

Abstract
 A Pd-catalyzed Larock annulation provides expedient access to a subset of resveratrol-derived natural products. The reported approach resulted in the structural revision of an intermediate en route to the natural product pauciflorol F, the total synthesis of which proceeded in two steps from the requisite pentannulation product.
A Pd-catalyzed Larock annulation provides expedient access to a subset of resveratrol-derived natural products. The reported approach resulted in the structural revision of an intermediate en route to the natural product pauciflorol F, the total synthesis of which proceeded in two steps from the requisite pentannulation product.
Polyphenolic natural products such as resveratrol and its derivatives (Figure 1)1 have emerged as important synthetic targets due to their diverse architectures and their potential medicinal utility. Many of these compounds are produced by various plants in response to infection or stress.2 Recent reports have demonstrated that resveratrol inhibits cancer growth in vitro3 and extends the lifespan of living organisms, including fruit flies and the turquoise killifish.4 To date, the specific mechanism of action of resveratrol in humans remains elusive due to its rapid metabolism and relatively low bioavailability.5 Several dimeric resveratrol metabolites (e.g., 2-5, Figure 1), which presumably arise from radical dimerizations of 1, have shown biological activities comparable to or greater than their parent monomer.6 For example, a mixture of resveratrol dimers and oligostilbenes isolated from Caragana sinica exhibited in vitro stimulation of osteoblast proliferation at concentrations as low as 100 pg/mL.6d However, most biological studies of resveratrol and its congeners have concentrated on plant models. Consequently, relatively little is known about the potential benefits of these molecules to human health. A more thorough exploration of the properties, mechanism of action and biochemical significance of resveratrol and its derivatives requires efficient and versatile syntheses of these molecules and their unnatural analogs.
Figure 1.

Selected resveratrol-derived natural products
Several research groups have recently reported syntheses of resveratrol-derived compounds. In 2006, Li et al. employed the biomimetic dimerization of a resveratrol derivative for the total synthesis of quadrangularin A (2a, Figure 1).7a In the same year, She, Pan and coworkers reported the total synthesis of pauciflorol F by employing a Pd-catalyzed 5-endo Heck cyclization strategy.7b Snyder et al. subsequently reported the syntheses of several polyphenolic natural products, including 2a, 3, and 5.7c,d
As a part of a program to develop concise synthetic routes to resveratrol-derived compounds, we recently reported a domino Heck cyclization/pentannulation process, which provided the scaffold of a subset of dimeric resveratrol-derived compounds (Scheme 1).8 The single-pot preparation of the dimeric resveratrol framework (see 8) employed the Pd-catalyzed reaction of bromostilbene derivative 6 and tolane 7. Oxidative cyclization of 8 using iron (III) chloride provided pentalene 9, which is the core for pallidol (3) and related fused [3.3.0] bicyclic molecules.
Scheme 1.

Heck pentannulation cascade
We envisioned that compounds such as pauciflorol F (5, Scheme 2) could arise from a 2,3-disubstituted indenone precursor (e.g., 10a) using a related carbocyclization strategy. Encouraged by the wide substrate scope of Larock's Pd-catalyzed annulation of alkynes to form 2,3-disubstituted-1-indenones,9 we imagined obtaining 10a from o-bromobenzaldehyde 1110 and tolane 7.8
Scheme 2.

Retrosynthetic analysis of 5
When a mixture of 11 and 7 was subjected to Larock's original annulation conditions,9 a 1:1 mixture of indenone regioisomers 10a and 10b was obtained, which could be separated by column chromatography (Scheme 3).
Scheme 3.

