

Abstract
SOX2 represents a High Mobility Group domain containing transcription factor that is essential for normal development in vertebrates. Mutations in SOX2 are known to result in a spectrum of severe ocular phenotypes in humans, also typically associated with other systemic defects. Ocular phenotypes include anophthalmia/microphthalmia (A/M), optic nerve hypoplasia, ocular coloboma and other eye anomalies. We screened 51 unrelated individuals with A/M and indentified SOX2 mutations in the coding region of the gene in 10 individuals. Seven of the identified mutations are novel alterations, while the remaining three individuals carry the previously reported recurrent 20-nucleotide deletion in SOX2, c.70del20. Among the SOX2-positive cases, seven patients had bilateral A/M and mutations resulting in premature termination of the normal protein sequence (7/38; 18% of all bilateral cases), one patient had bilateral A/M associated with a single amino acid insertion (1/38; 3% of bilateral cases), and the final two patients demonstrated unilateral A/M associated with missense mutations (2/13; 15% of all unilateral cases). These findings and review of previously reported cases suggest a potential genotype/phenotype correlation for SOX2 mutations with missense changes generally leading to less severe ocular defects. In addition, we report a new familial case of affected siblings with maternal mosaicism for the identified SOX2 mutation, which further underscores the importance of parental testing to provide accurate genetic counseling to families.
Keywords: Anophthalmia, microphthalmia, SOX2.
INTRODUCTION
Anophthalmia, defined as the absence of a globe in the orbit, and microphthalmia, defined as reduced size of the globe, are malformations of the eye seen in approximately 1–3/10,000 births. Anophthalmia and microphthalmia (A/M) are considered to represent a single continuum; the severity of microphthalmia varies significantly among affected individuals and clinical anophthalmia is believed to be the manifestation of extreme microphthalmia with ocular remnants visible only on imaging of the orbit. Chromosomal anomalies account for 23–30% of patients with A/M. Excluding those with chromosomal anomalies, 73–95% of individuals with A/M have other associated anomalies; only 20–45% of these can be attributed to a known syndrome [Clementi et al., 1996; Källén et al., 1996; Forrester and Merz, 2006].
Mutations in multiple genes have been implicated in A/M including SOX2, PAX6, SIX6, CHD7, CHX10, BCOR, RAX, PITX3, and OTX2 [Hever et al., 2006; Verma and Fitzpatrick, 2007]. Mutations in SOX2 appear to represent the most common cause of A/M. Previous studies have reported coding region mutations in 10–15% of individuals with bilateral severe eye malformations, including anophthalmia, microphthalmia, and coloboma[Fantes et al., 2003; Ragge et al., 2005]; one study also identified whole gene deletions in ~10% of patients who had severe A/M without coding region mutations in SOX2 [Bakrania et al., 2007].
Most of the SOX2 coding region mutations are represented by nucleotide substitutions or deletions/insertions resulting in a premature truncation of the normal protein. The majority of these truncating mutations are observed de novo in patients. Several missense mutations in SOX2 have also been identified and are predicted to affect the DNA-binding or transactivation domains of this protein, which are critical to its activity [Ragge et al, 2005; Faivre et al., 2006; Kelberman et al., 2006; Williamson et al., 2006; Sato et al., 2007; Wang et al., 2008]. Three familial cases of A/M with a SOX2 mutation (two truncating, one missense) identified in the proband and unaffected mother of affected siblings or half-siblings have been reported; maternal mosaicism was proposed for all three cases [Faivre et al., 2006; Chassaing et al., 2007; Schneider et al., 2008]. In addition, there have been three cases of milder eye defects associated with SOX2 missense mutations inherited from a father; two of the fathers were unaffected and the third had unilateral eye defects similar to those seen in his son [Kelberman et al., 2006; Wang et al., 2008].
The spectrum of phenotypes associated with SOX2 mutations ranges from severe forms of anophthalmia syndrome characterized by bilateral anophthalmia with mild dysmorphic facial features, genital anomalies in males, brain malformations, learning disabilities, and motor delay, to normal ocular phenotypes which may be observed in individuals with the same mutations [Zenteno et al., 2006; Chassaing et al., 2007]. Additional ocular features observed in SOX2-positive patients include cataract, coloboma, optic nerve hypoplasia, various forms of anterior segment dysgenesis, retinal and chorioretinal dystrophy, and high myopia [Kelberman et al., 2006; Bakrania et al., 2007]. The range of observed phenotypes is consistent with the critical role that Sox2 was shown to play in vertebrate embryonic development. Studies in animal models demonstrated that Sox2 deficiency leads to abnormal neurogenesis, defects in inner ear development, loss of pluripotency of embryonic stem cells [Ferri et al., 2004; Kiernan et al., 2005; Kelberman et al., 2006; Masui et al., 2007] and, most notably, a range of ocular phenotypes depending on the dosage of Sox2 in mice [Taranova et al., 2006].
In this report we present ten additional cases of mutations in SOX2 discovered in patients with anophthalmia/microphthalmia and a review of all SOX2 mutations reported to date. Our analysis suggests that missense mutations in SOX2 appear to result in milder phenotypes, similar to the dosage-dependent ocular phenotypes observed in mice. In addition, we report two new cases of familial anophthalmia associated with SOX2 mutations and parental mosaicism in one case.
MATERIALS AND METHODS
This human study was approved by the Institutional Review Boards of Children’s Hospital of Wisconsin and Albert Einstein Healthcare Network. Cases were identified and clinical data collected through the A/M Clinical Registry at Albert Einstein Medical Center, Philadelphia, or as part of the Genetic Studies of Human Developmental Disorders at the Medical College of Wisconsin, Milwaukee. Clinical data was assembled by review of medical records and/or clinical evaluation by one of the authors (AS).
DNA samples were extracted from blood or buccal samples obtained from 51 individuals with A/M. Of the 51 cases screened, 38 had bilateral A/M and 13 had unilateral A/M. The SOX2 coding region was amplified to produce two overlapping PCR products using the following primers: set 1 forward, 5′-AGTCCCGGCCGGGCCGAG-3′, and set 1 reverse, 5′-GGTAGCCCAGCTGGTCCTG-3′, and set 2 forward, 5′-CAAGACGCTCATGAAGAAGG-3′, and set 2 reverse, 5′-TACTCTCCTCTTTTGCACCC-3′. The PCR products were sequenced in both forward and reverse directions using Big Dye Terminator v3.1 with Applied Biosystems 3730 DNA sequencer. Chromatograms were examined manually and using Mutation Surveyor software (SoftGenetics). All initially identified changes were confirmed by additional independent PCR and sequencing experiments.
