

Abstract
Mutations in the collagen VI genes (COL6A1, COL6A2, and COL6A3) result in Ullrich congenital muscular dystrophy (UCMD), Bethlem myopathy (BM) or phenotypes intermediate between UCMD and BM. While UCMD can be caused by either recessively or dominantly acting mutations, BM has thus far been described as an exclusively autosomal dominant condition. We report two adult siblings with classic Bethlem myopathy who are compound heterozygous for a single nucleotide deletion (exon 23; c.1770delG), leading to in-frame skipping of exon 23 on the maternal allele, and a missense mutation p.R830W in exon 28 on the paternal allele. The parents are carriers of the respective mutations and are clinically unaffected. The exon skipping mutation in exon 23 results in a chain incapable of heterotrimeric assembly, while p.R830W likely ameliorates the phenotype into the Bethlem range. Thus, autosomal recessive inheritance can also underlie Bethlem myopathy, supporting the notion that UCMD and BM are part of a common clinical and genetic spectrum.
Keywords: Bethlem myopathy, collagen VI, COL6A2, autosomal recessive inheritance
Introduction
Bethlem myopathy (BM) was first described in 1976 by Bethlem and van Wijngaarden in 28 patients from 3 Dutch families manifesting a myopathy following autosomal dominant inheritance characterized by slowly progressive muscle weakness and typical flexion contractures of the long finger flexors, wrists, elbows, pectorales muscles and ankles, as well as of the spine [1]. Patients with Bethlem myopathy usually become symptomatic during the first or second decade of life although hypotonia in infancy can be seen [2, 3]. Two-thirds of patients over age 50 years are at least partially wheelchair dependent [3]. Findings on muscle imaging of an “outside-in” pattern of muscle degeneration are diagnostically helpful [4, 5]. Ullrich CMD is a more severe disorder of congenital onset with significant weakness, joint hyperlaxity as well as contractures [6]. The ability to ambulate may be achieved briefly and subsequently lost during the first decade of life, or may not be achieved at all [7].
Collagen VI constitutes a microfibrillar component in a number of extracellular matrices, including those in muscle, tendon and skin [8]. Collagen VI directly associates with basement membranes in various tissues [9], including skeletal muscle. Functions considered for collagen VI have included roles in differentiation and cell survival. Mutations in the collagen VI genes lead to an abnormal collagen VI matrix as well as a conspicuous loss of the tight connection of collagen VI with the basement membrane, visible in dermal fibroblast culture [10, 11].
In the majority of patients, both of the classical clinical phenotypes are caused by mutations in the three genes coding for collagen type VI, located on chromosome 21 (COL6A1 and COL6A2) as well as on chromosome 2 (COL6A3) [12]. Thus far, the mutations which have been described as associated with Bethlem myopathy have been exclusively dominant, with the in-frame skipping of exon 14 in COL6A1 and missense mutations affecting invariant glycine residues in the initial part of the triple helical domain, constituting the most common types of mutation [12]. Mutations causing Ullrich CMD, on the other hand, can be either autosomal recessive as initially described [13, 14] or follow a more severe dominant negative mechanism as recognized more recently [11, 15]. Recessive mutations seen in typical UCMD are often nonsense mutations, carrier status for which is not associated with a clinical phenotype. An increasing number of patients with phenotypes of intermediate severity between Bethlem and Ullrich are now being recognized with the availability of genetic testing in the collagen VI genes. The distinct phenotype of myosclerosis myopathy presenting with early, progressive contractures of proximal and distal joints in patients also has recently been associated with specific recessive collagen VI mutations [16].
In a family with two affected siblings we now provide evidence that the classic phenotype of Bethlem myopathy can also be caused by recessive mutations in collagen VI, suggesting the presence of a spectrum of collagen VI associated disorders, that is both clinical as well as genetic.
