

Abstract
CTP:phosphocholine cytidylyltransferase (CCT), critical for phosphatidylcholine biosynthesis, is activated by translocation to the membrane surface. The lipid activation region of Caenorhabditis elegans CCT is between residues 246 and 266 of the 347 amino acid polypeptide, a region proposed to form an amphipathic alpha helix. When leucine 246, tryptophan 249, isoleucine 256, isoleucine 257, or phenylalanine 260, on the hydrophobic face of the helix, were changed individually to serine low activity was observed in the absence of lipid vesicles, similar to wild-type CCT, while lipid stimulated activity was reduced compared to wild-type CCT. Mutational analysis of phenylalanine 260 implicated this residue as a contributor to auto-inhibition of CCT while mutation of L246, W249, I256, and I257 simultaneously to serine resulted in significantly higher activity in the absence of lipid vesicles and an enzyme that was not lipid activated. These results support a concerted mechanism of lipid activation that requires multiple residues on the hydrophobic face of the putative amphipathic alpha helix.
Keywords: cytidylyltransferase, phosphatidylcholine, mutagenesis, amphipathic, CDP-choline, lipid activation, C. elegans
The enzyme CTP:phosphocholine cytidylyltransferase (CCT, EC 2.7.7.15) is present in higher organisms and is part of the CDP-choline pathway that results in the biosynthesis of the phospholipid phosphatidylcholine (PC) [1-4]. PC is a major component of eukaryotic cell membranes, as well as the most abundant lipid of serum lipoproteins and pulmonary surfactant. In addition, PC is a precursor to vital components of signal transduction pathways such as diacylglycerol and phosphatidic acid. The CDP-choline pathway for PC synthesis was discovered fifty years ago by Eugene Kennedy [5], who soon thereafter showed CCT associated with biological membranes [6]. CCT was first reported to be lipid activated by Fiscus and Schneider forty years ago [7]. Since the enzyme catalyzes a reaction that can serve as a point of regulatory control for the CDP-choline pathway it is extensively regulated at the cellular level. The regulation of CCT activity is central to a variety of cellular processes, including the cell cycle [8], cell death [9], and vesicular traffic [10,11]. In many cells, activation of CCT occurs simultaneously with the translocation of the enzyme from a soluble form to membrane-associated form. In vitro CCT is activated in the presence of lipid vesicles, providing a test tube model for membrane association.
Modern molecular biological techniques and genome sequencing projects have facilitated the acquisition of a tremendous amount of nucleotide sequence data. A search of nucleotide and protein databases reveals putative isoforms of CCT from a variety of eukaryotic organisms, including plants, mammals, yeast, Drosophila, C. elegans, and the malaria parasite Plasmodium falciparum. Higher organisms such as humans, rat, and insects contain two isoforms of CCT, denoted CTα and CTβ in mammals [12] and CCT1 and CCT2 in Drosophila [13,14]. C. elegans may contain three isoforms of CCT, as judged by a search of wormbase at www.wormbase.org. CCTα from rat, the most extensively characterized form of CCT, exhibits a modular domain structure, containing an amino-terminal nuclear localization sequence (amino acids 8-28), a central catalytic domain (amino acids 72-236), and carboxy-terminal lipid-binding and phosphorylation regions (amino acids 240-367) [1]. Present within the catalytic domain are several highly conserved sequence motifs containing amino acids involved in catalysis or substrate binding, including 89HxGH92 [15], 140RYVDEV145 [16], and 196RTEGxSTS203 [17], where numbering refers to the rat CCTα amino acid sequence. The central catalytic domains are highly conserved, while the regulatory regions N-terminal and C-terminal to the catalytic domain are highly divergent. The amino acid sequence in the putative lipid binding region of various CCTs is not highly conserved, preventing accurate prediction of individual amino acids critical for lipid activation. The sequence similarity between the catalytic domains of rat, Drosophila, yeast and C. elegans CCT is approximately sixty percent while the sequence conservation C-terminal to the catalytic domain is less than five percent (Fig. 1).
Fig. 1.

Amino acid sequence alignment of CCT from C. elegans, Drosophila melanogaster, rat, and the yeast Saccharomyces cerevisiae, forms of CCT known to be activated in vitro by lipid vesicles. The boxed region corresponds to amino acids 246-266 of C. elegans CCT. Black shading indicates identity in at least three of the four sequences. Grey shading indicates similar amino acids between proteins with K=R, L=I=V, W=F=Y, E=D, T=S, and A=G. Celeg is C. elegans CCT (accession NP_001033540), Dros2 is D. melanogaster CCT (accession NP_647622), Ratα is Rat CCTα (accession NP_511177), and Yeast is S. cerevisiae CCT (accession NP_011718).