To obtain control data for normal variation in SOX2 in different ethnic populations, we obtained sequence data from 90 Caucasian, 87 African American and 90 Hispanic control individuals using the above approach. The control DNA samples were obtained from the Coriell Institute for Medical Research (Camden, NJ).
RESULTS
Sequencing of SOX2 in a cohort of 51 unrelated individuals affected with bilateral (38) or unilateral (13) anophthalmia/microphthalmia (Figure 1) revealed SOX2 mutations in ten patients (19.6%). These mutations were not seen in the ninety Caucasian, eighty-seven African American and ninety Hispanic control DNA samples. The phenotypic and genetic details of the affected families and identified mutations are presented below and in Table I and Figures 1 & 2.
Figure 1. Patient photographs.
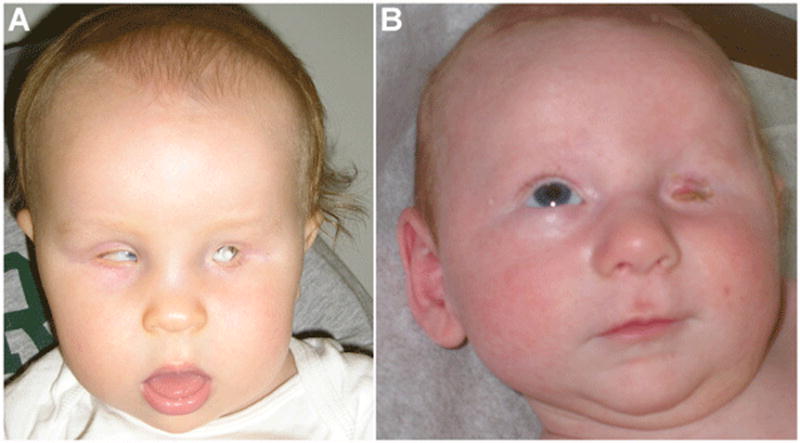
A) Patient 6 at 14 months of age with left anophthalmia (orbital expander in place) and right microphthalmia. B) Patient 9 at 2 months of age with left clinical anophthalmia and normal right eye.
Table I.
Summary of clinical features and SOX2 genotype for mutation-positive patients from this study.
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 | Patient 10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Female | Female | Male | Male | Male | Male | Male | Male | Male | Female |
| Race/ethnicity | Caucasian | Hispanic | Hispanic | Caucasian | Caucasian | Caucasian | Caucasian | Hispanic | Caucasian | Haitian (Black) |
| Age at eval | 2 | 2 | 9 | 2 | 7 | 4 | 8 | 9 | 3 | 3 |
| Ocular defects | ||||||||||
| Right | Anophthalmia | Severe Microphthalmia | Anophthalmia | Anophthalmia | Anophthalmia | Microphthalmia | Anophthalmia | Anophthalmia | Normal | Normal |
| Left | Microphthalmia, coloboma, glaucoma, cataract | Severe Microphthalmia | Anophthalmia | Anophthalmia | Anophthalmia | Anopthalmia | Anophthalmia | Anophthalmia | Anophthalmia | Severe Microphthalmia |
| Height | Unknown | 5th–10th | <3rd | 25th | <3rd | 10th–25th | 5th–10th | 3rd | 25th | 10th–25th |
| Motor Delay | Y | Y | Y | Y | Y | Y | Y | Y | Mild | N |
| Developmental Delay | Mild | Y | Autism Spectrum Disorder | Y | Y | Mild | Y | Autism Spectrum Disorder | Mild | N |
| Neuroimaging Results | Normal | Hamartoma of tuber cinereum | Normal | Normal | Cavum septum pellucidum, nonspecific periventricular white matter signal abnormality | Prominence of CSF spaces in posterior fossa; cavum septum pellucidum | Normal | Excess extra-axial fluid | Prominent lateral ventricles | Not available |
| Pituitary Anomalies | N | N | N | N | Abnormal configuration of pituitary and sellar structures | 9mm suprasellar mass posterior to pituitary stalk(likely benign teratoma) | N | Ectopic neurohypophysis underdeveloped pituitary, pan-hypopituitarism | N | N |
| Genital Anomalies | Vaginal adhesions | N | Micropenis, cryptorchidism | Foreskin adhesion | Micropenis | Micropenis, L cryptorchidism | N | N | Micropenis | N |
| Other | Small ears | Half-sister with unilateral anophthalmia and MR | Heart murmer | 2,3 toe syndactyly febrile seizures, mild pectus excavatum | Apnea, reflux, low growth hormone, mito accumulation on muscle bx | Seizure disorder, pyloric stenosis, feeding disorder | Cardiovascular defect, seizures, hernia | Acanthosis nigricans, TAb of half-brother with anophthalmia | Microcepahly2,3 toe syndactyly, hearing loss | Severe micrognathia |
| SOX2 mutation | ||||||||||
| nucleotide | c.16G>T | c.70del20 | c.70del20 | c.70del20 | c.487ins2 | c.540C>G | c.937del1 | c69ins3 | c.131C>G | c.859G>C |
| protein/effect | stop codon/trun-cation at 5 aa | frameshift/trun-cation at 23 aa | frameshift/trun-cation at 23 aa | frameshift/trun- cation at 23 aa | frameshift/trun-cation at 163 aa | stop codon/trun-cation at 180 aa | frameshift/trun-cation at 313 aa | 1 aa insertion, in frame/G23insG | missense/P44R | missense/A287P |
Figure 2.

