

Abstract
TIR domain-containing adaptor protein (TRIF) is an adaptor protein in Toll-like receptor (TLR) signaling pathways. Activation of TRIF leads to the activation of Interferon regulatory factor 3 (IRF3) and nuclear factor kappa B (NF-κB). While studies have shown that TLRs are implicated in cerebral ischemia/reperfusion (I/R) injury and in neuroprotection against ischemia afforded by preconditioning, little is known about TRIF’ role in the pathological process following cerebral I/R. The present study investigated the role that TRIF may play in acute cerebral I/R injury. In a mouse model of cerebral I/R induced by transient middle cerebral artery occlusion, we examined the activation of NFκB and IRF-3 signaling in ischemic cerebral tissue using ELISA and Western blots. Neurological function and cerebral infarct size were also evaluated 24 hours after cerebral I/R. NF-κB activity and phosphorylation of the inhibitor of kappa B (IκBα) increased in ischemic brains, but IRF3, inhibitor of κB kinase complex-ε (IKKε), and TANK-binding kinase1 (TBK1) were not activated after cerebral I/R in wild-type (WT) mice. Interestingly, TRIF deficit did not inhibit NF-κB activity or p-IκBα induced by cerebral I/R. Moreover, although cerebral I/R induced neurological and functional impairments and brain infarction in WT mice, the deficits were not improved and brain infarct size was not reduced in TRIF knockout mice compared to WT mice. Our results demonstrate that the TRIF-dependent signaling pathway is not required for the activation of NF-κB signaling and brain injury after acute cerebral I/R.
Keywords: TLRs, TRIF, cerebral ischemia/reperfusion, mouse
Introduction
Cerebral ischemia is the third leading cause of death and remains the leading cause of long-term disability in the United States. Current evidence suggests that the inflammatory responses to injury initiated by the innate immune system contribute to the pathogenesis of cerebral ischemia/reperfusion (I/R) [1, 2]. Many reports indicate that Toll-like receptors (TLRs) are implicated in the pathological processes of cerebral I/R injury, as well as in neuroprotection against ischemia afforded by preconditioning [3–7].
TLRs are a family of signaling molecules which play a critical role in mediating immunity and inflammation [8–10]. At present, at least ten human TLRs have been identified [11, 12] and eleven TLRs have been found in mice [13]. TLR signaling is mediated by cytosolic adaptor molecules that bind to the intracellular domain of TLRs. One of the intracellular adaptors, myeloid differentiation primary-response protein-88 (MyD88), recruits serial downstream kinases and then phosphorylates the inhibitor of κB (IκB), resulting in nuclear translocation of NFκB and the transcription of genes associated with innate immune and inflammation (Fig. 1). This MyD88-dependent pathway is shared by all the TLRs with the exception of TLR3 [14]. TLR signaling is also mediated through another adaptor, TIR domain-containing adaptor protein (TRIF), which activates TANK-binding kinase1 (TBK1) and inhibitor of κB kinase complex-ε (IKKε), leads to the activation of IFN regulatory factor 3 (IRF3), and thereby activates interferon-β (IFN-β) and IFN-β-inducible genes (Fig. 1). This TRIF-dependent pathway is unique to TLR3 and TLR4 signaling [15–17]. Thus, TLR4 is the only TLR that mediates both MyD88-dependent and TRIF-dependent pathways. Of all the TLRs, TLR4 has been the most extensively investigated and implicated in ischemic brain injury. Transcription and expression of TLR4 increased and TLR4-mediated NF-κB signaling was activated in ischemic brain, which induced in situ inflammatory responses and contributed to brain damage after cerebral I/R [4, 5]. Modulation of TLR4 inhibited the activation of NF-κB, decreased inflammatory responses and attenuated brain damage induced by cerebral I/R [4, 5, 7]. In contrast, Marsh et al. reported that activation of TLR4 by a specific ligand, Lipopolysaccharide (LPS), provides neuroprotection against subsequent cerebral ischemic injury which can be attributed to the activation of TLR4-mediated TRIF/IRF3 signaling, and activation of TRIF reduces neuronal death [18]. In this experiment we hoped to determine whether TRIF-dependent signaling plays a role in the acute brain damage that accompanies cerebral I/R.
Figure 1. Sketch diagram of TLR4 mediated signaling pathways.