Despite the lack of conservation of primary sequence in the putative lipid binding region, the enzymes from mammals [18,19], yeast [20,21], plants [22], Drosophila [13,14], C. elegans [23], and P. falciparum [24] are each activated by lipids. A comparison of previous lipid activation studies suggests that the concentration of lipid required for optimal activation of C. elegans CCT is significantly lower than mammalian CCTα [25-27]. Also, C. elegans CCT is very sensitive to lipid concentration and is inhibited at the higher concentration of lipid necessary for maximal activation of rat CCT [23]. If similarities exist in the mechanism of lipid activation of CCTs the conservation of primary sequence apparently is not required, suggesting that conservation of secondary structure may be important. Secondary structure predictions for the lipid binding region of rat CCTα predict a region of extensive alpha helix, from amino acids 237 to 293. In addition, the plotting of a helical wheel shows this helix to be amphipathic in nature, a structure postulated to function in lipid binding, resulting in activation of the enzyme [28]. In addition, the structure of peptides corresponding to the lipid binding region of rat CCT have been solved by NMR and shown to have extensive alpha helical character [29]. Using circular dichroism, the alpha helical character of the lipid binding region of rat CCTα was shown to increase upon association with lysophosphatidylcholine / phosphatidylglycerol mixed micelles, suggesting lipid association induces a shift in secondary structure, facilitating creation of an amphipathic helix whose hydrophobic face is inserted into the lipid bilayer [30].
Previous investigations have identified a lipid-binding domain in CCT from rat [31,32] and C. elegans [23] and have proposed this region is inhibitory in the absence of lipids, possibly due to direct interaction with the catalytic domain [26]. Localization of the lipid-binding region of C. elegans CCT to a twenty-one amino acid stretch was accomplished by characterizing a series of C-terminal truncation mutants. A truncated form of the 347 residue enzyme lacking amino acids beyond 225 or 245 was not activated in vitro by lipid vesicles while CCT truncated after amino acid 266, 281 or 319 was activated by lipid vesicles similar to wild-type enzyme [23]. Kinetic analysis revealed the lipid-independent CCT truncated after amino acid 245 had a kcat value 15-fold greater than either full-length CCT or CCT truncated after amino acid 266 in the absence of lipid. It was concluded, therefore, that elements critical for activation of C. elegans CCT by lipids are contained within amino acids 246 to 266. Consistent with previous observations with mammalian CCT, the region between 246 and 266 of C. elegans CCT was also predicted to form an amphipathic alpha helix, but the contribution of individual amino acids of the putative amphipathic alpha helix to lipid activation was not known.
The present research sought to investigate the contribution of individual amino acid side chains contained in the twenty-one residue putative amphipathic alpha helix of C. elegans CCT to activation in the presence of lipid vesicles as well as inhibition in the absence of added lipid. To provide insight into the mechanism of lipid activation leucine 246, tryptophan 249, isoleucine 256, isoleucine 257, and phenylalanine 260 on the hydrophobic face of the putative amphipathic alpha helix were changed individually and in combination to serine, a small amino acid with a polar hydroxyl group. After site-directed mutagenesis, expression, and purification the ability of each mutant enzyme to be activated by lipid vesicles was compared to wild-type C. elegans CCT.
Materials and methods
Chemicals and reagents
Escherichia coli strain DH10BAC, Bac-to-Bac baculovirus expression system, serum free Spodoptera frugiperda (Sf9) cells, Sf900II media, and Pfx DNA polymerase were from Invitrogen. Oligonucleotides were synthesized by Integrated DNA Technologies. Restriction endonucleases and T4 DNA ligase were from New England Biolabs. Amersham Biosciences was the source of [14C]phosphorylcholine chloride. Cytidine 5′-triphosphate (CTP), phosphocholine, oleate, phosphatidylcholine and protease inhibitors were obtained from Sigma. TALON metal affinity resin was from Clontech and BigDye sequencing kit from Applied Biosystems. Protein Assay kit was from Bio-Rad.