A) Nucleotide sequences for the identified SOX2 mutations. Fragments of chromatograms obtained for different regions of the SOX2 gene using patient and parent (mosaic mother, family 2) DNA are shown. Mutation positions are indicated with red arrows. Mosaicism is demonstrated in the mother of Patient 2 by the small peaks corresponding to the frameshift at position 70 under the large peaks corresponding to the normal sequence, as opposed to the relatively equal peaks seen in Patient 2 with heterozygote status. The region corresponding to the start of the maternal mosaic sequence is enlarged for easier viewing. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.] B) Sequence alignment of vertebrate SOX2 proteins and human mutations. Human, mouse and frog sequences are shown; positions and details of identified human mutations are indicated at the bottom of the alignment; High Mobility Group (HMG) DNA-binding domain (comprising seventy-two amino acids) is shown in grey; regions of high homology shared by all Group B SOX proteins, SOX1-3, are marked with a black line at the top of the alignment. fs- frameshift, *stop codon, ins- insertion, pt- patient.
Patient 1 is a 2-year-old Caucasian female with right anophthalmia and left microphthalmia, coloboma, glaucoma, and cataract with some vision in the left eye. Clinical descriptions were obtained through available medical records. Growth parameters were not available for review. The patient had adhesions of the vagina treated with progesterone cream. She is noted to have small ears. Brain MRI, cardiac exam and renal ultrasound were normal. Speech was delayed with good comprehension and walking was late with a “drunken” gait. This patient was found to have a c.16G>T mutation (please see chromatogram in Figure 2A and mutation position in Figure 2B) which creates a stop codon and leads to truncation of the protein product at 5 amino acids. Parents have not been tested.
Patient 2 is a 2-year-3 month-old Hispanic female with bilateral severe microphthalmia. At 2 years 3 months of age, her weight was 13 kg (50th–75th centile), height was 84.5 cm (5th–10th centile) and head circumference 46.5 cm (10th–25th centile). Renal ultrasound was normal. MRI of the head revealed a hamartoma of the tuber cinereum. Motor development is delayed and she has a wide based stance; she crawled at 12 months and is not yet walking. She babbles, has better receptive than expressive language, and is friendly and interactive. Family history is remarkable for a 14 year old maternal half-sister with mental retardation and unilateral anophthalmia. There is no known consanguinity. The patient was found to have the previously reported c.70del20 mutation (Figure 2), a 20-nucleotide deletion, which results in frameshift and truncation of the protein product at 23 amino acids. The mother was found be mosaic for the c.70del20 mutation in three independent sequencing reactions and clinical testing confirmed that the affected sibling also carries this mutation.
Patient 3 is a 9-year-old Hispanic male with bilateral clinical anophthalmia and no vision. He was first evaluated at 10 months of age. At that time height and head circumference were below the 3rd centile (67.5 cm and 42 cm, respectively). Dysmorphic features including microcephaly, slightly cupped ears, mild dolicocephaly, lateral flaring of the eyebrows, and widely-spaced central incisors were noted. He has a single café-au-lait mark. He has micropenis and cryptorchidism. Cardiology evaluation is pending following the new observation of a grade 2/6 ejection murmur at the left sternal border. Head CT was normal. He has significant motor and cognitive delay and he has been diagnosed with autism spectrum disorder. Based on clinical examination, a possible diagnosis of Lenz syndrome was discussed with the family prior to SOX2 studies. This patient was also found to be heterozygous for the previously reported c.70del20 mutation (see above and Figure 2B). The patient’s mother was tested and does not carry this mutation. The patient’s father is unavailable for testing.
Patient 4 is an 8-year-old Caucasian male with bilateral clinical anophthalmia who was evaluated at 2 years 9 months of age. His weight was 25th–50th centile (13.7 kg), height was 25th centile (91.5 cm) and head circumference was 10th –25th centile (47.5 cm). Clinical examination revealed hypotonia, foreskin adhesions, bilateral cutaneous 2–3 toe syndactyly and mild pectus excavatum. Brain MRI was normal. He has significant global developmental delay. He was sitting and crawling at around 3 years of age and is still unable to walk. He has very limited speech and has been diagnosed with verbal apraxia. He has a history of febrile seizures which have resolved. This patient also carries the previously reported c.70del20 mutation (see above and Figure 2B). Both parents were tested clinically and are negative for the mutation seen in their son.
Patient 5 is an almost 7-year-old Caucasian male with bilateral anophthalmia. Clinical descriptions were obtained through available medical records. His height is below the 3rd centile (104.9 cm) with weight at the 3rd to 5th centile (17.9 kg) and he demonstrated low growth hormone levels (IGF-1 and IGF-binding protein 3). The patient has a history of micropenis at birth treated with testosterone. He has reflux and has been fed via gastrojejunal tube since age 15 months. In addition, he has had recurrent episodes of apnea. Muscle biopsy at 20 months of age showed mitochondrial accumulation with secondary glycogen and lipid accumulation. Brain MRI showed a cavum septum pellucidum, non-specific periventricular white matter signal abnormality, and abnormal configuration of the pituitary and sellar structures. He has significant global developmental delay; he rolls, but is unable to sit, crawl, or walk, and has limited speech. Muscle biopsy showed mitochondrial accumulation. This patient was found to have a c.487ins2 mutation (Figure 2) which leads to frameshift and truncation of the protein product at 163 amino acids. Both parents were tested and did not carry the mutation seen in their son.
Patient 6 is a 4-year-old Caucasian male with left anophthalmia and right microphthalmia (Figure 1A). Clinical descriptions were obtained through available medical records. Height and weight are in the normal range (97.5 cm at the 10th–25th centile and 16.6 kg at the 50th–75th centile, respectively). The patient has a history of micropenis at birth, treated with testosterone, and left undescended testicle repaired surgically. He has a history of pyloric stenosis and was fed via gastrostomy tube from age 2 months to 1 year due to poor oral intake. Renal ultrasound was normal. Brain MRI showed a 9 mm suprasellar mass posterior to the pituitary stalk (likely a benign teratoma), prominent cerebrospinal fluid spaces in the posterior fossa, and a cavum septum pellucidum. A tonic-clonic seizure disorder was diagnosed at age 3 years. His gross motor skills are significantly delayed; he first walked at 3½ years of age and currently takes only 8–10 steps independently. He also has mild delays in speech and fine motor milestones. This patient was found to have a c.540C>G mutation (Figure 2) which creates a stop codon and results in truncation of the protein product at 180 amino acids. Both parents were tested and were negative for the mutation seen in their son.