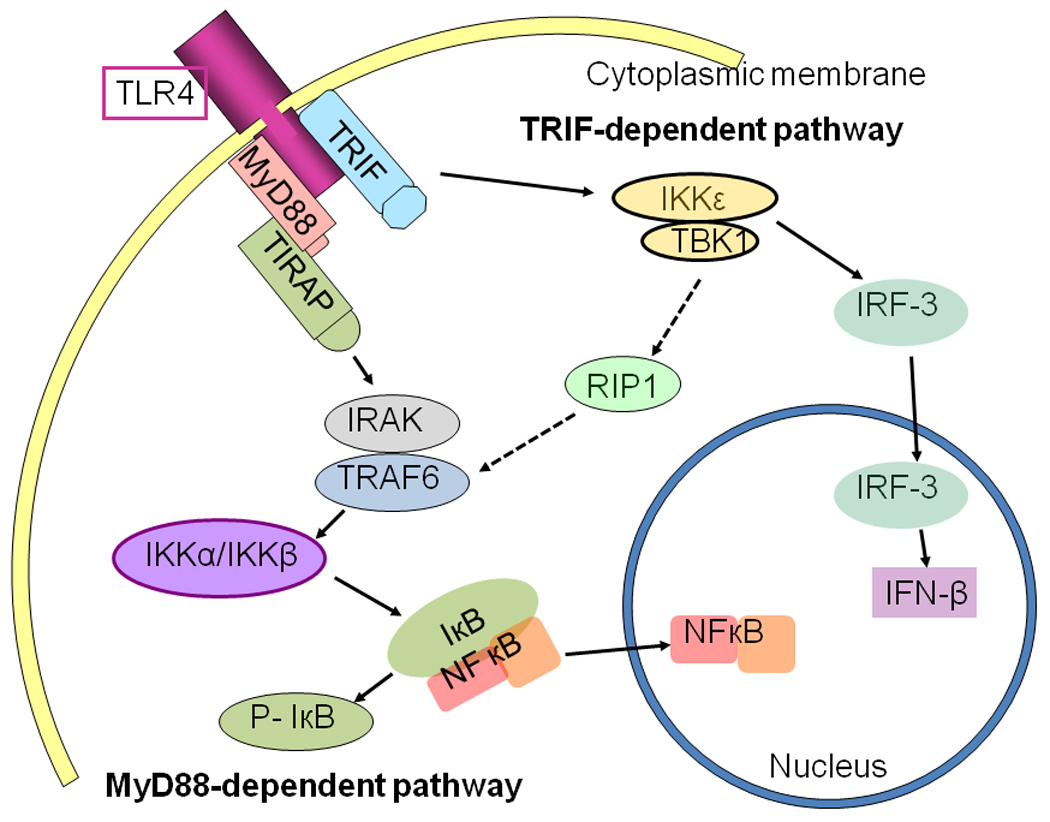
MyD88-dependent pathway: TLR4 recruits MyD88, which recruits IRAKs interacting with TRAF6, thereby activating IKKα/β and phosphorylating IκB, resulting in NFκB nuclear translocation and initiation of gene transcription associated with innate immune responses and inflammation (left in Fig. 1). TRIF-dependent pathway: TLR4 recruits TRIF, which in turn activates TBK1/IKKε that interacts with TRAF6, resulting in NFκB nuclear translocation and activating IRF-3-inducing interferon-β (IFN-β) (right in Fig. 1).
Materials and Methods
Animals
TRIF gene knockout mice (TRIFKO, C57BL/6J-Ticam1LPS2/J, n=20, male) and wild-type mice (WT, C57BL/6J, n=19, male) with body weights between 25–30g were obtained from the Jackson Laboratory and maintained in the Division of Laboratory Animal Resources at Emory University. The experiments outlined in this manuscript conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, revised 1996). Animal care and experimental protocols (248-2008) were approved by the Emory University Committee on Animal Care. Mice were assigned to the following four groups: wild-type sham control (W-S, n=6), wild-type mice subjected to focal cerebral ischemia/reperfusion (W-I/R, n=13), TRIF knockout sham control (TRIF-S, n=6) and TRIF knockout mice subjected to focal cerebral ischemia/reperfusion (TRIF-I/R, n=14).
Induction of focal cerebral ischemia/reperfusion (I/R)
Focal cerebral ischemia was induced by occlusion of the middle cerebral artery (MCAO) on the left side according to previously published methods [6]. Briefly, mice were subjected to anesthesia by inhalation of 1.5% to 2% isoflurane driven by 100% oxygen flow. The trachea was intubated and the lungs were mechanically ventilated (110 breaths/min, tidal volume: 0.5ml). Body temperature was regulated at 37.0°C with a temperature control system. Following the skin incision, a coated 6-0 filament (Doccol Corp., Redlands, CA, USA) was introduced into the internal carotid artery (ICA) via external carotid artery (ECA) and advanced 11 mm from the carotid bifurcation (cerebral ischemia started). After ischemia for 60 minutes, the filament was withdrawn and the reperfusion started. The ischemic and reperfusion conditions were confirmed by regional cerebral blood flow (rCBF) detected by a Doppler rCBF monitor. The ECA was tightened, the skin was closed and anesthesia was discontinued. The mice were allowed to recover in pre-warmed cages. Control mice underwent the same process without MCAO.