Site-directed mutagenesis
The plasmid pFASTBACHTa-CelCT, which contains the cDNA encoding 6×-His-tagged C. elegans CCT [23], was used as a template for the overlap extension method of site-directed mutagenesis [33]. This procedure consists of three PCR reactions utilizing four oligonucleotides primers. For mutagenesis of the cDNA encoding C. elegans CCT, primer 1 (5′-AAATATTCCGGATTATTC-3′) anneals to the bottom strand (non-coding strand) at the 5′ end of the cDNA. Primer 2 (5′-TAGCGGCATGGATCCTTTCCTCTTCCAGTGC-3′) anneals to the top strand (coding strand) near the 3′ end of the cDNA and contains 10 mismatched nucleotides at the 5′ end, which are underlined in the sequence. Primer 3 (5′-CTCTACAAATGTGGTATGGC-3′) anneals to a region of the top strand (coding strand) that is 3′ with respect to the region where primer 2 anneals. The fourth primer (primer M) contains the desired mutation. Primer M for L246S had the sequence 5′-GGGAATCGAGCTTTCCTCCACCTGGAAAAG-3′, for W249S 5′-CGAGCTTCTGTCCACCTCCAAAAGCAAATC-3′, for I256S 5′-GCAAATCAGACGATTCCATCCGGGACTTTATAG-3′, and for I257S 5′-GCAAATCAGACGACATCTCCCGGGACTTTATAG-3′. In each case the altered codon is underlined. For generating mutant L246S/W249S/I256S/I257S the oligonucleotide had the sequence 5′-GGGAATCGAGCTTTCCTCCACCTCCAAAAGCAAATCAGACGACTCCTCCCGGGACTT TATAG-3′. Mutation of phenylalanine 260 utilized an oligonucleotide with a mixture of G, A, T, and C at each position in the F260 codon. Mutant enzymes F260I, F260V, F260L, F260K, and F260D were selected for characterization.
PCR reaction #1 utilized primers 1 and 2 and resulted in a DNA fragment that has a mismatch at one end encoded in primer 2, preventing extension from its 3′ end in subsequent PCR reactions, providing the basis for selection of mutant CCT clones. PCR #2 utilized primer 3 and mutant primer M and resulted in a DNA fragment complementary to the PCR #1 fragment. After purification, the PCR #1 and PCR #2 products were combined in a third PCR reaction with primers 1 and 3 to produce the final mutant CCT construct. The PCR #3 product was ligated into expression vector pFASTBACHTa-CelCT using restriction endonucleases SalI and HindIII. DNA constructs were sequenced using automated dideoxy chain termination DNA sequencing.
Expression and purification of C. elegans CCT
A baculovirus expression system was previously optimized for expression of C. elegans CCT in Spodoptera frugiperda (Sf9) insect cells [23]. High-titer virus was used to infect one liter cultures of Sf9 cells at 1 × 106 cells/ml. Sf9 cells were harvested 48 hours post-infection for protein purification. C. elegans CCT was expressed with a 6×-His-tag at the amino terminus, incorporating an additional 28 amino acids encoded in pFASTBACHTa (MSYYHHHHHHDYDIPTTENLYFQGAMDP). Purification of 6×-His-tag enzymes utilized TALON Co2+ metal affinity column chromatography. Sf9 insect cells expressing the desired form of C. elegans CCT were collected, lysed by Dounce homogenization and centrifuged at 100,000 × g for 20 minutes. The Sf9 cell supernatant liquid was loaded directly onto a 3 ml column equilibrated with 20 mM Tris-Cl, pH 8.0, 100 mM NaCl. The column was washed with 10 column volumes of 20 mM Tris-Cl, pH 8.0, 500 mM NaCl, 200 mM galactose, 10 mM imidazole, 1% glycerol, 1% Nonidet P-40 followed by 10 column volumes of 20 mM Tris-Cl, pH 8.0, 100 mM NaCl, 10 mM imidazole. The enzyme was eluted from the metal affinity column with 20 mM Tris-Cl, pH 8.0, 100 mM NaCl, 150 mM imidazole. Fractions were collected into an equal volume of 20 mM Tris-Cl, pH 7.5, 300 mM NaCl, 2 mM DTT, 1 mM EDTA. Protein concentration was determined by the method of Bradford [34] using BSA as standard.
CCT enzyme assay
Activity of purified enzymes was determined with a charcoal binding assay that employs [14C]phosphocholine [35]. Enzyme (240 nanomolar final concentration) was incubated with 10 mM CTP and 4 mM [14C]phosphocholine for 15 minutes at 37°C and reactions were stopped by the addition of 10% TCA containing 150 mM unlabeled phosphocholine. Reaction mixtures were incubated on ice with an equal volume of 0.5 mg/ml charcoal for one hour. After two washes with 0.6 ml water, bound product, [14C]CDP-choline, was quantitated by liquid scintillation counting. Assays contained various amounts of lipid vesicles comprised of a 1:1 molar ratio of PC:oleate, as indicated.
Circular dichroism
Circular dichroism was conducted at 25°C using a AVIV model 215 CD spectrometer. Scans were run three times and averaged. Data were acquired from 190 nm to 260 nm at 1 nm intervals in a 1 mM rectangular quartz cuvette. Protein samples were 2.4 micromolar in 50 mM potassium phosphate pH 7.