Patient 7 is an 8-year-10 month-old Caucasian male with bilateral anophthalmia. Clinical examination revealed normal growth: head circumference at the 10th–25th centile (51 cm), height at 5th–10th centile (122.4cm) and weight at 10th–25th centile (26.6kg). Dysmorphic features including short, narrow palpebral fissures, slightly cupped ears, maxillary overbite with crowded teeth and prominent central incisors, retrognathia, long chest, 5th finger clinodactyly and hyperextensible joints were noted. He has a deep sacral dimple with normal ultrasound of the lumbosacral spine. A spigelian hernia was noted. Echocardiogram shortly after birth revealed a patent ductus arteriosus, mild tricuspid regurgitation, atrial septum bowing into right atrium and moderately severe pulmonary hypertension. Cranial MRI was normal. He has a history of complex partial seizures. He has gross motor delay with balance difficulties, but can walk independently. His speech was significantly delayed and he continues to have an expressive communication disorder. This patient was found to be heterozygous for the c.937del1 (Figure 2), a 1-nucleotide deletion which causes frameshift and leads to truncation of the protein product at 313 amino acids followed by 57 erroneous residues due to frameshift. Parental samples were not available for testing.
Patient 8 is a 9-year-old Hispanic male with bilateral anophthalmia. His height is at the 3rd centile (11.8cm), weight is at the 3rd centile (20.5kg) and head circumference is at the 3rd–5th centile (50.4 cm), despite treatment with growth hormone. He demonstrated low growth hormone levels (IGF-1 and IGF-binding protein 3) at 1 year 6 months of age and has been treated since. Dysmorphic features including coarse facial features with deep set, widely spaced eyes, full eye brows, thick eye lashes, fleshy ear lobes, and a large mouth with widely spaced teeth. Acanthosis nigricans was noted. He has normal hearing but little conversational speech. MRI of the brain showed excess extra-axial fluid and an ectopic neurohypophysis with an underdeveloped pituitary gland. At approximately 7 years of age he was diagnosed with panhypopituitarism and began treatment with thyroxin in addition to growth hormone. He has significant insomnia, global developmental delay and autistic features. He walked at age 5½ years; currently gait is described as “shuffling” and he needs support when walking outside. The mother had a subsequent pregnancy, a half-sibling to the patient, with a prenatal diagnosis of a male fetus with right anophthalmia which was terminated. Autopsy revealed left retinal dysplasia and right microphthalmia with absence of the optic nerve. The patient was found to be heterozygous for a c.69ins3 mutation (Figure 2), an in-frame insertion adding a single glycine at position 23. Clinical testing of autopsy tissue performed on the affected half-sibling did not identify the mutation, but allele drop-out could not be ruled-out. Testing of a blood sample from the mother did not show any signs of mosaicism. The father is unavailable for testing.
Patient 9 is a 3-year-6-month-old Caucasian male with unilateral (left) clinical anophthalmia (Figure 1B). His current height and weight are in the normal range (96 cm at the 25th centile and 13.6 kg at the 10th centile, respectively), but his head circumference is less than the 3rd centile (47.4 cm). On clinical examination he was noted to have microcephaly, mild 2,3 toe syndactyly and sutural ridging. He has micropenis and a normal renal ultrasound. Brain MRI revealed that the lateral ventricles are mildly prominent with no evidence of hydrocephalus. Initial newborn hearing screening was normal but he has failed subsequent hearing screens. Speech and motor development are mildly delayed and he has an unusual gait. He walked at 20 months of age but continues to exhibit an unsteady gait. His receptive language skills have tested in the normal range but he exhibits about a 25% delay in expressive speech. Endocrinology evaluation was performed with no pituitary deficiency identified. The patient was found to be heterozygous for a c.131C>G mutation (Figure 2) that results in the substitution of arginine for proline at position 44. Both parents were tested and were negative for the mutation seen in their son.
Patient 10 is a 3-year-old Haitian (Black) female with unilateral (left) microphthalmia. She is adopted from Haiti and no medical history is available until she was taken to an orphanage at age 2 years. Her growth parameters are in the normal range with height of 90 cm (10th–25th centile), weight of 12.6 kg (10th–25th centile) and head circumference of 49.2 cm (25th–50th centile). Dysmorphic features include facial asymmetry and marked micrognathia with difficulty opening her mouth fully. She has had surgery to break and expand her jaw with follow-up stretching exercises. Teeth are unremarkable. She has a leg-length discrepancy of 3 cm on the left with an area of scar tissue on the thigh, thought to be due to a bone infection while in the orphanage. Renal ultrasound and cervical, thoracic and lumbar spine X-rays were normal; head imaging studies were not performed. Development is typical. She has no motor delays or ataxia and speaks in full sentences with no difficulties. This patient was found to have a heterozygous mutation, c.859G>C (Figure 2), a missense mutation which results in the substitution of proline for alanine at position 287. Parental samples were not available for testing.
DISCUSSION
Screening of SOX2 in fifty-one unrelated patients with bilateral or unilateral anophthalmia/microphthalmia identified mutations in ten individuals, 21% of bilateral A/M and 15% of unilateral A/M. Out of these ten mutations, seven represent new alterations in SOX2 with the c.16G>T substitution in Patient 1 being the most 5′ SOX2 mutation reported to date. Interestingly, despite a dramatic difference in the predicted effects on the amino acid composition of mutant proteins between Patient 1 (1.6% of the normal SOX2 protein) and Patient 7 (99% of the normal SOX2 protein), the phenotypes of both patients appear to be similar and severe, with bilateral anophthalmia/microphthalmia seen in both cases. The phenotypes of the other five patients with nonsense/frameshift mutations (Patients 2–6) also appear to be consistent with the previously reported cases with mutations resulting in truncation of the SOX2 protein (Table II). Of special interest is the 20-nucleotide deletion, c.70del20, which was identified in three unrelated families with severe microphthalmia/anophthalmia syndrome. This deletion has been previously reported and we present further evidence of its high recurrence. This deletion accounts for 30% of SOX2 mutations in our population and 18% of all SOX2 cases reported to date; this mutation alone explains 8% of bilateral anophthalmia/microphthalmia cases in this study and, therefore, development of additional diagnostic tests for an accurate detection of this deletion may be warranted.