Case reports
Patient A, age 43 years, and his biological brother (patient B), age 41 years, were seen for evident weakness and contractures. Patient A was born full-term following an uncomplicated pregnancy and delivery. There were no abnormal findings at the time of birth. The patient had no delay in motor milestones; however, he reported that he was never able to run as fast as other children his age. He reported a history of obligate toe walking since approximately 6 years of age. Bilateral Achilles tendon release surgery was performed at 10 years of age. He noticed gradual progressive weakness over time. In his 30s, he clearly noticed significant proximal weakness, and by age 40 years he started to use a cane for ambulating outside of the home. At present, he is unable to arise from the floor without holding onto furniture. A muscle biopsy performed at 8 years of age was described as inconclusive (not available for our review).
Physical examination of patient A at 43 years of age revealed proximal muscle weakness in the MRC grade 4-/5 to 4/5 range and contractures of the pectorales muscles, elbows, long finger flexors and Achilles tendons bilaterally (Figure 1b). There was mild hyperlaxity of the distal fingers along the DIP joints. The patient walked with a marked Trendelenburg gait but was able to walk unassisted across the examination room. Deep tendon reflexes were 1+ at the bilateral brachioradialis, biceps, triceps and Achilles and trace at the bilateral patellae. There was stiffness of the lumbar spine. Skin examination revealed keratosis pilaris along the extensor surfaces of the bilateral arms and legs. There was evidence of a poorly-healing scar just inferior to the right clavicle, at the site of a recent surgical excision of a basal cell nevus.
Fig. 1.

Patient genotype and phenotype. Pedigree with inheritance of the two mutations (a). Clinical photographs of patients A and B demonstrating typical finger flexor contractures (b).
Patient B (patient A’s biological brother) was born full-term following an uncomplicated pregnancy and delivery. There were no abnormal findings at the time of birth. The patient was noted to show obligate toe walking as a toddler and underwent bilateral Achilles tendon release surgery between 7 and 8 years of age, which was repeated at 14 years of age along with concomitant tendon transfer to the dorsal surface of the feet. The patient had no delay in motor milestones; however, he reports never being able to run as fast as other children his age. He noticed a gradual progression of proximal weakness over time. Now at 41 years of age, he is able to ambulate independently without the use of mobility aids. He is unable to arise from the floor without holding onto furniture, however.
Physical examination of patient B at 41 years of age revealed proximal muscle weakness in the MRC grade 4/5 range and contractures of the bilateral pectorales muscles, elbows, long finger flexors and Achilles tendons bilaterally (Figure 1b). There was mild hyperlaxity of the distal fingers along the DIP joints. The patient walked with a Trendelenburg gait. He was able to walk across the examination room without assistance. Deep tendon reflexes were 1+ at the bilateral brachioradialis, biceps, triceps, patellae and Achilles tendons. Skin examination revealed evidence of keratosis pilaris along the extensor surfaces of the arms and legs as well as along the back.
Muscle ultrasounds performed at the time of patient A and patient B’s clinic evaluation revealed evidence of a “central cloud” phenomenon in the rectus femoris, reflecting degeneration around the central fascia in the rectus femoris [4], as well as a peripheral increase in echogenicity, similar to the pattern described on CT and MRI in Bethlem myopathy (not shown) [2, 5].