Results and discussion
Working model for CCT activation by lipid vesicles
A major cellular control point regulating flux through the CDP-choline pathway is alteration of the location and activity of the enzyme CTP:phosphocholine cytidylyltransferase (CCT). The working hypothesis is that the lipid binding region interacts with the catalytic domain and is inhibitory in the absence of lipid vesicles. Interaction with the cellular membrane induces a conformational change that removes the inhibitory region and activates the enzyme. The lipid binding region of C. elegans CCT, however, may be necessary for the enzyme to adopt its optimal, fully activated structure since removal of the lipid binding region, amino acids beyond 245 of C. elegans CCT, results in a lipid-independent enzyme that catalyzes the reaction less efficiently than wild-type CCT [23]. Therefore it is possible that the region of CCT that supplies the amino acids that cause inhibition in the absence of lipid also provide the elements necessary for activation upon membrane association. If this is the case, then construction of a lipid-independent mutant enzyme that has the same catalytic efficiency as wild-type enzyme would not be possible.
Since previous investigations suggest an entire helical hydrophobic face must be inserted into the membrane for activation of CCT [30] it is likely that multiple hydrophobic amino acids of the putative amphipathic alpha helix are critical for lipid activation. The relative contribution of individual hydrophobic amino acids to membrane binding, however, is not known, providing a rationale for the present study in which residues on the hydrophobic face of the putative amphipathic alpha helix are mutated individually. Previous investigations have also suggested that one or more amino acids within the membrane binding region may play a role in auto-inhibition of CCT in the absence of lipids through interaction with the catalytic domain [26]. If a single amino acid mediates inhibition of CCT then mutation of this amino acid would result in a form of CCT that has increased activity in the absence of lipid when compared to wild-type CCT. However, the possibility exists that amino acids of CCT that interact with the catalytic domain also associate with lipid. In this case the mutant enzymes may be constitutively active and possess significant activity in the absence of lipid but would not be activated by lipid to the same extent as wild-type CCT. In this study forms of CCT were characterized in which a single amino acid or multiple amino acids on the hydrophobic face of the amphipathic alpha helix were mutated to shed light on the mechanism of CCT activation by lipids.
Leucine 246, tryptophan 249, isoleucine 256, and isoleucine 257 contribute to membrane binding
Wild-type C. elegans CCT exhibited an average activity of 400 ± 150 nmoles CDP-choline / min / mg in the absence of added lipid vesicles and was activated approximately 20-fold in the presence of 5 μM PC:oleate vesicles (1:1 molar ratio) (Fig 2). Leucine 246, tryptophan 249, isoleucine 256, isoleucine 257, and phenylalanine 260, amino acids on the hydrophobic face of the putative amphipathic alpha helix, were mutated individually to serine to determine the contribution of each residue to lipid activation. Mutant enzyme W249S was activated by lipid to a lesser extent than wild-type and catalyzed the reaction at 10% of the wild-type rate. Previous analyses suggest that lipid activation is well correlated with membrane binding, thus enzyme activation was used as an indirect read-out of the CCT - membrane binding event [32,36]. The data, therefore, suggest W249 may play a greater role in membrane association than in inhibition of the catalytic domain since removal of this amino acid results in an enzyme with decreased activity in the presence of lipid. Mutating amino acids L246, I256, or I257 resulted in enzymes that retained between 25% and 40% of the wild-type activity in the presence of optimal concentrations of lipid. In the case of W249S, L246S, I256S, and I257S activity in the absence of lipid was still low, comparable to wild-type CCT activity without added lipid vesicles.
Fig. 2.
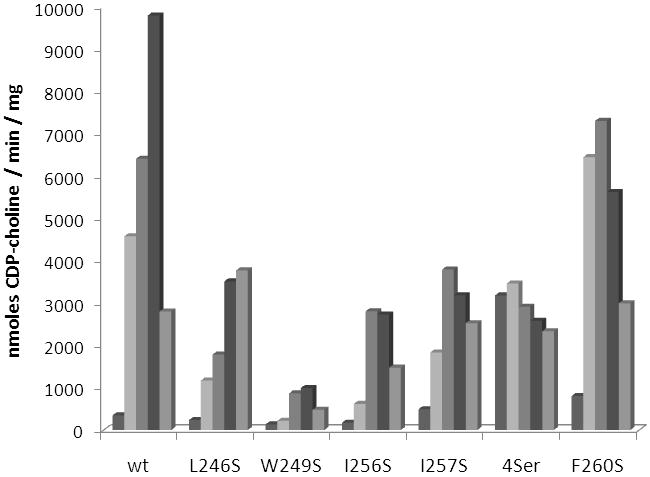
Activity of wild-type and mutant forms of C. elegans CCT. Shown is the activity of each enzyme in the presence of increasing concentration of PC:oleate vesicles. Concentrations, from left to right, are 0 μM, 2.5 μM, 5 μM, 10 μM, and 20 μM PC:oleate. 4Ser denotes a mutant enzyme where leucine 246, tryptophan 249, isoleucine 256, and isoleucine 257 were replaced simultaneously by serine. Experiments were repeated using similar lipid concentrations and different preparations of enzyme. Data are representative of the trends consistently observed.