Synthesis of indenones 10a and 10b
Regiocontrol in the Larock pentannulation to form indenones remains a challenge,11 as confirmed by our observations in the reaction of 7 and 11. However, either regioisomer (i.e., 10a or 10b) provides access to a resveratrol-derived compound (i.e., pauciflorol F, 5, or its regioisomer isopauciflorol F7d).
Our initial studies examined the conversion of 10a (Scheme 4) to the resveratrol-derived natural product pauciflorol F. Reduction of the indenone double bond was accomplished using heterogenous catalytic hydrogenation conditions (Pd/C, H2), which yielded cis indanone 12. Epimerization of the C(2) stereocenter of 12 was attempted using substoichiometric amounts of K2CO3 in a 3:1 mixture of MeOH and CH3CN. However, instead of the expected trans-indanone isomer (i.e., 14), the α-hydroxy indanone 13 was isolated. The structure of 13 was corroborated by IR, which showed an absorption band at 3434 cm−1 (consistent with an O-H stretch). Additionally, the 13C NMR spectrum of 13 showed a signal at 85.7 ppm, suggestive of a quaternary carbon bound to oxygen.
Scheme 4.

Hydrogenation studies of indenone 10a
Further support for α-hydroxy indanone 13 was obtained by a series of chemical conversions, as illustrated in Scheme 5. Reduction of the carbonyl group of 13 with NaBH4 furnished diol 15, whose relative stereochemistry was assigned by analogy to a previous report.12a Esterification of the secondary hydroxyl group of 15 with p-nitrobenzoylchloride gave 16 in 30% yield, along with unreacted 15. Alternatively, methanolysis of 1 3 in the presence of HBr gave a 1:1 mixture of 13a and 13.
Scheme 5.

Chemical transformations of 13
The ease with which α-ketol 13 forms from 12 under the indicated conditions is somewhat surprising, given that simple indanones seldom undergo rapid α-hydroxylation in air. However, perarylated indanones are known to readily undergo oxygenation upon formation of the corresponding enolate.12
We were successful in circumventing the formation of α-ketol 13 by performing the hydrogenation of 10a and subsequent epimerization at C(2) to yield 14 in one pot (Scheme 4), following the precedent of Zimmerman.13 This one-pot protocol avoided the exposure of the incipient indenolate to oxidants such as molecular oxygen, which may promote α-hydroxylation.
During the course of our studies, careful inspection of the literature describing the two existing total syntheses of pauciflorol F7b-d exposed some discrepancies regarding the previously established structural assignments of three compounds related to the natural product.
The first manifestation of an inconsistency arose from comparison of the previously reported 1H NMR data for permethylated pauciflorol F (14) and its cis-disposed analog (i.e., 12).7d It was reported that enolization of 1 4 with KHMDS and subsequent quenching with water afforded the product of kinetic proton capture (i.e., 12), drawing from an analysis of a related system by Zimmerman.13 However, the two doublets characteristic of the aliphatic hydrogens at C(2) and C(3) (see 14) are absent from the data reported for 12.7d Instead, two singlets are reported, one at 2.99 ppm (OH), which is shifted significantly upfield from what would be expected, and one at 4.62 ppm (CH). The spectral data associated with the compound reported as 12 were thus in good agreement with α-ketol 13.14
Interestingly, it was also indicated that treatment of the compound previously assigned as 12 (which we now believe to be 13) with BBr3 did effect global phenol deprotection,7d and yielded pauciflorol F (i.e., both 13 and 14 furnished pauciflorol F when treated with BBr3, Scheme 6), albeit in low yield.15 This surprising observation is also consistent with data presented by She, Pan and coworkers in their communication of the first total synthesis of pauciflorol F.7b Both reports suggested the feasibility of converting a compound consistent with 13 to pauciflorol F upon treatment with BBr3. This transformation would require both global methyl ether cleavage and reductive removal of the C(2) hydroxyl group. Although we have also been successful in accomplishing the conversion of 13 and 14 to 5 using the reported conditions,7b the reported yield for the conversion of 13 to 5 by She and Pan (87% yield) was far greater than what we obtained (trace amounts).
Scheme 6.

BBr3-mediated formation of pauciflorol F
To gain insight into the surprising conversion of 13 to 5 using BBr3 in the absence of an added reductant,16 we have conducted a series of studies to better understand this transformation, as illustrated in Scheme 7. The addition of Et3SiH significantly increases the efficiency of the transformation, suggesting an ionic reduction mechanism may be operative. Importantly, the use of 1 equiv of Et3SiH and excess BBr3 proved to be beneficial and converted 14 to pauciflorol F (5) in 65% yield.
Scheme 7.