Results
Genomic DNA was extracted from serum samples from patients A, B and their parents, and direct genomic sequencing of all exons and exon-intron boundaries of COL6A1, COL6A2 and COL6A3 was performed on patient A using SCAIP technology [17]. Mutations were discovered in the COL6A2 gene: a single nucleotide deletion in exon 23 (c.1770delG), predicted to result in a premature termination codon (p.Thr590ThrfsX4), was in cis with a missense change in exon 26 (c.2351G>A, p.R784H) and was inherited from the mother (figure 2a). The c.1770delG mutation also converted the exon 23 splice donor site from ‘acgGTAGGT’ to ‘cacGTAGGT’, decreasing the MaxENT score from 10.15 to 6.85 [18]. Exon 23 skipping would lead to a 12 amino acid in-frame deletion of the C-terminal TH domain (p.Gly579_Thr590del). RT-PCR analysis on RNA isolated from a dermal fibroblast culture from patient A using primers localized on exon 16 and 26 thus amplifying a fragment containing exon 23 respectively revealed the presence of two bands, the smaller of which on sequencing had skipped exon 23, confirming the effect of the c.1770delG mutation on splicing (figure 2b). The other allele, inherited from the patient’s father, showed a missense mutation in exon 28 (c.2488C>T), causing a change from arginine to tryptophan at position 830 (p.R830W) (figure 2a, c). This residue is located next to a MIDAS motif and shows evolutionary conservation (Figure 2c). Modeling of the mutation using ProtScale with the Zimerman-Eliezer-Simha polarity scales [19] on the Expasy server [20] demonstrated a clear change in the polarity scale of the protein centered around the mutation, consistent with the change from a positively charged polar (R) to a nonpolar (W) sidechain (Suppl Figure 1). Patient B was confirmed to carry both the maternal c.1770delG and p.R784H alleles and the paternal p.R830W allele; both parents were found to be carriers of the respective mutations and are clinically unaffected.
Fig. 2.
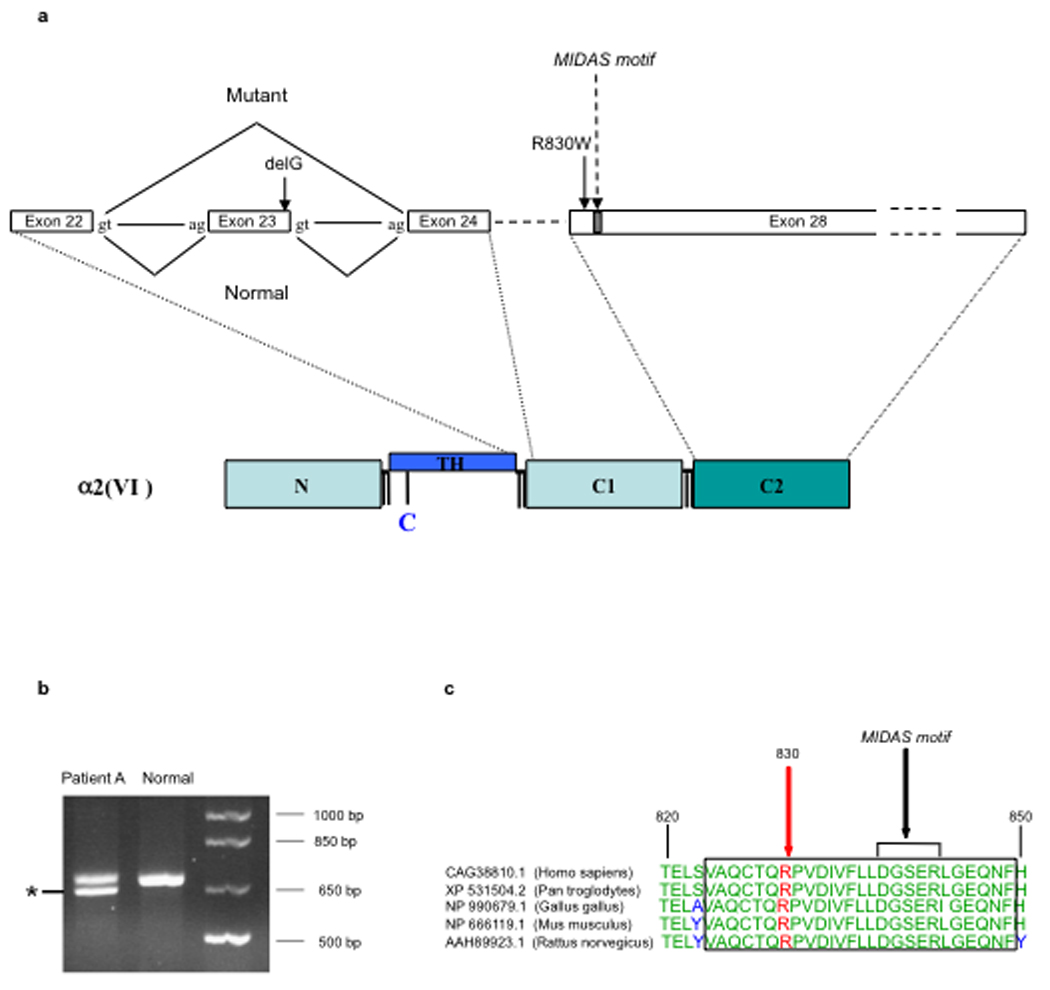
Collagen VI alpha 2 (COL6A2) mutations and consequences. (a) Diagrammatic representation of the two mutations in the collagen VI alpha 2 (COL6A2) gene, demonstrating the effect of c.1770delG on exon 23 splicing, causing in-frame skipping of the entire exon 23, which will result in exclusion of the chain from assembly; and the position of the exon 28 missense mutation c.2488C>T (R830W) relative to the modified MIDAS motif in the C2 domain. (b) Gel electrophoresis