Phenylalanine 260 plays a role in auto-inhibition of catalysis
The most active of the mutant enzymes was F260S, which displayed maximal activity approximately 75% of the wild-type maximal activity and had more than 2-fold greater activity in the absence of lipid (Fig. 2). These data suggest phenylalanine 260 is more involved in inhibiting the catalytic domain and not as critical for membrane association. To further explore the role of F260 additional mutations were made at this position (Fig 3). Phenylalanine 260 was replaced by isoleucine, valine, leucine, lysine, and aspartate. If the hydrophobic nature of the phenylalanine side chain is mediating inhibition of CCT in the absence of lipid, possibly through association with the catalytic domain, then introduction of a hydrophilic or charged amino acid side chain at that position should result in a mutant enzyme that has increased activity in the absence of lipid. Without added lipid vesicles mutant enzymes F260I, F260V, and F260L did not have activity significantly different than wild-type enzyme. However, F260K and F260D possessed 18-fold and 38-fold greater activity than wild-type C. elegans CCT in the absence of lipid vesicles while still retaining activity similar to wild-type enzyme in the presence of 5 μM lipid vesicles. The significant increase in activity in the absence of lipid upon introduction of a charged amino acid at position 260 supports the hypothesis that the hydrophobic character of F260 is contributing to inhibition of the catalytic domain in the absence of lipid.
Fig. 3.
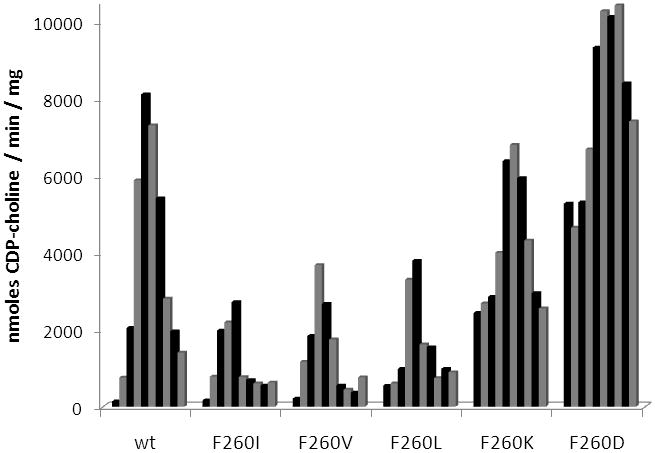
Activity of wild-type C. elegans CCT and phenylalanine 260 mutant enzymes. Shown is the activity of each enzyme in the presence of increasing concentration of PC:oleate vesicles. Concentrations, from left to right, are 0 μM, 0.5 μM, 1 μM, 3 μM, 5 μM, 10 μM, 15 μM, 20 μM, 25 μM, and 30 μM PC:oleate. Experiments were repeated using similar lipid concentrations and different preparations of enzyme. Data are representative of the trends consistently observed.
Lipid activation requires multiple hydrophobic amino acids
When amino acids 246 - 266 are modeled as an idealized helix the amphipathic nature of this segment is apparent (Fig. 4). Amino acids W249, L246, I256, I257, and F260 occur in the primary sequence with a periodicity that results in clustering of hydrophobic side chains on one face of the alpha helix. This secondary structure element has been postulated to mediate interaction with membrane lipids in rat CCT [28,37,38].
Fig. 4.
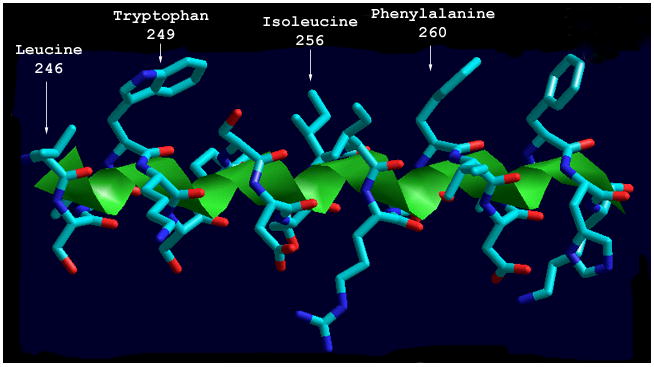
Structural model of the putative amphipathic alpha helix of C. elegans CCT. Amino acids 246-266 of C. elegans CCT were modeled as a three-dimensional amphipathic alpha helix. Shown is a side view of the amphipathic alpha helix with hydrophobic amino acid side chains projected up. The figure was generated using Hyperchem (Hypercube, Inc.).