Western Blots
Four hours after I/R, mice were killed for brain harvesting. Cellular proteins were prepared from the parietal cortex in ischemic cerebral hemispheres (left hemispheres), electrophoresed with SDS-polyacrylamide gel and transferred onto Hybond ECL membranes (Amersham Pharmacia, Piscataway, NJ). The ECL membranes were incubated with the appropriate primary antibody followed by incubation with peroxidase-conjugated secondary antibodies. Signals were detected with the ECL system (Amersham Pharmacia) and quantified by scanning densitometry and computer-assisted image analysis. The primary antibodies used were anti-phospho-IκBα, anti-IκBα, anti-IKKε and anti-TAB1 (Cell Signaling Technology, Danvers, MA 01923). The secondary antibodies were also purchased from Sigma-Aldrich and Cell Signaling Technology.
Enzyme-linked immunosorbent assay (ELISA)
Nuclear proteins were isolated from parietal cortex in the ischemic cerebral hemispheres as previously described [5]. NFκB activity and IRF-3 activity were examined by ELISA using an NFκB-p65 assay kit and an IRF-3 activity kit (Active Motif. Inc.). For each sample, 15µg of nuclear proteins was used for detection according to manufacturer instructions. The absorbance was read on a spectrophotometer with a wavelength of 450nm with a reference wavelength of 655nm.
Evaluation of neurological score
An investigator blinded to group identity scored all mice with a neurological evaluation instrument described previously [19]. Briefly, the scoring system included five principal tasks: spontaneous activity over a 3-minute period (0–3), symmetry of movement (0–3), open-field path linearity (0–3), beam walking on a 3cm × 1 cm beam (0–3), and response to vibrissae touch (1–3) [6, 20].
Assessment of cerebral infarct volume size
Cerebral infarct size was evaluated according to previously applied methods [21]. Twenty four hours after reperfusion, mice were given an overdose of Pentobarbital (75 mg/kg, i.p.) and then transcardiacally perfused with cold saline followed by 4% paraformaldehyde in PBS (PH=7.4) via the ascending aorta. Brains were removed and post-fixed in 4% paraformaldehyde for 48 hrs and then stored at 4°C in a solution of 30% sucrose-PBS for two days. The brains were embedded in OCT and sectioned coronally to 10 µm-thick slices starting from the frontal pole at an interval of 1mm. The sections were stained with 0.5% cresyl violet (Nissl staining). The infarct areas defined as the area showing reduced Nissl staining under light microscope were traced and quantified with an image-analysis system. The areas of infarction in both hemispheres were calculated for each brain slice. Infarct volume adjusted for edema was calculated by dividing the infarct volume by the edema index, which was calculated by dividing the total volume of the left hemisphere by the total volume of the right hemisphere. Infarct volumes are expressed as percentage of the total brain volume ± S.E.M [6].
Histological observation
Brain sections taken in the coronal plane 1.5 mm behind Bregma were stained with hematoxylin–Eosin (H–E) using standard methods. Morphology and ischemic damage were verified by an investigator unaware of group identity.
Statistical analysis
Continuous scale measurements were expressed by the mean and standard error of the mean (S.E.M) for each group. Group comparisons for neurological function (neurological score) and infarct size were done with the t-test (applied to score values). One-way ANOVA and the least significant difference procedure were used to assess the effectiveness of intervention and group differences for levels of proteins and activity of NFκB and IRF-3. Probability levels of 0.05 or smaller were considered significant.
Results
Activity of NFκB in brain tissue after acute cerebral I/R injury
ELISA results showed that, in WT mice, NFκB activity was increased after cerebral I/R, compared to sham controls (0.283 ± 0.005 vs 0.096 ± 0.003, p<0.05, Fig. 2A). In TRIFKO mice, NFκB activity also increased after cerebral I/R, and was not significantly different from WT mice (0.279 ± 0.009 vs 0.100 ± 0.001, p<0.05, Fig. 2A).
Figure 2. Activation of NFκB signaling in mice brains subjected to cerebral I/R.