of RT-PCR product from RNA isolated from fibroblasts from patient A amplified with primers on exon 16 and 26 in collagen VI alpha 2 demonstrating a smaller PCR product lacking exon 23 (asterix). (c) Evidence of sequence conservation of the 830 residue with nearby MIDAS motif indicated. (Alignment was carried out using the BLAST program, http://www.ncbi.nlm.nih.gov/BLAST/download.shtml.)
Dermal fibroblasts were cultured from the skin biopsy obtained from patient A to subconfluency and supplemented with ascorbic acid for another three days to allow for optimal formation of the collagenous extracellular matrix, followed by staining for collagen VI using polyclonal and monoclonal antibodies [21]. Microscopic analysis showed that the fibroblasts were able to deposit a collagen VI extracellular matrix; however, compared to the matrix deposited by normal fibroblasts, it seemed more sparse and less smooth with a “speckled” appearance (Figure 3b), as can be seen in cultures from patients with Bethlem myopathy [22]. There also was evidence of some intracellular retention of immunoreactive material after cells were permeabilized with triton (Figure 3d).
Fig. 3.
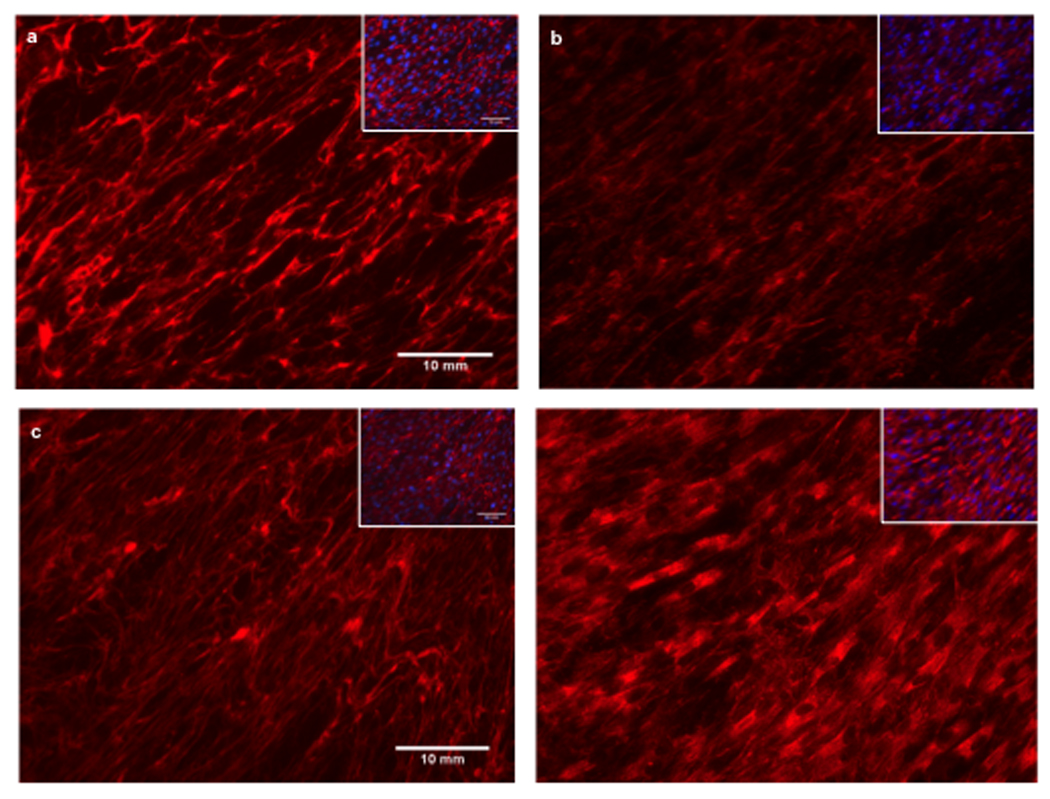
Immunocytochemistry for collagen VI on cultured fibroblasts derived from patient A and normal control fibroblasts with monoclonal anti-collagen VI antibody. The matrix in the patient (b) is sparse and has a “speckled” appearance compared to control (a) which has a dense, well-formed matrix. Addition of Triton X-100 reveals significant intracellular retention of immunoreactive material in the patient’s fibroblasts (d) but not in the control (c). Insets with DAPI nuclear staining to demonstrate equal cell density. Images were taken at x20 magnification. Bar = 10µm.
Discussion
Here we report two adult brothers presenting with the classic phenotype of Bethlem myopathy, including childhood onset, expected progression, patterns of weakness and contractures, as well as typical skin findings and changes on muscle imaging. Within the Bethlem phenotype these patients can be considered to be affected fairly typically. Both siblings are compound heterozygous for mutations in the COL6A2 gene, while each parent is a clinically unaffected carrier of one of these mutations.