An original goal of this study was to produce a lipid-independent C. elegans CCT by site-directed mutagenesis similar to the truncation mutants characterized previously [23]. Since single amino acid changes at positions 246, 249, 256, and 257 resulted in small changes in lipid activation, the next approach was to characterize a mutant enzyme with all four amino acids replaced by serine. It was thought that each of the changes seen with single mutant enzymes may be additive and multiple mutations may be sufficient to produce a full-length lipid-independent form of CCT. Upon mutation of all four amino acids (L246, W249, I256, and I257) to serine C. elegans CCT indeed was no longer activated by the addition of lipid vesicles (Fig. 2). However, the activity in the presence of lipid was lower than wild-type enzyme, approximately 35%, similar to what was seen previously for the truncation mutant that removed amino acids beyond 245 [23]. These data support the notion that the inhibitory region of CCT may also function as an activator region, and removal of inhibitory amino acids will not only relieve inhibition in the absence of lipid vesicles but will also prevent full activation in the presence of lipids.
To address whether the overall fold of the quadruple serine mutant was altered relative to wild-type C. elegans CCT circular dichroism (CD) spectrometry was conducted. The CD spectrum of the mutant enzyme in which L246, W249, I256, and I257 were each mutated to serine was essentially identical to wild-type enzyme (Fig. 5), suggesting the overall folding of the mutant enzyme was not significantly altered.
Fig. 5.
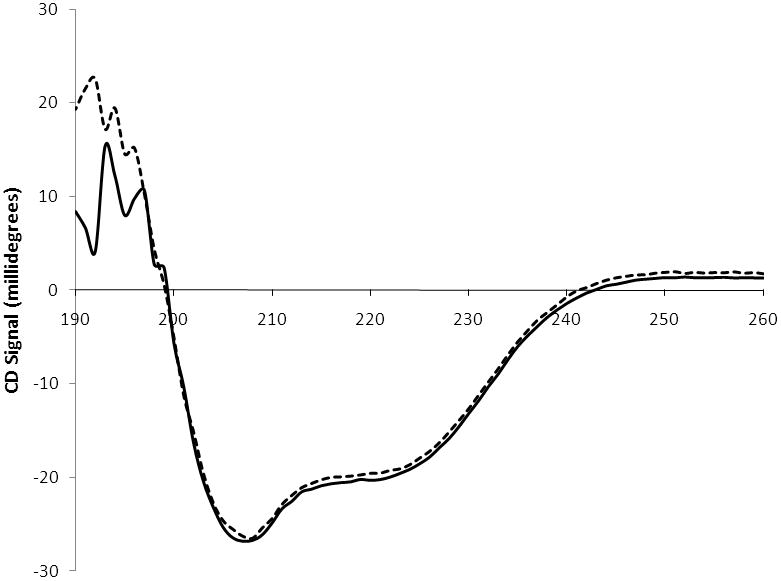
Circular dichroism spectra. A CD spectrum is shown for wild-type C. elegans CCT (solid line) and a mutant enzyme in which L246, W249, I256, and I257 were mutated simultaneously to serine (dashed line). Data were acquired at 1 nm intervals and spectra represent the accumulation of 3 scans.
Conservation of hydrophobic amino acids
It would be useful to correlate the data from this study of a nematode CCT to mammalian CCT isoforms. It is difficult to conclude whether the mechanism of lipid activation of C. elegans CCT is similar to mammalian CCTs since C. elegans CCT requires less lipid for activation and is inhibited at higher concentrations of lipid necessary to activate mammalian CCTs [23]. As shown in Fig. 1, lack of amino acid sequence identity beyond the catalytic domain of CCT from C. elegans, rat, Drosophila, and yeast makes it difficult to predict amino acids critical for lipid activation. Nonetheless, analysis of amino acid sequence of rat and yeast CCT beyond the catalytic domain reveals putative amphipathic alpha helices similar to C. elegans CCT amino acids 246-266. A sequence alignment compiled using amino acid sequences of CCTs from mammals, the fruit fly, fungi, plants, and a protozoan indicates each contains a region C-terminal to the catalytic domain that illustrates the periodic conservation of hydrophobicity indicative of an amphipathic alpha helix, suggesting they are lipid-activated CCTs (Fig. 6). The mammalian sequences analyzed were unique in that they contained a much longer amphipathic alpha helical region of approximately 45 amino acids, shown as two separate halves in Fig. 6. It is likely that there is a necessary amphipathic alpha helix length as well as a minimal hydrophobic binding energy required for CCT to bind to a membrane. The demonstration that removal of just four hydrophobic amino acids abolished lipid activation does not necessarily mean that these residues are sufficient to mediate lipid activation. A reasonable assumption may be that replacing two isoleucines, a leucine, and a tryptophan with serines lowers the binding energy below the threshold required for efficient membrane interaction.