Ionic reductions of 13
Additionally, studies of several α-ketol compounds (see 17-20, Figure 2) have revealed that the C(2) aryl substituent must possess a para-methoxy group (as in 17 and 18) in order for the ionic reduction to proceed. α-Ketols 17 and 18 yielded 87% and 95% of the corresponding indanones upon exposure to BBr3 and Et3SiH,17 whereas 19 and 20 were both unreactive under these conditions.
Figure 2.

α-Ketol model compounds
In conclusion, we have applied a palladium-catalyzed cascade reaction to the synthesis of the resveratrol-derived natural product pauciflorol F. The synthetic sequence is highly convergent and proceeds in three steps from tolane 7 and bromobenzaldehyde 11. This simple resveratrol-derived compound should serve as a building block for more complex, functionally diverse analogs. Additionally, we have revised the structural assignments for the precursors of permethylated pauciflorol F, which demonstrate that hydroxylation of perarylated indanones is a facile process. Current efforts are focused on the optimization of the regiocontrol in the Larock annulation and its application to the preparation of more complex resveratrol-derived natural products.
Supplementary Material
Acknowledgment
The authors are grateful to UC Berkeley, the NIH (NIGMS RO1 GM84906-01), and the American Cancer Society (RSG-09-017-01-CDD) for generous financial support. R.S. is a 2009 Alfred P. Sloan Foundation Fellow and a 2009 Camille Dreyfus Teacher-Scholar.
Footnotes
Supporting Information Available. Experimental details and characterization data for all new compounds are available free of charge via the Internet at http://pubs.acs.org
References
- 1.Takaoka M. Proc. Imp. Acad. Tokyo. 1940;16:405–407. [Google Scholar]
- 2.Langcake P, Pryce RJ. Physiol. Plant Pathol. 1976;9:77–86. [Google Scholar]
- 3.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CWW, Fong HHS, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 4.Valenzano DR, Terzibasi E, Genade T, Cattaneo A, Domenici L, Cellerino A. Curr. Biol. 2006;16:296–300. doi: 10.1016/j.cub.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 5.Bertelli AAE, Giovannini L, Stradi R, Bertelli A, Tillement JP. Int. J. Tiss. Reac. 1996;18:67–71. [PubMed] [Google Scholar]
- 6.a Ohyama M, Tanaka T, Iinuma M. Phytochemistry. 1995;38:733–740. [Google Scholar]; b Ohyama M, Tanaka T, Iinuma M. Chem. Pharm. Bull. 1994;42:2117–2120. [Google Scholar]; c Adesanya SA, Nia R, Martin TM, Boukamcha N, Montagnac A, Pais M. J. Nat. Prod. 1999;62:1694–1695. [Google Scholar]; d Luo H–F, Zhang L–P, Hu C–Q. Tetrahedron. 2001;57:4849–4854. [Google Scholar]; e Li WW, Ding LS, Li BG, Chien YZ. Phytochemistry. 1996;42:1163–1165. [Google Scholar]; f Tanaka T, Ito T, Nakaya K, Iinuma M, Riswan S. Phytochemistry. 2000;54:63–69. doi: 10.1016/s0031-9422(00)00026-1. [DOI] [PubMed] [Google Scholar]; g Coggon P, Janes NF, King TJ, Wallwork SC. J. Chem. Soc. 1965:406–408. [Google Scholar]; h Ito T, Tanaka T, Iinuma M, Iliya I, Nakaya K, Ali Z, Takahashi Y, Sawa R, Shirataki Y, Murata J, Darnaedi D. Tetrahedron. 2003;59:5347–5363. [Google Scholar]; i Supudompol B, Likhitwitayawuid K, Houghton PJ. Phytochemistry. 