The single nucleotide deletion in exon 23 (c.1770delG) on the maternal allele is an leads to the in-frame skipping of exon 23 at the N-terminal end of the triple-helical domain. In-frame deletions in this location of the triple helical domain render the chain incapable of incorporation into the heterotrimer, leading to complete exclusion of the mutant allele from the assembly process [22]. Thus, the mutation effectively results in a recessively acting null allele [21]. Our non-quantitative RT-PCR analysis suggests that this exon skipping occurs at a high rate (Figure 2b), but even if the skipping were to occur at a less complete rate, the single nucleotide deletion would result in a frameshift and premature stop, since the nucleotide is part of the coding region. Carriers for null mutations in COL6A2 are clinically unaffected [13, 14]. In the homozygous state the mutation would be predicted to lead to the total lack of assembly-competent alpha2 (VI) chain allowing for no collagen VI basic heterotrimer to form [21], which would lead to the phenotype of Ullrich congenital muscular dystrophy [7, 12].
The missense mutation p.R830W on the paternal allele thus acts on this virtual null background from the maternal allele. The fact that this mutation leads to the less severe phenotype of Bethlem myopathy suggests that it must be considerably milder in terms of its effect on collagen VI function compared to a null mutation. Pathogenic missense mutations in the COL6A2 C2 domain have been observed only rarely (L837P [15], R876S [23]). R830W has not been reported before as a mutation or as a polymorphism in the available databases (www.dmd.nl), nor has it been seen in our own unpublished experience. The arginine at position 830 is conserved between species (Figure 2c) and localizes at the beginning of the C2 domain, in close proximity to a modified metal ion-dependent adhesion site (MIDAS) motif (position 839–843) (Figure 2c), which is of importance in metal-ion mediated protein-protein interactions, likely coordinating the formation of the collagen VI antiparallel dimer [24]. The p.R830W amino acid change is nonconservative, changing the polarity of the protein in the region (Suppl Figure 1), conceivably changing the functionality of the MIDAS motif, although we have not formally demonstrated this. The missense change p.R784H coinherited in cis with c.1770delG on the maternal allele will not be part of assembled collagen VI because the preceding exon skipping renders the chain incompetent for assembly. It likely qualifies as a polymorphism as it has been observed before without clear disease association [17] (and personal observation).