Fig. 6.

Putative amphipathic alpha helices of various CCT isoforms. Hydrophobic amino acids that plot on the hydrophobic face of the putative amphipathic alpha helix are boxed in black. The serine residues boxed in grey also plot on the hydrophobic face of the alpha helix. Rat and human CCTs contain an extended putative alpha helix with sequence repeats that is depicted in two halves. Celeg is C. elegans CCT (accession NP_001033540), Dros2 is D. melanogaster CCT2 (accession NP_647622), Dros1 is D. melanogaster CCT1 (accession NP_728628), Ratα is rat CCTα (accession NP_511177), Ratβ is rat CCTβ (accession NP_775174), Humanα is human CCTα (accession NP_005008), Humanβ is human CCTβ (accession NP_004836), Yeast is S. cerevisiae CCT (accession NP_011718), Pfalc is P. falciparum CCT (accession P49587), Ecun is E. cuniculi CCT (accession NP_586276), pea is P. sativum CCT (accession CAA70317), rapeseed is B. napus CCT (accession BAA09642), and Athal is A. thaliana CCT (accession NP_193249).
Summary and conclusion
Ever since the initial report more than forty years ago that CCT is lipid activated [7] the mechanism of this regulatory process has been intently studied. Prior research has localized specific regions of primary structure as critical for lipid binding [23,31,32] and provided the structure of peptides corresponding to the lipid binding region [29]. Investigations have implicated an amphipathic alpha helix that inserts into the hydrophobic lipid bilayer as central to membrane interaction [30]. In this study the potential role of individual amino acids within the putative amphipathic alpha helix of C. elegans CCT has been investigated. Results support the hypothesis that multiple amino acids contained within the hydrophobic face of a putative amphipathic alpha helical region that includes Leu246, W249, I256, and I257 are required for membrane binding and activation upon translocation to the membrane. In addition, Phe260 on the hydrophobic face of the putative amphipathic alpha helix in C. elegans CCT appears to play a role in mediating inhibition of catalysis of the soluble form of the enzyme.
Acknowledgments
This work was supported by National Institutes of Health grant R15 GM064512.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kent C. Biochim Biophys Acta. 1997;1348:79–90. doi: 10.1016/s0005-2760(97)00112-4. [DOI] [PubMed] [Google Scholar]
- 2.Kent C. Biochim Biophys Acta. 2005;1733:53–66. doi: 10.1016/j.bbalip.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Cornell RB, Northwood IC. Trends Biochem Sci. 2000;25:441–447. doi: 10.1016/s0968-0004(00)01625-x. [DOI] [PubMed] [Google Scholar]
- 4.Jackowski S, Fagone P. J Biol Chem. 2005;280:853–856. doi: 10.1074/jbc.R400031200. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy EP, Weiss SB. J Biol Chem. 1956;222:193–214. [PubMed] [Google Scholar]
- 6.Wilgram GF, Kennedy EP. J Biol Chem. 1963;238:2615–2619. [PubMed] [Google Scholar]
- 7.Fiscus WG, Schneider WC. J Biol Chem. 1966;241:3324–3330. [PubMed] [Google Scholar]
- 8.Jackowski S. J Biol Chem. 1994;269:3858–3867. [PubMed] [Google Scholar]
- 9.Baburina I, Jackowski S. J Biol Chem. 1998;273:2169–2173. doi: 10.1074/jbc.273.4.2169. [DOI] [PubMed] [Google Scholar]
- 10.Skinner HB, McGee TP, McMaster CR, Fry MR, Bell RM, Bankaitis VA. Proc Natl Acad Sci USA. 1995;92:112–116. doi: 10.1073/pnas.92.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feldman DA, Weinhold PA. J Biol Chem. 1998;273:102–109. doi: 10.1074/jbc.273.1.102. [DOI] [PubMed] [Google Scholar]