Table II.
Genotype and phenotype information for all patients with SOX2 mutations reported to date.
| Reference | Mutation | R Eye | L Eye | Delay | Other | Family History |
|---|---|---|---|---|---|---|
| NONSENSE/FRAMESHIFT/TRUNCATION MUTATIONS | ||||||
| this study patient 1 | c.16G>T | AN | MI, CA, COL, GL | MD | GU | |
| Bakrania 2007 | c.53C>A | AN | AN | MD | EN, GI, MA | |
| Pedace 2009 | c.59_60insGG | AN | MI, COL | MD | GU | |
| Kelberman 2006 | c.60insG | AN | AN | GD | BA, EA, MA | Parents DNA normal |
| Ragge 2005 | c.67del23 | AN | AN | GD | BA, SS, SZ | Parents DNA normal |
| Chassaing 2007 | c.70del17 | AN | AN | EA | Mutation seen in affected sibling, unaffected mother (mosaicism proposed) | |
| Chassaing 2007 | c.70del17 | NOR | NOR | NOR | BA, VA | Sibling of above |
| Zenteno 2005 | c.70del20 | AN | AN | BA | ||
| Bakrania 2007 | c.70del20 | AN | ASD, CA, COL, GL | GD | EA, GU, TEF | |
| Bakrania 2007 | c.70del20 | AN | AN | NOR | ||
| Kelberman 2006 | c.70del20 | MI | AN | GD | BA, EA | Parents DNA normal |
| Zenteno 2006 | c.70del20 | NOR | AN | TEF, GU | Monozygotic twin below;Parents DNA normal | |
| Zenteno 2006 | c.70del20 | NOR | NOR | TEF | Monozygotic twin of above | |
| Kelberman 2008 | c.70del20 | AN | AN | NOR | EN | Parents DNA normal |
| this study patient 2 | c.70del20 | MI | MI | MD | BA | Half-sister is affected; unaffected mother carries mutation (mosaic) |
| this study patient 3 | c.70del20 | AN | AN | GD | GU, SS | Mother’s DNA normal |
| this study patient 4 | c.70del20 | AN | AN | GD | GU, SZ | Parents DNA normal |
| Williamson 2006 | c.163C>T | AN | AN | EA, GU, TEF | ||
| Kelberman 2008 | c.181C>T | AN | AN | GD | EN | Parents DNA normal |
| Bakrania 2007 | c.188delA | CA, COL, CR, ET, MC, MY, PU | MY, ONH, PU | NOR | BA | |
| Fantes 2003; Ragge 2005 | c.248C>A | AN | MI, CA, PU | GD | BA, MA, SZ | Parents DNA normal |
| Fantes 2003; Ragge 2005 | c.277G>T | AN | MI, SC | MD | MA | Parents DNA normal |
| Bakrania 2007 | c.285dupG | AN | AN | |||
| Zhou 2008 | c.310G>T | AN | AN | GD | BA, SS | |
| Kelberman 2006 | c.387delC | COL | MI | GD | BA, EA, GU, SD | Parents DNA normal |
| Hagstrom 2005 | c.463C>T | AN | AN | GD | BA, HL | Parents DNA normal |
| Kelberman 2006 | c.479delA | AN | AN | GD | BA, EA, GU, HL | Parents DNA normal |
| Bakrania 2007 | c.480C>G | MI | MI | GI, GU | ||
| Kelberman 2006 | C.480insA | MI | MI | GD | BA, EA, GU, MA | Parents DNA normal |
| this study patient 5 | c.487ins2 | AN | AN | GD | BA, GI, GU, MA, EN, SS | Parents DNA normal |
| Fantes 2003; Ragge 2005 | c.529C>T | AN | AN | GD | GU, MA, SZ, | Parents DNA normal |
| Fantes 2003; Ragge 2005 | c.529C>T | AN | AN | GD | HL, GU, MA, SZ | Parents DNA normal |
| Kelberman 2006 | c.529C>T | AN | AN | GD | EA, GU | Parents DNA normal |
| this study patient 6 | c.540C>G | MI | AN | MD | BA, GI, GU, SZ | Parents DNA normal |
| Schneider 2008; Zhou 2008 | c.551delC | AN | MI | GD | BA, EA, GI, GU, SZ, | Mutation seen in affected sibling, unaffected mother (mosaicism proposed) |
| Schneider 2008; Zhou 2008 | c.551delC | AN | AN | GD | BA, EA | Sibling of above |
| Ragge 2005 | c.628delA | AN | AN | NOR | BA, SZ | Parents DNA normal |
| this study patient 7 | c.937del1 | AN | AN | GD | CV, SZ | Parents DNA normal |
| Ragge 2005 | c.943del2 | AN | AN | GD | BA | Parents DNA normal |
| this study patient 8 | c.69ins3 | AN | AN | GD | BA, EN, SS | Affected half-brother;mother’s DNA normal |
| Total | 40 | AN=30 MI=5 O=2 NOR=3 | AN=27 MI=9 O=2 NOR=2 | |||
| GENE DELETION | ||||||
| Bakrania 2007 | 3′ deletion | AN | AN | NOR | ||
| Bakrania 2007 | deletion | AN | CA | GD | BA, GU, SZ | |
| Bakrania 2007 | deletion | MI, RD | MI, RD | GI | ||
| Bakrania 2007 | deletion | MI, RD | MI, RD | MD | SS | |
| Bakrania 2007 | deletion | NOR | AN | GD | EA, GU | |
| Bakrania 2007 | deletion | AN | MI, SC | |||
| Williamson 2006 | deletion | AN | AN | EA, GU, TEF | Parents DNA normal | |
| Fantes 2003; Ragge 2005 | deletion | AN | AN | GD | BA, MA, SS | Parents DNA normal |
| Kelberman 2008 | deletion | AN | MI | GD | BA, CV, EN | |
| Guichet 2004 | deletion | AN | AN | BA, EN, SS, GI | ||
| Total | 10 | AN=7 MI=2 N=1 | AN=5 MI=4 O=1 | |||
| MISSENSE MUTATIONS | ||||||
| this study patient 9 | c.131C>G (p.P44R) | NOR | AN | mild GD | BA, GU, HL | Parents DNA normal |
| Faivre 2006 | c.138T>G (p.N46K) | AN | AN | BA | Affected sibling; mutation seen in unaffected mother(mosaicism proposed) | |
| Williamson 2006 | c.221G>C (p.R74P) | MI | MI | EA, TEF | ||
| Sato 2007 | c.224T>A (p.L75Q) | AN | NOR | NOR | EN | |
| Ragge 2005 | c.290T>C (p.L97P) | MI, COL, SC | SC, AK | GD | MA, SZ | Parents DNA normal |
| Kelberman 2006 | c.389G>C (p.G130A) | ONH | ONH | BA | Mutation also seen in unaffected father | |
| Kelberman 2006 | c.389G>C (p.G130A) | NOR | NOR | NOR | Father of above | |
| Kelberman 2006 | c.571G>A (p.A191T) | ONH | ONH | BA, EA | Mutation also seen in unaffected father | |
| Kelberman 2006 | c.571G>A (p.A191T) | NOR | NOR | NOR | Father of above | |
| Wang 2008 | c.695C>A (p.T232N) | MC, IH, PU | COL | NOR | BA, GU | Mutation also seen in affected father |
| Wang 2008 | c.695C>A (p.T232N) | CA, COL | NOR | NOR | Father of above | |
| this study patient 10 | c.859G>C (p.A287P) | NOR | MI | NOR | ||
| Total | 12 | AN=2 MI=2O=4 NOR=4 | AN=2 MI=2O=4 NOR=4 | |||