The patients presented here provide evidence that classic Bethlem myopathy can be inherited in an autosomal recessive manner. Thus, Bethlem myopathy joins Ullrich congenital muscular dystrophy in that both dominant and recessive genetic mechanisms have now been established as underlying these two classic disorders which lie at the ends of the phenotypic spectrum of collagen VI related myopathies. The fact that an “Ullrich mutation” in compound heterozygosity with a presumably milder “Bethlem mutation” results in a Bethlem myopathy phenotype leads to the conclusion that the collagen VI related myopathies are best understood as existing along a clinical and genetic spectrum, in which the severity of the mutations and the resulting functional abnormality of the collagen VI matrix dictate a phenotypic spectrum, ranging from severe Ullrich to mild Bethlem with phenotypes of intermittent severity connecting the two. Consequently, a fundamentally different genetic or biochemical mechanism distinguishing Ullrich congenital muscular dystrophy from Bethlem myopathy can no longer be assumed.
This observation significantly impacts genetic counseling and the interpretation of genetic testing in a patient with Bethlem myopathy, since the finding of one missense mutation of unclear significance now should prompt consideration of the possibility of an additional second mutation on the other allele, consistent with recessive inheritance. Furthermore, studies in the extended family are imperative to determine the mode of inheritance as well as disease association of a given genetic change of unclear significance. Obviously, the determination of recessively acting mutations in a patient with Bethlem myopathy drastically reduces the risk of disease recurrence in the patients own offspring.
Supplementary Material
Polarity scale of a 41 AA region around AA 830 (marked by arrow head) using the Zimerman-Eliezer-Simha algorithm in ProtScale [19] on the Expasy server [20]. Note change of polarity score around the mutation R830W.
Acknowledgements
We would like to thank the family for their encouragement to carry out and report this study. We would like to thank Livija Medne, MS, CGC, for helpful comments and discussions. C.G.B. is supported by grants from NIH/NIAMS (R01AR051999) and from MDA USA (MDA3896).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bethlem J, Van Wijngaarden GK. Benign myopathy, with autosomal dominant inheritance: a report on three pedigrees. Brain. 1976;99:91–100. doi: 10.1093/brain/99.1.91. [DOI] [PubMed] [Google Scholar]
- 2.Merlini L, Morandi L, Granata C, Ballestrazzi A. Bethlem myopathy: early-onset benign autosomal dominant myopathy with contractures. Description of two new families. Neuromuscular Disorders. 1994;4:503–511. doi: 10.1016/0960-8966(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 3.Jöbsis GJ, Boers JM, Barth PG, de Visser M. Bethlem myopathy: a slowly progressive congenital muscular dystrophy with contractures. Brain. 1999;122:649–655. doi: 10.1093/brain/122.4.649. [DOI] [PubMed] [Google Scholar]
- 4.Bönnemann CG, Brockmann K, Hanefeld F. Muscle ultrasound in bethlem myopathy. Neuropediatrics. 2003;34:335–336. doi: 10.1055/s-2003-44665. [DOI] [PubMed] [Google Scholar]
- 5.Mercuri E, Lampe A, Allsop J, Knight R, Pane M, Kinali M, et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15:303–310. doi: 10.1016/j.nmd.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Ullrich O. Kongenitale, atonisch-sklerotische Muskeldystrophie. Monatsschr Kinderheilkd. 1930;47:502–510. [Google Scholar]
- 7.Bertini E, Pepe G. Collagen type VI and related disorders: Bethlem myopathy and Ullrich scleroatonic muscular dystrophy. Eur J Paediatr Neurol. 2002;6:193–198. doi: 10.1053/ejpn.2002.0593. [DOI] [PubMed] [Google Scholar]