- 12.Lykidis A, Murti KG, Jackowski S. J Biol Chem. 1998;273:14022–14029. doi: 10.1074/jbc.273.22.14022. [DOI] [PubMed] [Google Scholar]
- 13.Helmink BA, Friesen JA. Biochim Biophys Acta. 2004;1683:78–88. doi: 10.1016/j.bbalip.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Tilley DM, Evans CR, Larson TM, Edwards KA, Friesen JA. Biochemistry. 2008;47:11838–11846. doi: 10.1021/bi801161s. [DOI] [PubMed] [Google Scholar]
- 15.Veitch DP, Gilham D, Cornell RB. Eur J Biochem. 1998;255:227–234. doi: 10.1046/j.1432-1327.1998.2550227.x. [DOI] [PubMed] [Google Scholar]
- 16.Weber CH, Park YS, Sanker S, Kent C, Ludwig ML. Structure. 1999;7:1113–1124. doi: 10.1016/s0969-2126(99)80178-6. [DOI] [PubMed] [Google Scholar]
- 17.Helmink BA, Braker JD, Friesen JA. Biochemistry. 2003;42:5043–5051. doi: 10.1021/bi027431+. [DOI] [PubMed] [Google Scholar]
- 18.Choy PC, Lim PH, Vance DE. J Biol Chem. 1977;252:7673–7677. [PubMed] [Google Scholar]
- 19.Kalmar GB, Kay RJ, LaChance AC, Cornell RB. Biochim Biophys Acta. 1994;1219:328–334. doi: 10.1016/0167-4781(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 20.Johnson JE, Kalmar GB, Sohal PS, Walkey CJ, Yamashita S, Cornell RB. Biochem J. 1992;285:815–820. doi: 10.1042/bj2850815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friesen JA, Park YS, Kent C. Prot Expr Purif. 2001;21:141–148. doi: 10.1006/prep.2000.1354. [DOI] [PubMed] [Google Scholar]
- 22.Kinney AJ, Moore TS., Jr Arch Biochem Biophys. 1987;259:15–21. doi: 10.1016/0003-9861(87)90464-4. [DOI] [PubMed] [Google Scholar]
- 23.Friesen JA, Liu MF, Kent C. Biochim Biophys Acta. 2001;1533:86–98. doi: 10.1016/s1388-1981(01)00145-7. [DOI] [PubMed] [Google Scholar]
- 24.Yeo HJ, Larvor MP, Ancelin ML, Vial HJ. Biochem J. 1997;324:903–910. doi: 10.1042/bj3240903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feldman DA, Rounsifer ME, Weinhold PA. Biochim Biophys Acta. 1985;833:429–437. doi: 10.1016/0005-2760(85)90100-6. [DOI] [PubMed] [Google Scholar]
- 26.Friesen JA, Campbell HA, Kent C. J Biol Chem. 1999;274:13384–13389. doi: 10.1074/jbc.274.19.13384. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, MacDonald JIS, Kent C. J Biol Chem. 1993;268:5512–5518. [PubMed] [Google Scholar]
- 28.Cornell RB. Biochem Soc Trans. 1998;26:539–544. doi: 10.1042/bst0260539. [DOI] [PubMed] [Google Scholar]
- 29.Dunne SJ, Cornell RB, Johnson JE, Glover NR, Tracey AS. Biochemistry. 1996;35:11975–11984. doi: 10.1021/bi960821+. [DOI] [PubMed] [Google Scholar]
- 30.Taneva S, Johnson JE, Cornell RB. Biochemistry. 2003;42:11768–11776. doi: 10.1021/bi035234k. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Kent C. J Biol Chem. 1995;270:18948–18952. doi: 10.1074/jbc.270.32.18948. [DOI] [PubMed] [Google Scholar]
- 32.Cornell RB, Kalmar GB, Kay RJ, Johnson MA, Sanghera JS, Pelech SL. Biochem J. 1995;310:699–708. doi: 10.1042/bj3100699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 34.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 35.MacDonald JIS, Kent C. Prot Expr Purif. 1993;4:1–7. doi: 10.1006/prep.1993.1001. [DOI] [PubMed] [Google Scholar]
- 36.Arnold RS, DePaoli-Roach AA, Cornell RB. Biochemistry. 1997;36:6149–6156. doi: 10.1021/bi970023z. [DOI] [PubMed] [Google Scholar]
- 37.Johnson JE, Aebersold R, Cornell RB. Biochim Biophys Acta. 1997;1324:273–284. doi: 10.1016/s0005-2736(96)00233-7. [DOI] [PubMed] [Google Scholar]
- 38.Johnson JE, Rao M, Hui SK, Cornell RB. Biochemistry. 1998;37:9509–9519. doi: 10.1021/bi980340l. [DOI] [PubMed] [Google Scholar]