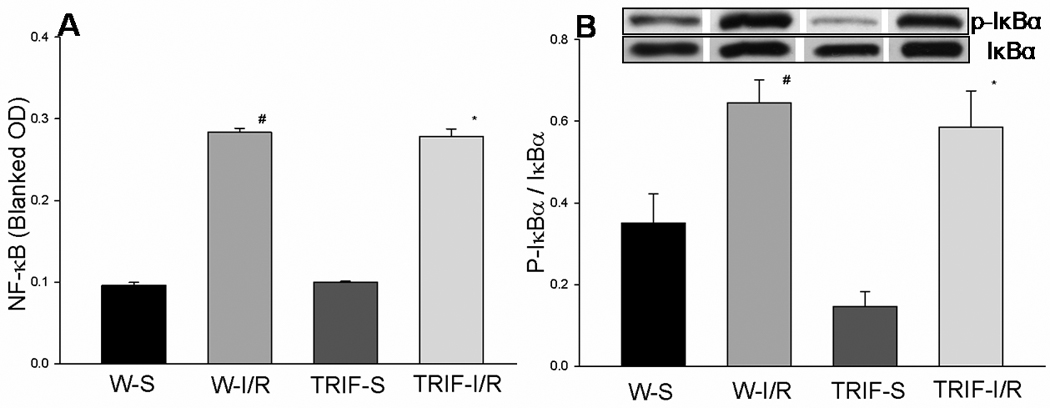
(A): Cellular proteins were isolated from brain tissues and NFκB activity was evaluated by ELISA. Results showed that NFκB activity was significantly increased after cerebral I/R compared with sham controls (p<0.05). NFκB activity in TRIFKO mice was not significantly different from that in WT mice. (B) Cellular proteins were isolated from brain tissues and evaluated by Western blots. Levels of p-IκBα were significantly increased in WT and TRIFKO mice after cerebral I/R. There was no significant difference in p-IκBα levels between WT and TRIFKO mice. W-S (n=3): WT mice sham control; W-I/R (n=5): WT mice subjected to cerebral I/R; TRIF-S (n=3): TRIF knockout mice sham control; TRIF-I/R (n=6): TRIF knockout mice subjected to cerebral I/R. # = p<0.05 compared with W-S; *= p<0.05 compared with TRIF-S.
Phosphorylation of the inhibitor of kappa B (IκBα) in brain tissue after acute cerebral I/R injury
The IκB proteins are characterized by the presence of a variable number of ankyrin repeats, which reside primarily in the cytoplasm and function to prevent NF-κB translocation to the nucleus. Activation of NF-κB requires the proteasomal degradation (phosphorylation) of IκBα. We assessed the levels of phosphorylated IκBα (p-IκB) in brain tissue. Compared to sham controls, p-IκB in WT mice was significantly increased after cerebral I/R (0.646 ± 0.055 vs 0.35 ± 0.073, p<0.05, Fig. 2B). Levels of p-IκBα in TRIFKO mice also increased significantly compared to sham control (0.585 ± 0.090 vs 0.146 ± 0.037, p<0.05, Fig. 2B), which were not different from those in WT mice after cerebral I/R.
Activity of IRF-3 in brain tissue after acute cerebral I/R injury
IRF-3, a member of the interferon regulatory transcription factor (IRF) family, is an important transcription factor mediated through the TLR-mediated TRIF-dependent pathway. In WT mice, IRF-3 activity was not significantly increased in brain tissue 4 h after cerebral I/R, compared to sham controls. In TRIFKO mice, IRF-3 activity was not increased compared to controls. There was no significant difference in IRF-3 between WT mice and TRIFKO mice 4 h after cerebral I/R (0.260 ± 0.011 vs 0.239 ± 0.019, Fig.3A).
Figure 3. Activation of IRF-3 signaling in mice brains subjected to cerebral I/R.
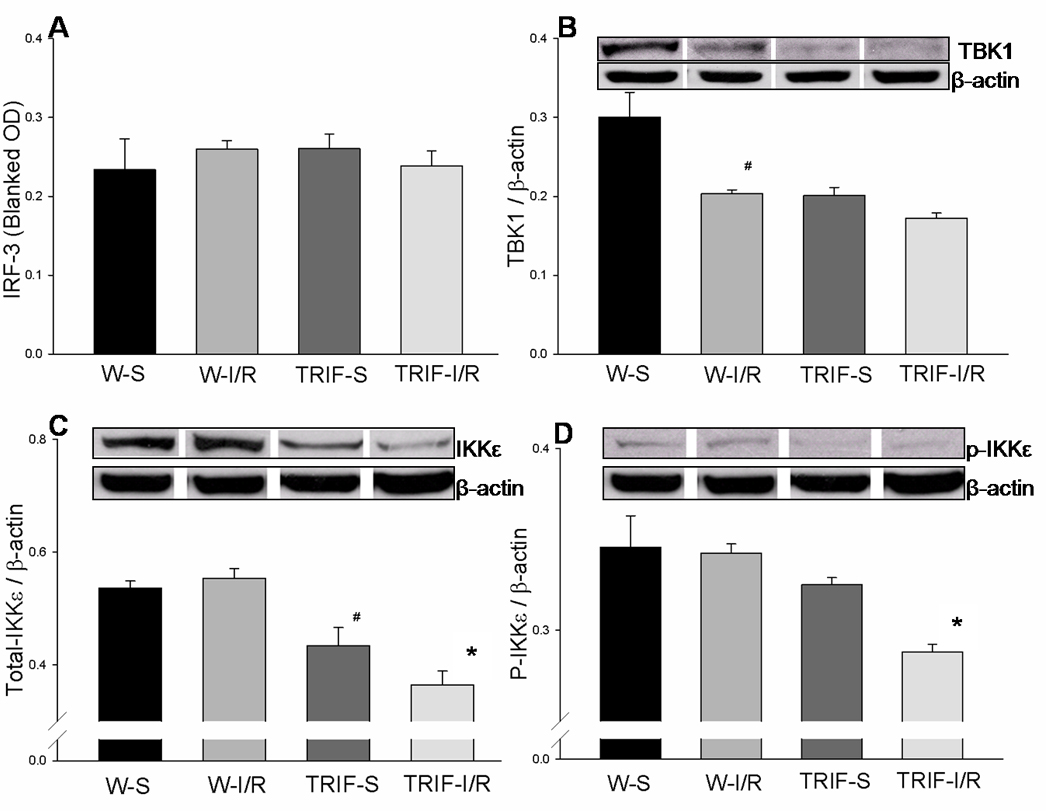