AN, Anophthalmia; MI, Microphthalmia; AK, Aphakia; ASD, Anterior segment dysgenesis; CA, Cataract; COL, Coloboma; CR, Chorioretinal dystrophy; ET, Esotropia; GL, Glaucoma; IH, Iris hypoplasia; MC, Microcornea; MY, High myopia; ONH, Optic nerve hypoplasia; PU, Pupillary abnormality; RD, Retinal detachment; SC, Sclerocornea
BA, Brain anomalies; CV, Cardiovascular anomalies; EA, Esophageal atresia; EN, Endocrinologic anomalies; FD, Feeding disorder/reflux; GD, Global delay; GU, Genitourinary anomalies; HL, hearing loss; MA, Muscle abnormality; MD, Motor delay; NOR, Normal; SS, Short stature; SZ, Seizures; VA, Vertebral anomaly
The other identified changes include a one amino acid-insertion and two novel missense mutations. The one amino acid insertion involving glycine at position 23 in Patient 8, c.69ins3, may represent a new type of SOX2 mutation with a potentially unique mechanism. The normal human protein has 5 glycine residues in its N-terminal region; one additional residue is inserted in the mutant protein. This mutation was not found in any of the control samples of diverse ethnic origins including ninety individuals of Hispanic decent, matching this patient’s ethnicity. At the same time, this region does not appear to be conserved in different species as the mouse SOX2 protein contains seven and the Xenopus protein has two glycines in the same area. The proband was reported to have an affected half-sibling, but clinical genetic testing of the available autopsy tissue proved to be difficult and failed to produce a definitive answer in regards to presence/absence of this mutation in the half-sibling sample. Examination of a blood sample from the mother did not show any signs of mosaicism while the father is not available for testing. The c.69ins3 insertion may represent a causative or contributing mutation in this family or it could be a very rare polymorphism. A contributing mutation is not sufficient to cause a phenotype alone, but together with another mutation in a different gene, results in the observed disease. This type of digenic inheritance involving a gene which is typically implicated in autosomal dominant disease has been shown for the L185P mutation in RDS, which results in retinitis pigmentosa when combined with a heterozygous mutation in ROM1 (Kajiwara et al., 1994; Dryja et al. 1997). Functional studies of this mutation as well as additional screenings of the SOX2 gene in patients with A/M are likely to provide further insight into effects of this mutation.
Based upon their location, the missense changes in Patient 9 (P44R) and Patient 10 (A287P) are predicted to affect the high mobility group domain that mediates SOX2 binding to DNA and the C-terminal region involved in transactivation and interactions with co-activators and other proteins, respectively [Kamachi et al., 2000; Nowling et al., 2000]. Therefore, these changes are predicted to disrupt the regulatory activities of this transcription factor. Both the arginine at position 44 and the alanine at position 287 were found to be conserved in different vertebrate species, which further supports their importance for the normal functioning of this protein.
Six cases of inherited SOX2 mutations, three involving anophthalmia/microphthalmia, have been reported in the literature, with maternal mosaicism proposed for the families with A/M[Faivre et al., 2006; Kelberman et al., 2006; Chassaing et al., 2007; Schneider et al., 2008; Wang et al., 2008]. We report one additional case involving familial anophthalmia/microphthalmia associated with parental mosaicism (Family 2). This suggests a relatively high rate of familial SOX2 mutations and underscores the importance of careful analysis of parental samples whenever a SOX2 mutation is identified in a child.
Interestingly, missense mutations in our sample corresponded to less severe ocular phenotypes than nonsense or frameshift mutations. Both missense mutations were associated with unilateral anophthalmia and normal development of the contralateral eye. Analysis of the literature revealed that the association observed in our sample appears to be even more evident when all previously reported SOX2 mutations are taken into consideration (Table II). Out of forty reported nonsense or frameshift mutations, 89% of eyes were anophthalmic or microphthalmic, 5% demonstrated some other, milder, ocular phenotype, and 6% of eyes were normal; similar frequencies were observed for partial/complete gene deletions, 90%, 5% and 5%, correspondingly. In contrast, patients carrying missense mutations were equally likely to display A/M (33% of eyes), other phenotypes (33%) or normal ocular development (33%). In addition, patients with missense mutations demonstrated fewer other developmental or systemic abnormalities. In our sample, Patient 10 showed no developmental issues and Patient 9 exhibited only mild delays in motor and speech development, as opposed to the significant speech and motor delays typically described in SOX2 anophthalmia syndrome. Again, this correlation held true when analyzing previously reported SOX2 mutations with global delay noted in only one out of eight patients. Considering all patients with SOX2 mutations, patients with missense mutations averaged one additional major systemic feature, while patients with deletions, nonsense, or frameshift mutations averaged two additional major systemic features.