2004;65:2589–2594. doi: 10.1016/j.phytochem.2004.08.003. [DOI] [PubMed] [Google Scholar]; j Sahadin EH, Hakim LD, Juliawaty YM, Syah LBD, Ghisalberti EL, Latip J, Said IM, Achmad SAZ. Naturforsch., C: Biosci. 2005;60:723–727. doi: 10.1515/znc-2005-9-1011. [DOI] [PubMed] [Google Scholar]; k Ito T, Tanaka T, Iinuma M, Nakaya K, Takahashi Y, Sawa R, Murata J, Darnaedi D. J. Nat. Prod. 2004;67:932–937. doi: 10.1021/np030236r. [DOI] [PubMed] [Google Scholar]
- 7.For selected synthetic investigations of resveratrol-derived natural products, see: Li W, Li H, Li Y, Hou Z. Angew. Chem., Int. Ed. 2006;45:7609–7611. doi: 10.1002/anie.200603097.Bo C, Lu J-P, Xie X-G, She X-G, Pan X-F. Chin. J. Org. Chem. 2006;26:1300–1302.Snyder SA, Zografos AL, Lin Y. Angew. Chem., Int. Ed. 2007;46:8186–8191. doi: 10.1002/anie.200703333.Snyder SA, Breazzano SP, Ross AG, Lin Y, Zografos AL. J. Am. Chem. Soc. 2009;131:1753–1765. doi: 10.1021/ja806183r.
- 8.Jeffrey JL, Sarpong R. Tetrahedron Lett. 2009;50:1969–1972. doi: 10.1016/j.tetlet.2009.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larock RC, Doty MJ. J. Org. Chem. 1993;58:4579–4583. [Google Scholar]; For earlier examples of palladium-catalyzed syntheses of indenones, see also: Tao W, Silverberg LJ, Rheingold AL, Heck RF. Organometallics. 1989;8:2550–2559.Vicente J, Abad J–A, Gil-Rubio J. J. Organomet. Chem. 1992;436:C9–C12.Liebeskind LS, South MS. J. Org. Chem. 1980;45:5426–5429.
- 10.Mattson AE, Scheidt KA. J. Am. Chem. Soc. 2007;129:4508–4509. doi: 10.1021/ja068189n. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
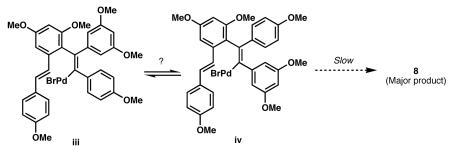
11.On the basis of our previous observations during the synthesis of 8 (ref. 8), we expected the reaction of 7 and 11 to proceed with good regiocontrol. The observed 1:1 regioselectivity in the formation of 10a and 10b may be the result of a fast cyclization of organopalladium intermediates i and ii (Eq. 1). If the analogous cyclization of organopalladium intermediates iii or iv is slow, a fast equilibration via de-carbopalladation may be favorable, which could have led to improved regioselectivity in the formation of 8.
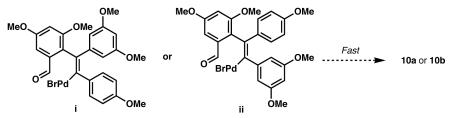
(1) 
(2) - 12.a Alesso EN, Finkielsztein LM, Lantaño B, Bianchi DE, Moltrasio Iglesias GY, Aguirre JM. Aust. J. Chem. 1997;50:149–152. [Google Scholar]; b Marco JL. Synth. Commun. 1996;26:4225–4231. [Google Scholar]
- 13.Zimmerman HE. J. Am. Chem. Soc. 1956;78:1168–1173. [Google Scholar]
- 14.For a detailed analysis and comparison of the 1H NMR spectral data for 12, 13 and 14, see the Supporting Information.
- 15.Following our own studies in this area, it came to our attention that the Snyder obtained low yields in the BBr3-mediated phenol deprotection of the compound we now believe to be 13. Personal communication with Prof. S. A. Snyder (Email correspondence on 9/16/09).
- 16.One possible mechanism for the reductive removal of the C(2) hydroxyl group of 13 involves a BBr3-mediated disproportionation.
- 17.For details, see the Supporting Information.


Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.