(A): Cellular proteins were isolated from brain tissues and IRF-3 activity was evaluated by ELISA. IRF-3 activity was not significantly increased in either WT or TRIFKO mice after cerebral I/R compared with their sham controls. (B) Cellular proteins were isolated from brain tissues and evaluated by Western blots. In WT mice, the expression of TBK1 decreased in brain tissue 4 hours after cerebral I/R, compared with sham controls (p<0.05). The basal level of TBK1 in TRIFKO mice was lower than that in WT mice (p<0.05). However, the level of TBK1 in TRIFKO mice did not change after cerebral I/R, and was not different from that in WT mice subjected to cerebral I/R. (C) In WT mice, cerebral I/R did not increase the expression of IKKε. The basal level of IKKε in TRIFKO mice was lower than that in WT mice (p<0.05). IKKε was decreased in TRIFKO mice after cerebral I/R, and significantly lower than that in WT mice (p<0.05). (D) In WT mice, cerebral I/R did not increase the phosphorylation of IKKε (p-IKKε). P-IKKε was decreased in TRIFKO mice after cerebral I/R, and significantly lower than that in WT mice (p<0.05). W-S (n=3): WT mice sham control; W-I/R (n=5): WT mice subjected to cerebral I/R; TRIF-S (n=3): TRIF knockout mice sham control; TRIF-I/R (n=6): TRIF knockout mice subjected to cerebral I/R. # = p<0.05 compared with W-S; * = p<0.05 compared with W-I/R.
Expression of TANK-binding kinase-1 (TBK1) in brain tissue after acute cerebral I/R injury
TBK1 has been implicated in IRF-3 and NFκB activation and is required for activation of IRF3 by TRIF [21].In WT mice, the expression of TBK1 decreased in brain tissue 4 h after cerebral I/R, compared to sham controls (0.203 ± 0.005 vs 0.301 ± 0.031, p<0.05; Fig. 3B). The basal level of TBK1 in TRIFKO mice was lower than that in WT mice (0.201 ± 0.010 vs 0.301 ± 0.031, p<0.05; Fig. 3B). However, the level of TBK1 in TRIFKO mice did not change after cerebral I/R, and was not significantly different from WT mice subjected to cerebral I/R. (Fig. 3B)
Expression of IκB kinase-ε (IKKε) in brain tissue after acute cerebral I/R injury
IKKε is an IκB kinase homolog essential for the activation of IRF-3 through the MyD88-independent TRIF pathway [21]. In WT mice, cerebral I/R did not increase the expression and phosphorylation of IKKε (p- IKKε) [Fig. 3C and 3D]. The basal level of IKKε in TRIFKO mice was lower than that in WT mice (0.434 ± 0.032 vs 0.536 ± 0.013, p<0.05; Fig. 3C). In addition, IKKε and p-IKKε were decreased in TRIFKO mice after cerebral I/R, and were significantly lower than in WT mice (IKKε: 0.365 ± 0.024 vs 0.554 ± 0.017, p<0.05; p-IKKε 0.288 ± 0.004 vs 0.343 ± 0.005, p<0.05; Fig. 3C and 3D).
Neurological deficits associated with focal cerebral I/R
There were no neurological deficits in the control mice. Figure 4A shows that cerebral I/R decreased the neurological score in both WT (5.0 ± 0.57) and TRIFKO (4.3 ± 0.73) mice, and there was no significant difference in neurological score between WT and TRIFKO mice 24 hours after cerebral I/R (Fig. 4A).