The phenotypic variability observed for SOX2 mutations may be explained by the critical and broad role of this factor in ocular development as well as certain unique features of this protein. The SOX proteins contain the highly conserved High Mobility Group (HMG) domain, which interacts with DNA, and a C-terminal transactivation domain, which cooperates with other proteins to activate expression of downstream genes. As many other HMG-containing proteins, SOX2 was shown to have low DNA-binding affinity and therefore requires additional proteins (co-DNA-binding factors) to facilitate this interaction and achieve efficient and specific binding to target DNA sites [Scaffidi and Bianchi, 2001]. It is plausible to assume that due to this required interaction with multiple additional factors to execute both its DNA-binding and transactivation functions, performance of the mutant SOX2 proteins may be greatly modified based upon these additional interactions. Missense mutations in SOX2 may affect certain interactions/functions of this protein to a greater extent than others and, therefore, may result in variable phenotypes based on the specific requirement/role of these interactions in eye development. Some of the identified members of the SOX2 pathway are also associated with A/M. The SOX2 and OTX2 proteins were recently shown to co-regulate expression of the RAX gene, thus connecting three known A/M genes in the same pathway [Danno et al., 2008].
Another possible explanation for the milder phenotypes seen in some patients is that missense mutations result in only partial loss-of-function and the less severe phenotypes may be due to a variable requirement of SOX2 for different developmental processes. A similar phenomenon was observed in animal models: Sox2-deficiency in mice has been shown to be associated with variable phenotypes (Kiernan et al., 2005; Kelberman et al., 2006; Taranova et al., 2006]. Most notably, Taranova et al. (2006) generated a Sox2 allelic series in the mouse and revealed dosage-dependent variation in ocular phenotypes. Variable expressivity of Sox2 deficiency in mice in relation to the ocular phenotype was also demonstrated by Kelberman and coauthors (2006) who reported anterior pituitary defects and male impaired fertility but normal eye development in mutant mice heterozygous for a Sox2 deletion.
Functional analyses of human SOX2 mutations may provide new insights into potential disease mechanisms as well as the normal function of this important transcription factor. Also, further studies involving examination of SOX2 in a variety of ocular phenotypes may identify additional mutations and help to define the role of this gene in human eye disease.
Acknowledgments
We are grateful to the families for their participation in this study. This project was supported by awards EY013606 and EY015518 from the National Eye Institute and Children’s Research Institute Foundation at Children’s Hospital of Wisconsin grant (EVS), Albert B. Millett Memorial Fund, A Mellon Mid-Atlantic Charitable Trust, Rae S. Uber Trust, A Mellon Mid-Atlantic Charitable Trust and Gustavus and Louis Pfeiffer Research Foundation (AS) and General Clinical Research Center grant M01 RR00058 from the National Center for Research Resources, NIH.
References
- Bakrania P, Robinson DO, Bunyan DJ, Salt A, Martin A, Crolla JA, Wyatt A, Fielder A, Ainsworth J, Moore A, Read S, Uddin J, Laws D, Pascuel-Salcedo D, Ayuso C, Allen L, Collin JR, Ragge NK. SOX2 anophthalmia syndrome: 12 new cases demonstrating broader phenotype and high frequency of large gene deletions. Br J Ophthalmol. 2007;91:1471–6. doi: 10.1136/bjo.2007.117929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing N, Gilbert-Dussardier B, Nicot F, Fermeaux V, Encha-Razavi F, Fiorenza M, Toutain A, Calvas P. Germinal mosaicism and familial recurrence of a SOX2 mutation with highly variable phenotypic expression extending from AEG syndrome to absence of ocular involvement. Am J Med Genet Part A. 2007;143A:289–91. doi: 10.1002/ajmg.a.31524. [DOI] [PubMed] [Google Scholar]
- Clementi M, Tenconi R, Bianchi F, Botto L, Calabro A, Calzolari E, Cianciulli D, Mammi I, Mastroiacovo P, Meli P, Spagnolo A, Turolla L, Volpato S. Congenital eye malformations: A descriptive epidemiologic study in about one million newborns in Italy. Birth Defects Orig Artic Ser. 1996;30:413–24. [PubMed] [Google Scholar]
- Danno H, Michiue T, Hitachi K, Yukita A, Ishiura S, Asashima M. Molecular links among the causative genes for ocular malformation: Otx2 and Sox2 coregulate Rax expression. Proc Nat Acad Sci. 2008;105:5408–5413. doi: 10.1073/pnas.0710954105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, Hahn LB, Kajiwara K, Berson EL. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1997;38:1972–82. [PubMed] [Google Scholar]
- Faivre L, Williamson KA, Faber V, Laurent N, Grimaldi M, Thauvin-Robinet C, Durand C, Mugneret F, Gouyon JB, Bron A, Huet F, Hayward C, Heyningen VV, Fitzpatrick DR. Recurrence of SOX2 anophthalmia syndrome with gonosomal mosaicism in a phenotypically normal mother. Am J Med Genet. 2006;140A:636–639. doi: 10.1002/ajmg.a.31114. [DOI] [PubMed] [Google Scholar]
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause anophthalmia. Nat Genet. 2003;33:461–3. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- Ferri AL, Cavallaro M, Braida D, Di Cristofano A, Canta A, Vezzani A, Ottolenghi S, Pandolfi PP, Sala M, DeBiasi S, Nicolis SK. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development. 2004;131:3805–19. doi: 10.1242/dev.01204. [DOI] [PubMed] [Google Scholar]