- 8.Timpl R, Chu ML. Microfibrillar collagen type VI. In: Mecham RP, editor. Extracellular matrix assembly and structure. Orlando: Academic Press; 1994. pp. 207–242. [Google Scholar]
- 9.Kuo HJ, Maslen CL, Keene DR, Glanville RW. Type VI collagen anchors endothelial basement membranes by interacting with type IV collagen. Journal of Biological Chemistry. 1997;272:26522–26529. doi: 10.1074/jbc.272.42.26522. [DOI] [PubMed] [Google Scholar]
- 10.Ishikawa H, Sugie K, Murayama K, Ito M, Minami N, Nishino I, et al. Ullrich disease: Collagen VI deficiency: EM suggests a new basis for muscular weakness. Neurology. 2002;59:920–923. doi: 10.1212/wnl.59.6.920. [DOI] [PubMed] [Google Scholar]
- 11.Pan TC, Zhang RZ, Sudano DG, Marie SK, Bonnemann CG, Chu ML. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet. 2003;73:355–369. doi: 10.1086/377107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lampe AK, Bushby KM. Collagen VI related muscle disorders. J Med Genet. 2005;42:673–685. doi: 10.1136/jmg.2002.002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camacho Vanegas O, Bertini E, Zhang RZ, Petrini S, Minosse C, Sabatelli P, et al. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci U S A. 2001;98:7516–7521. doi: 10.1073/pnas.121027598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higuchi I, Shiraishi T, Hashiguchi T, Suehara M, Niiyama T, Nakagawa M, et al. Frameshift mutation in the collagen VI gene causes Ullrich’s disease. Ann Neurol. 2001;50:261–265. doi: 10.1002/ana.1120. [DOI] [PubMed] [Google Scholar]
- 15.Baker NL, Morgelin M, Peat R, Goemans N, North KN, Bateman JF, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. 2005;14:279–293. doi: 10.1093/hmg/ddi025. [DOI] [PubMed] [Google Scholar]
- 16.Merlini L, Martoni E, Grumati P, Sabatelli P, Squarzoni S, Urciuolo A, et al. Autosomal recessive myosclerosis myopathy is a collagen VI disorder. Neurology. 2008;71:1245–1253. doi: 10.1212/01.wnl.0000327611.01687.5e. [DOI] [PubMed] [Google Scholar]
- 17.Lampe AK, Dunn DM, von Niederhausern AC, Hamil C, Aoyagi A, Laval SH, et al. Automated genomic sequence analysis of the three collagen VI genes: applications to Ullrich congenital muscular dystrophy and Bethlem myopathy. J Med Genet. 2005;42:108–120. doi: 10.1136/jmg.2004.023754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 19.Zimmerman JM, Eliezer N, Simha R. The characterization of amino acid sequences in proteins by statistical methods. J Theor Biol. 1968;21:170–201. doi: 10.1016/0022-5193(68)90069-6. [DOI] [PubMed] [Google Scholar]
- 20.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; 2005. [Google Scholar]
- 21.Lampe AK, Zou Y, Sudano D, O'Brien KK, Hicks D, Laval SH, et al. Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance. Hum Mutat. 2008;29:809–822. doi: 10.1002/humu.20704. [DOI] [PubMed] [Google Scholar]
- 22.Hicks D, Lampe AK, Barresi R, Charlton R, Fiorillo C, Bonnemann CG, et al. A refined diagnostic algorithm for Bethlem myopathy. Neurology. 2008;70:1192–1199. doi: 10.1212/01.wnl.0000307749.66438.6d. [DOI] [PubMed] [Google Scholar]
- 23.Petrini S, Tessa A, Stallcup WB, Sabatelli P, Pescatori M, Giusti B, et al. Altered expression of the MCSP/NG2 chondroitin sulfate proteoglycan in collagen VI deficiency. Mol Cell Neurosci. 2005;30:408–417. doi: 10.1016/j.mcn.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Ball S, Bella J, Kielty C, Shuttleworth A. Structural basis of type VI collagen dimer formation. J Biol Chem. 2003;278:15326–15332. doi: 10.1074/jbc.M209977200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Polarity scale of a 41 AA region around AA 830 (marked by arrow head) using the Zimerman-Eliezer-Simha algorithm in ProtScale [19] on the Expasy server [20]. Note change of polarity score around the mutation R830W.