Figure 4. Neurological deficits and brain damage associated with focal cerebral I/R.
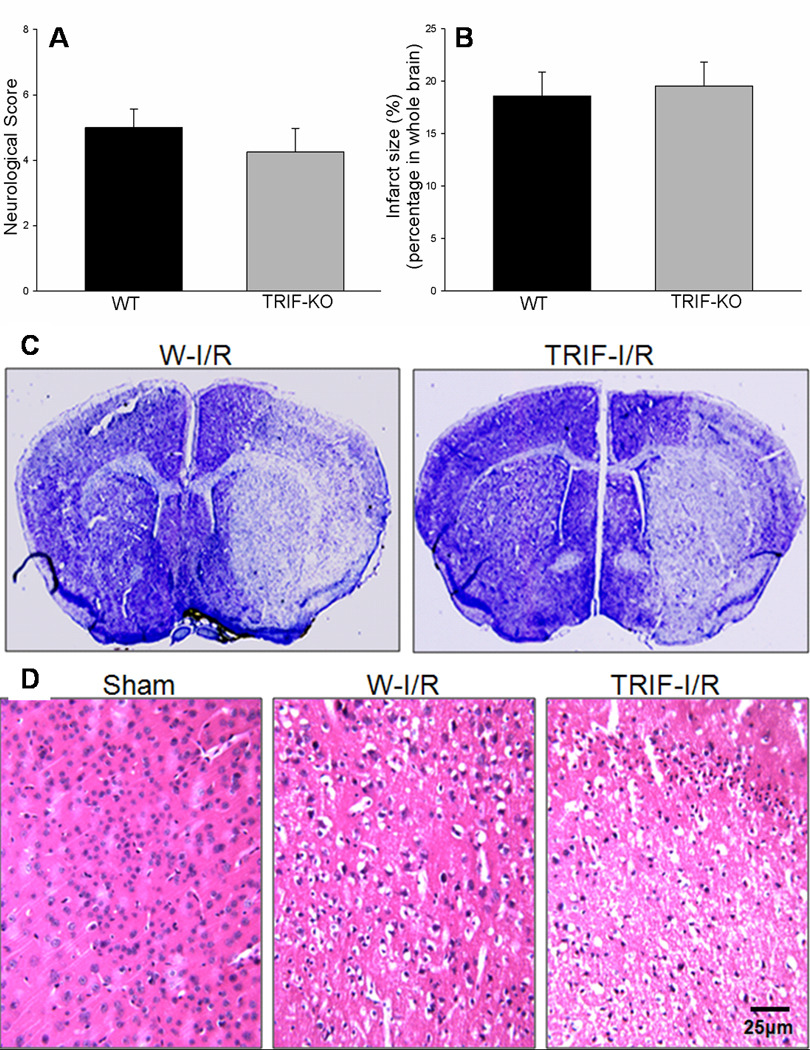
Cerebral I/R decreased neurological function in both WT and TRIFKO mice. There was no difference in neurological score between WT and TRIFKO mice 24 hours after cerebral I/R (Fig. 4A). In addition, cerebral I/R induced obvious brain infarction. There was no significant difference in infarct size between WT mice and TRIFKO mice 24 hours after cerebral I/R (Fig. 4B). WT (n=8): wild type mice; TRIFKO (n=8): TRIF knockout mice. Figure 4C showed the representative pictures for brain infarction. The infarct areas were defined as the area showing reduced Nissl staining under light microscope. Figure 4D showed H&E-stained sections of brains (cortex). Mice subjected to sham surgery showed normal histology. WT mice subjected to I/R showed acute pan-necrosis. TRIFKO mice subjected to I/R also showed pan-necrosis throughout the ischemic areas.
Cerebral infarct size after I/R
Infarct size was evaluated in the mice 24 hours after cerebral I/R. As shown in Figure 2, cerebral I/R induced obvious brain infarction. Infarct size was not significantly different between WT mice (18.60 ± 2.23%) and TRIFKO mice (19.55 ± 2.26%) 24 h after cerebral I/R (Fig.4B). The representative pictures for infarct size are shown in Fig. 4C.
Observation on brain histopathology following acute I/R
H&E-stained sections of brains from mice subjected to sham surgery reveal normal histology (Fig. 4D). All WT mice subjected to I/R showed acute pan-necrosis throughout the MCA. The consistent infarction areas of brain in WT mice were thalamus and caudate-putamen, with more variable involvement of the overlying neocortex and the hippocampus. TRIFKO mice subjected to I/R also showed pan-necrosis throughout the areas supplied by middle cerebral artery (Fig. 4D).