- Forrester MB, Merz RD. Descriptive epidemiology of anophthalmia and microphthalmia, Hawaii, 1986–2001. Birth Defects Res A Clin Mol Teratol. 2006;76:187–92. doi: 10.1002/bdra.20237. [DOI] [PubMed] [Google Scholar]
- Guichet A, Triau S, Lepinard C, Esculapavit C, Biquard F, Descamps P, Encha-Razavi F, Bonneau D. Prenatal diagnosis of primary anophthalmia with a 3q27 interstitial deletion involving SOX2. Prenat Diagn. 2004;24:828–32. doi: 10.1002/pd.997. [DOI] [PubMed] [Google Scholar]
- Hagstrom SA, Pauer GJ, Reid J, Simpson E, Crowe S, Maumenee IH, Traboulsi EI. SOX2 mutation causes anophthalmia, hearing loss, and brain anomalies. Am J Med Genet A. 2005;138:95–8. doi: 10.1002/ajmg.a.30803. [DOI] [PubMed] [Google Scholar]
- Hever AM, Williamson KA, van Heyningen V. Developmental malformations of the eye: the role of PAX6, SOX2 and OTX2. Clin Genet. 2006;69:459–70. doi: 10.1111/j.1399-0004.2006.00619.x. [DOI] [PubMed] [Google Scholar]
- Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994;264:1604–8. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- Källén B, Robert E, Harris J. The descriptive epidemiology of anophthalmia and microphthalmia. Int J Epidemiol. 1996;25:1009–16. doi: 10.1093/ije/25.5.1009. [DOI] [PubMed] [Google Scholar]
- Kamachi Y, Uchikawa M, Kondoh H. Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet. 2000;16:182–7. doi: 10.1016/s0168-9525(99)01955-1. [DOI] [PubMed] [Google Scholar]
- Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. 2006;116:2442–55. doi: 10.1172/JCI28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelberman D, de Castro SCP, Huang S, Crolla JA, Palmer R, Gregory JW, Taylor D, Cavallo L, Faienza MF, Fischetto R, Achermann JC, Martinez-Barbera JP, Rizzoti K, Lovell-Badge R, Robinson ICAF, Gerrelli D, Dattani MT. SOX2 plays a critical role in the pituitary, forebrain, and eye during human embryonic development. J Clin Endocrinol Metab. 2008;93:1865–73. doi: 10.1210/jc.2007-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan AE, Pelling AL, Leung KKH, Tang AS, Bell DM, Tease C, Lovell-Badge R, Steel KP, Cheah KS. SOX2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434:1031–1035. doi: 10.1038/nature03487. [DOI] [PubMed] [Google Scholar]
- Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K, Okochi H, Okuda A, Matoba R, Sharov AA, Ko MS, Niwa H. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol. 2007;9:625–35. doi: 10.1038/ncb1589. [DOI] [PubMed] [Google Scholar]
- Nowling TK, Johnson LR, Wiebe MS, Rizzino A. Identification of the transactivation domain of the transcription factor Sox-2 and an associated co-activator. J Biol Chem. 2000;275:3810–8. doi: 10.1074/jbc.275.6.3810. [DOI] [PubMed] [Google Scholar]
- Pedace L, Castori M, Binni F, Pingi A, Grammatico B, Scommegna S, Majore S, Grammatico P. A novel heterozygous SOX2 mutation causing anophthalmia/microphthalmia with genital anomalies. Eur J Med Genet. 2009;52:273–6. doi: 10.1016/j.ejmg.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Ragge N, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, Salt A, Collin JR, Vivian AJ, Free SL, Thompson P, Williamson KA, Sisodiya SM, van Heyningen V, Fitzpatrick DR. SOX2 Anophthalmia Syndrome. Am J Med Genet. 2005;135A:1–7. doi: 10.1002/ajmg.a.30642. [DOI] [PubMed] [Google Scholar]
- Sato N, Kamachi Y, Kondoh H, Shima Y, Morohashi K, Horikawa R, Ogata T. Hypogonadotropic hypogonadism in an adult female with a heterozygous hypomorphic mutation of SOX2. Eur J Endocrinol. 2007;156:167–71. doi: 10.1530/EJE-06-0606. [DOI] [PubMed] [Google Scholar]
- Scaffidi P, Bianchi ME. Spatially precise DNA bending is an essential activity of the sox2 transcription factor. J Biol Chem. 2001;276:47296–47302. doi: 10.1074/jbc.M107619200. [DOI] [PubMed] [Google Scholar]
- Schneider A, Bardakjian TM, Zhou J, Hughes N, Keep R, Dorsainville D, Kherani F, Katowitz J, Schimmenti LA, Hummel M, FitzPatrick DR, Young TL. Familial recurrence of SOX2 anophthalmia syndrome: Phenotypically normal mother with two affected daughters. Am J Med Genet A. 2008;146A:2794–8. doi: 10.1002/ajmg.a.32384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, Pevny LH. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006;20:1187–1202. doi: 10.1101/gad.1407906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma AS, FitzPatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007;2:47. doi: 10.1186/1750-1172-2-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Liang X, Yi J, Zhang Q. Novel SOX2 mutation associated with ocular coloboma in a Chinese family. Arch Ophthalmol. 2008;126:709–13. doi: 10.1001/archopht.126.5.709. [DOI] [PubMed] [Google Scholar]
- Williamson KA, Hever A, Rainger J, Rogers RC, Magee A, Fiedler Z, Keng WT, Sharkey FH, McGill N, Hill CJ, Schneider A, Messina M, Turnpenny PD, Fantes JA, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause Anophthalmia-Esophageal-Genital (AEG) syndrome. Hum Molec Genet. 2006;15:1413–1422. doi: 10.1093/hmg/ddl064. [DOI] [PubMed] [Google Scholar]
- Zenteno JC, Gascon-Guzman G, Tovilla-Canales JL. Bilateral anophthalmia and brain malformations caused by a 20-bp deletion in the SOX2 gene. Clin Genet. 2005;68:564–6. doi: 10.1111/j.1399-0004.2005.00518.x. [DOI] [PubMed] [Google Scholar]
- Zenteno JC, Perez-Cano HJ, Aguinaga M. Anophthalmia-Esophageal Atresia Syndrome Caused by a SOX2 Gene Deletion in Monozygotic Twin Brothers with Markedly Discordant Phenotype. Am J Med Gen. 2006;140A:1899–1903. doi: 10.1002/ajmg.a.31384. [DOI] [PubMed] [Google Scholar]
- Zhou J, Kherani F, Bardakjian TM, Katowitz J, Hughes N, Schimmenti LA, Schneider A, Young TL. Identification of novel mutations and sequence variants in the SOX2 and CHX10 genes in patients with anophthalmia/microphthalmia. Mol Vis. 2008;14:583–92. [PMC free article] [PubMed] [Google Scholar]