thomas WG. Mol Pharmacol. 2005;67:375–382. doi: 10.1124/mol.104.004721. [DOI] [PubMed] [Google Scholar]
- 32.Gurevich VV, Benovic JL. Methods Enzymol. 2000;315:422–437. doi: 10.1016/s0076-6879(00)15859-8. [DOI] [PubMed] [Google Scholar]
- 33.Gurevich VV. Methods Enzymol. 1996;275:382–397. doi: 10.1016/s0076-6879(96)75023-1. [DOI] [PubMed] [Google Scholar]
- 34.Shinohara T, Dietzschold B, Craft CM, Wistow G, Early JJ, Donoso LA, Horwitz J, Tao R. Proc Nat Acad Sci USA. 1987;84:6975–6979. doi: 10.1073/pnas.84.20.6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gurevich VV. J Biol Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- 36.Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. J. Biol. Chem. 1999;274:11451–11454. doi: 10.1074/jbc.274.17.11451. [DOI] [PubMed] [Google Scholar]
- 37.Gurevich VV, Benovic JL. J Biol Chem. 1992;267:21919–21923. [PubMed] [Google Scholar]
- 38.Gurevich VV, Benovic JL. J. Biol. Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 39.Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. J Biol Chem. 2006;281:9765–9772. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanson SM, Van Eps N, Francis DJ, Altenbach C, Vishnivetskiy SA, Klug CS, Hubbell WL, Gurevich VV. EMBO J. 2007;26:1726–1736. doi: 10.1038/sj.emboj.7601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanson SM, Dawson ES, Francis DJ, Van Eps N, Klug CS, Hubbell WL, Meiler J, Gurevich VV. Structure. 2008;16:924–934. doi: 10.1016/j.str.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altenbach C, Froncisz W, Hemker R, McHaourab H, Hubbell WL. Biophys J. 2005;89:2103–2112. doi: 10.1529/biophysj.105.059063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeschke G. Chemphyschem. 2002;3:927–932. doi: 10.1002/1439-7641(20021115)3:11<927::AID-CPHC927>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 44.Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. J Magn Reson. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- 45.Jeschke G, Chechik V, Ionita P, Godt A, Zimmermann H, Banham J, Timmel CR, Hilger D, Jung H. Appl Magn Reson. 2006;30:473–498. [Google Scholar]
- 46.Xu J, Baase WA, Baldwin E, Matthews BW. Protein Sci. 1998;7:158–177. doi: 10.1002/pro.5560070117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song X, Vishnivetskiy SA, Gross OP, Emelianoff K, Mendez A, Chen J, Gurevich EV, Burns ME, Gurevich VV. Curr Biol. 2009;19:700–705. doi: 10.1016/j.cub.2009.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Isas JM, Langen R, Haigler HT, Hubbell WL. Biochemistry. 2002;41:1464–1473. doi: 10.1021/bi011856z. [DOI] [PubMed] [Google Scholar]
- 49.Altenbach C, Greenhalgh DA, Khorana HG, Hubbell WL. Proc Natl Acad Sci U S A. 1994;91:1667–1671. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohguro H, Palczewski K, Walsh KA, Johnson RS. Protein Sci. 1994;3:2428–2434. doi: 10.1002/pro.5560031226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dinculescu A, McDowell JH, Amici SA, Dugger DR, Richards N, Hargrave PA, Smith WC. J Biol Chem. 2002;277:11703–11708. doi: 10.1074/jbc.M111833200. [DOI] [PubMed] [Google Scholar]
- 52.Feuerstein SE, Pulvermüller A, Hartmann R, Granzin J, Stoldt M, Henklein P, Ernst OP, Heck M, Willbold D, Koenig BW. Biochemistry. 2009 doi: 10.1021/bi900544p. in press. [DOI] [PubMed] [Google Scholar]
- 53.Smith WC, McDowell JH, Dugger DR, Miller R, Arendt A, Popp MP, Hargrave PA. Biochemistry. 1999;38:2752–2761. doi: 10.1021/bi982643l. [DOI] [PubMed] [Google Scholar]
- 54.Fotiadis D, Jastrzebska B, Philippsen A, Muller DJ, Palczewski K, Engel A. Curr Opin Struct Biol. 2006;16:252–259. doi: 10.1016/j.sbi.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 55.Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. J. Biol. Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- 56.Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. J. Biol. Chem. 2002;277(11):9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 57.Smith WC, Gurevich EV, Dugger DR, Shelamer CL, Vishnivetskiy SA, McDowell H, Gurevich VV. Invest. Ophthalmol. Vis. Sci. 2000;41:2445–2455. [PubMed] [Google Scholar]
- 58.Shi H, Rojas R, Bonifacino JS, Hurley JH. Nat Struct Mol Biol. 2006;13:540–548. doi: 10.1038/nsmb1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collins BM, Norwood SJ, Kerr MC, Mahony D, Seaman MN, Teasdale RD, Owen DJ. Traffic. 2008;9:366–379. doi: 10.1111/j.1600-0854.2007.00688.x. [DOI] [PubMed] [Google Scholar]
- 60.Key TA, Foutz TD, Gurevich VV, Sklar LA, Prossnitz ER. J Biol Chem. 2003;278:4041–4047. doi: 10.1074/jbc.M204687200. [DOI] [PubMed] [Google Scholar]
- 61.Ren XR, Reiter E, Ahn S, Kim J, Chen W, Lefkowitz RJ. Proc Nat Acad Sci USA. 2005;102:1448–1453. doi: 10.1073/pnas.0409534102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, Lefkowitz RJ. Proc Nat Acad Sci USA. 2005;102:1442–1447. doi: 10.1073/pnas.0409532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song X, Coffa S, Fu H, Gurevich VV. J Biol Chem. 2009;284:685–695. doi: 10.1074/jbc.M806124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song X, Gurevich EV, Gurevich VV. J Neurochem. 2007;103:1053–1062. doi: 10.1111/j.1471-4159.2007.04842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krupnick JG, Gurevich VV, Benovic JL. J. Biol. Chem. 1997;272:18125–18131. doi: 10.1074/jbc.272.29.18125. [DOI] [PubMed] [Google Scholar]
- 66.Wu N, Hanson SM, Francis DJ, Vishnivetskiy SA, Thibonnier M, Klug CS, Shoham M, Gurevich VV. J Mol Biol. 2006;364:955–963. doi: 10.1016/j.jmb.2006.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Song X, Raman D, Gurevich EV, Vishnivetskiy SA, Gurevich VV. J Biol Chem. 2006;281:21491–21499. doi: 10.1074/jbc.M603659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hanson SM, Cleghorn WM, Francis DJ, Vishnivetskiy SA, Raman D, Song S, Nair KS, Slepak VZ, Klug CS, Gurevich VV. J Mol Biol. 2007;368:375–387. doi: 10.1016/j.jmb.2007.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Experimental (black trace) and simulated (red trace) spectra of R1 in position 111. B. Space-filling model of arrestin1 (1CF1, D-chain) with 111R1 shown as a stick model to illustrate its projection into the solvent. The C-tail and β-strand I are shown in magenta and dark green, respectively.