Discussion
After the onset of cerebral ischemia, a multifaceted inflammatory reaction emerges over the next few hours. Numerous inflammatory mediators are induced, including cytokines and chemokines, which then release further inflammatory mediators contributing to the inflammatory reaction. Most of the genes that encode inflammation factors are mediated by NFκB, an important nuclear transcription factor [22, 23]. Experimental studies have demonstrated that NF-κB plays a detrimental role in cerebral ischemia [24–26]. In the ischemic brain, NF-κB can be activated by extracellular and intracellular stimuli as well as through several receptor-mediated signaling pathways. TLRs, functioning as upstream regulators of NF-κB signaling, mediate the activation of NF-κB. TLRs can be activated by microbial pathogens as well as endogenous ligands released from injured tissue, and then induce NFκB activation and other inflammatory responses. Many studies have demonstrated that TLR2, TLR4 and TLR9 are involved in cerebral I/R injury and in protection against ischemic brain damage afforded by preconditioning [3–7, 27].
We have previously reported that TLR4 signaling was induced in ischemic brain and contributed to cerebral I/R injury [5]. TLR4 deficit decreased infarct size, reduced neuronal apoptosis and improved neurological outcome after cerebral I/R. In addition, TLR4-deficient mice showed less NF-κB DNA-binding activity and lower levels of several inflammatory mediators (IL-6, TNF, Fas ligand (FasL) and high-mobility group box 1 (HMGB1)) compared to WT mice after cerebral I/R [5]. Although TLR4 activation after stroke exacerbates injury, several investigators have reported that activation of TLR4 with the specific ligand LPS, given before cerebral ischemia (preconditioning), protects the brain from damage [18, 28]. Activation of TLR4 with LPS may cause cells to switch their dominant signaling pathway and, through TRIF, induce expression of IFNβ via activation of the interferon regulatory factor IRF3. Enhanced TLR4 signaling to TRIF–IRF3–IFNβ would be expected to contribute to neuroprotection [18]. Since TRIF-dependent signaling also mediates NFκB activation, it is important to know whether TRIF signaling contributes to ischemia-induced brain injury, and further work needs to be done to answer this question.
Here we show that in WT mice, NFκB activity and phosphorylation of IκBα increased 4 hours after cerebral I/R an observation consistent with our previous report [7]. In contrast, the activity of IRF-3 was not significantly changed compared to sham controls. The levels of TBK1 and IKKε, the essential components of the IRF-3 signaling pathway, were not activated by cerebral I/R; in fact, the level of TBK1 decreased in WT mice after cerebral I/R (Fig 3). These results can be taken to indicate that cerebral I/R induces the activation of NFκB signaling but not TRIF/IRF-3 signaling after ischemic insult. In TRIFKO mice, basal levels of TBK1 and IKKε were lower than in WT mice and IRF-3 activity was changed significantly after cerebral I/R. Interestingly, NFκB activity and phosphorylation of IκBα in TRIFKO mice still increased significantly compared to controls and were not different from that in WT mice with cerebral I/R (Fig. 2). MyD88 and TRIF both induce activation of NF-κB, My88 in the early phase [14], and TRIF later [29]. We take our results to demonstrate that cerebral I/R-induced NFκB signaling is not attenuated by the lack of TRIF. The TRIF-dependent signaling pathway does not contribute to the activation of NFκB signaling in ischemic brain, at least in the early stage of the injury cascade.
We and others have reported that TLR4 deficiency can reduce infarct size and neuronal death induced by cerebral I/R [4, 5, 7]. Whether TRIF, contributes to ischemia-induced brain injury still remains unknown. To investigate the role of TRIF in cerebral I/R injury further, we measured neurological function and infarct size 24 hours after focal cerebral ischemia induced by MCAO. We observed that MCAO for 60 minutes followed by 24 hours reperfusion induced substantial neurological functional deficits and brain infarction in WT mice. Compared to WT mice, there was neither improved neurological function nor reduced brain damage and infarct size in TRIFKO mice.
In summary, the most significant findings of the present study were that acute cerebral I/R activated NFκB signaling, but not IRF-3 signaling; and that TRIF deficiency neither inhibits the activation of NFκB induced by cerebral ischemia, nor attenuates cerebral I/R injury. We interpret our results to suggest that TRIF-independent signaling is not required for acute cerebral I/R injury. However, considering that activation of TRIF signaling induces expression of IFNβ via IRF3 and is expected to contribute to neuroprotection, the possible neuroprotective roles of TRIF at later stages after cerebral ischemia cannot be ruled out and need further investigation.
Acknowledgements
This work was supported by AHA National Program SDG 0830481N to FH, Emory University URC Grant (2009-002) to FH, research funding from the Department of Pediatric Cardiovascular Surgery at the Children’s Hospital of Atlanta and NIH RO1 NS04851 to DS.
Abbreviations
- I/R
Ischemia/reperfusion
- TRIF
TIR domain-containing adaptor protein
- TLR
Toll-like receptor
- IRF3
Interferon regulatory factor 3
- NF-κB
nuclear factor kappa B
- I-κBε
inhibitor of kappa B
- IKKε
inhibitor of κB kinase complex-ε
- TBK1
TANK-binding kinase 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Simoni MG, Milia P, Barba M, De Luigi A, Parnetti L, Gallai V. The inflammatory response in cerebral ischemia: focus on cytokines in stroke patients. Clin Exp Hypertens. 2002;24:535–542. doi: 10.1081/ceh-120015330. [DOI] [PubMed] [Google Scholar]
- 2.Stoll G. Inflammatory cytokines in the nervous system: multifunctional mediators in autoimmunity and cerebral ischemia. Rev Neurol. 2002;158:887–891. [PubMed] [Google Scholar]
- 3.Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 4.Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 5.Hua F, Ma J, Ha T, Xia Y, Kelley J, Williams DL, Kao RL, Browder IW, Schweitzer JB, Kalbfleisch JH, Li C. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol. 2007;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hua F, Ma J, Ha T, Kelley YJ, Williams DL, Kao RL, Kalbfleisch JH, Browder IW, Li C. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol. 2008;199:75–82. doi: 10.1016/j.jneuroim.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hua F, Ma J, Ha T, Kelley JL, Kao RL, Schweitzer JB, Kalbfleisch JH, Williams DL, Li C. Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Res. 2009;1262:100–108. doi: 10.1016/j.brainres.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 9.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 10.Anderson KV. Toll signaling pathways in the innate immune response. Current Opinion in Immunology. 2000;12:13–19. doi: 10.1016/s0952-7915(99)00045-x. [DOI] [PubMed] [Google Scholar]
- 11.Chuang TH, Ulevitch RJ. Identification of hTLR10: a novel human Toll-like receptor preferentially expressed in immune cells. Biochim. Biophys. Acta. 2001;1518:157–161. doi: 10.1016/s0167-4781(00)00289-x. [DOI] [PubMed] [Google Scholar]
- 12.Du X, Poltorak A, Wei Y, Beutler B. Three novel mammalian toll-like receptors: gene structure, expression, and evolution. Eur. Cytokine Netw. 2000;11:362–371. [PubMed] [Google Scholar]
- 13.Sandor F, Buc M. Toll-like receptors. I. structure, function and their ligands. Folia Biol (Praha) 2005;51:148–157. [PubMed] [Google Scholar]
- 14.Akira S, Takeda K. Toll-like receptor signaling. Nat. Rev. Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 15.Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-beta in Toll-like receptor-stimulated dendritic cell activation. Int Immunol. 2002;14:1225–1231. doi: 10.1093/intimm/dxf089. [DOI] [PubMed] [Google Scholar]
- 16.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 17.Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B. Identification of Lps2 as a key transducer of MyD88-independent TIR signaling. Nature. 2003;424:743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 18.Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel-Poore MP. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29:9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- 20.Shimamura N, Matchett G, Yatsushige H, Calvert JW, Ohkuma H, Zhang J. Inhibition of integrin alphavbeta3 ameliorates focal cerebral ischemic damage in the rat middle cerebral artery occlusion model. Stroke. 2006;37:1902–1909. doi: 10.1161/01.STR.0000226991.27540.f2. [DOI] [PubMed] [Google Scholar]
- 21.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 22.Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-beta in Toll-like receptor-stimulated dendritic cell activation. Int Immunol. 2002;14:1225–1231. doi: 10.1093/intimm/dxf089. [DOI] [PubMed] [Google Scholar]
- 23.Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 24.Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, Weih F, Frank N, Schwaninger M, Koistinaho J. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35:987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- 25.Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28:2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- 27.Stevens SL, Ciesielski TM, Marsh BJ, Yang T, Homen DS, Boule JL, Lessov NS, Simon RP, Stenzel-Poore MP. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2008;28:1040–1047. doi: 10.1038/sj.jcbfm.9600606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hickey EJ, You X, Kaimaktchiev V, Stenzel-Poore M, Ungerleider RM. Lipopolysaccharide preconditioning induces robust protection against brain injury resulting from deep hypothermic circulatory arrest. J Thorac Cardiovasc Surg. 2007;133:1588–1596. doi: 10.1016/j.jtcvs.2006.12.056. [DOI] [PubMed] [Google Scholar]
- 29.Kawai T, Takeuchi O, Fujita T, Inoue J, Muhlradt PF, Sato S, Hoshino K, Akira S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 2001;167:5887–5894. doi: 10.4049/jimmunol.167.10.5887. [DOI] [PubMed] [Google Scholar]